
Abstract
Mutations involving epigenetic regulators (TET2~60% and ASXL1~40%) and splicing components (SRSF2~50%) are frequent in chronic myelomonocytic leukemia (CMML). On a 27-gene targeted capture panel performed on 175 CMML patients (66% males, median age 70 years), common mutations included: TET2 46%, ASXL1 47%, SRSF2 45% and SETBP1 19%. A total of 172 (98%) patients had at least one mutation, 21 (12%) had 2, 24 (14%) had 3 and 30 (17%) had >3 mutations. In a univariate analysis, the presence of ASXL1 mutations (P=0.02) and the absence of TET2 mutations (P=0.03), adversely impacted survival; while the number of concurrent mutations had no impact (P=0.3). In a multivariable analysis that included hemoglobin, platelet count, absolute monocyte count and circulating immature myeloid cells (Mayo model), the presence of ASXL1 mutations (P=0.01) and absence of TET2 mutations (P=0.003) retained prognostic significance. Patients were stratified into four categories: ASXL1wt/TET2wt (n=56), ASXL1mut/TET2wt (n=31), ASXL1mut/TET2mut (n=50) and ASXL1wt/TET2mut (n=38). Survival data demonstrated a significant difference in favor of ASXL1wt/TET2mut (38 months; P=0.016), compared with those with ASXL1wt/TET2wt (19 months), ASXL1mut/TET2wt (21 months) and ASXL1mut/TET2mut (16 months) (P=0.3). We confirm the negative prognostic impact imparted by ASXL1 mutations and suggest a favorable impact from TET2 mutations in the absence of ASXL1 mutations.
Similar content being viewed by others

Introduction
Gene mutations are common (>90%) in chronic myelomonocytic leukemia (CMML) and involve epigenetic regulators (TET2~60% and ASXL1~40%), spliceosome components (SRSF2~50%) and cell signaling (RAS~30% and CBL~15%).1, 2, 3, 4 Mutations involving ASXL1, TET2, RUNX1, CBL, SRSF2, RAS and IDH2 have demonstrated prognostic relevance on univariate survival analyses.1, 5, 6 However, on multivariable analyses that have included additional CMML relevant factors, only ASXL1 mutations (frameshift and nonsense) have been shown to be prognostically detrimental.1, 2 This has led to the incorporation of ASXL1 mutations into molecular prognostic models such as the Molecular Mayo Model and the Groupe Francais des Myelodysplasies model.1, 2
TET2 mutations (chromosome 4q24) are frequent and are thought to be the driver mutations in CMML.7 TET2 catalyzes the conversion of 5-methyl-cytosine to 5-hydroxymethyl-cytosine, regulating methylation and transcription.8 The prognostic relevance of TET2 mutations remains unclear with some studies demonstrating favorable,9 unfavorable10 and no impact1 on overall survival (OS). In vitro studies have shown that ASXL1 mutations enhance the de-ubiquitinase activity of the ASXL1–BAP1 (BRCA associated protein 1) complex, which then cooperates with loss of TET2 to skew towards myeloid development.11 However, the mechanisms behind this effect and the prognostic interplay between TET2 and ASXL1 mutations remain unknown.
In the current study, we used a 27-gene panel assay to: (i) identify additional prognostically-relevant mutations in CMML, (ii) to determine if the number of mutations carries prognostic relevance and (iii) to study the prognostic interplay between TET2 and ASXL1 mutations.
Materials and methods
One-hundred and seventy five patients with CMML were included in the study. All patients had bone marrow biopsies and cytogenetic studies performed at diagnosis. The diagnosis of CMML, including subclassification into CMML-1 or CMML-2, and leukemic transformation were according to the 2008 World Health Organization criteria.12 Risk stratification was per the Mayo-French cytogenetic system,13 the Mayo model,14 the Groupe Francais des Myelodysplasies model1 and the Molecular Mayo model.2 Twenty-seven gene panel targeted capture assays were carried out on bone marrow DNA specimens obtained at diagnosis for the following genes: TET2, DNMT3A, IDH1, IDH2, ASXL1, EZH2, SUZ12, SRSF2, SF3B1, ZRSR2, U2AF1, PTPN11, Tp53, SH2B3, RUNX1, CBL, NRAS, JAK2, CSF3R, FLT3, KIT, CALR, MPL, NPM1, CEBPA, IKZF and SETBP1.
Paired-end indexed libraries were prepared from individual patient DNA in the Mayo Clinic Genomic Sequencing Core Laboratory using the NEBNext Ultra Library prep protocol on the Agilent Bravo liquid handler (NEB, Ipswich, MA, USA/Agilent Technologies Inc., Santa Clara, CA, USA). Capture libraries were assembled according to the Nimblegen standard library protocol (Roche Nimblegen, Inc., Basel, Switzerland). A panel including the regions of 27 heme-related genes was selected for custom target capture using the Agilent SureSelect Target Enrichment Kit (Agilent Technologies Inc, Santa Clara, CA, USA). Capture libraries were pooled at equimolar concentrations and loaded onto paired end flow cells at concentrations of 7–8 pM to generate cluster densities of 600 000–800 000/mm2 following Illumina’s standard protocol using the Illumina cBot and HiSeq Paired end cluster kit version 3, in batches of 48 samples per lane (Illumina Incorporated, San Diego, CA, USA). The flow cells were sequenced as 101 × 2 paired end reads on an Illumina HiSeq 2000 using TruSeq SBS sequencing kit version 3 (Illumina Incorporated) and HiSeq data collection version 2.0.12.0 software (Illumina Incorporated). Base-calling was performed using Illumina’s RTA version 1.17.21.3 (Illumina Incorporated).
Genesifter software was utilized (PerkinElmer, Danvers, MA, USA) to analyze targeted sequence data. Reads from the sequencing in fastq format were aligned using the Burrows-Wheeler Aligner against the genomic reference sequence for Homo sapiens (Build 37.2; NCBI http://www.ncbi.nlm.nih.gov/). An additional alignment, post-processing set of tools were then used to do local realignment, duplicate marking and score recalibration to generate a final genomic aligned set of reads. Nucleotide variants were called using the Genome Analysis Toolkit (GATK -Broad Institute, Cambridge, MA, USA) that identified single nucleotide and small insertion/deletion events using default settings. Specific variants were deemed as mutations if they were associated with a heme malignancy (as identified by COSMIC database), or if they have not been associated with a single nucleotide polymorphism database.
Based on prior observations, only frame shift and nonsense ASXL1 mutations were considered pathogenic.2, 14 For TET2, frame shift, nonsense, missense, insertions and deletions were considered pathogenic. Previously annotated single nucleotide polymorphisms (http://www.hapmap.org) in all the aforementioned genes were considered nonpathogenic.
All statistical analyses considered parameters obtained at time of referral to the Mayo Clinic, which in most instances coincided with time of bone marrow biopsy. Differences in the distribution of continuous variables between categories were analyzed by either Mann–Whitney (for comparison of two groups) or Kruskal–Wallis (comparison of three or more groups) test. Patient groups with nominal variables were compared by the chi-square test. Overall survival was calculated from the date of first referral to date of death (uncensored) or last contact (censored). Leukemia-free survival (LFS) was calculated from the date of first referral to date of leukemic transformation (uncensored) or death/last contact (censored). Overall and LFS curves were prepared by the Kaplan–Meier method and compared by the log-rank test. Cox proportional hazard regression model was used for multivariable analysis. P <0.05 were considered significant. The Stat View (SAS Institute, Cary, NC, USA) statistical package was used for all calculations.
Results
Among the 175 study patients, 115 (66%) were males with a median age of 70 years (range, 18–90). One hundred and forty-six (83%) patients were subclassified as CMML-1 and the remainder had CMML-2. At a median follow-up of 23 months, 146 (83%) deaths and 25 (14%) leukemic transformations were documented. Median survivals were 24 months for CMML-1 and 16 months for CMML-2 (P=0.38). Cytogenetic risk stratification was carried out using the Mayo-French cytogenetic model,13 with the following distribution: 118 (78%) low, 21 (10%) intermediate and 18 (12%) high risk. Overall risk stratification was based on Mayo prognostic model:14 25% high, 32% intermediate and 43% low risk; Molecular Mayo Model:2 30% high, 30% intermediate-2, 31% intermediate-1 and 9% low risk; and the Groupe Francais des Myelodysplasies model:1 19% high, 37% intermediate and 44% low risk. Baseline laboratory values and risk stratification are detailed in Table 1.
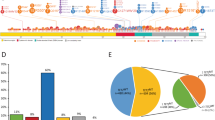
Mutational frequencies were as follows: TET2 46%, ASXL1 47%, SRSF2 45%, SETBP1 19%, CBL 14%, RUNX1 14%, NRAS 12%, U2AF1 8%, SF3B1 6%, ZRSR2 6%, Tp53 5%, DNMT3A 5%, IDH2 5%, PTPN11 5%, SH2B3 5%, JAK2V617F 4%, NPM1 3%, CSF3R 2%, IDH1 2%, EZH2 1%, SUZ12 1%, KIT 1%, FLT3 1% and CALR 1% (Figure 1 and Table 1). No mutations were detected in MPL or IKZF. One hundred and seventy two patients (98%) had at least one mutation, 21 (12%) had 2, 24 (14%) had 3, 20 (11%) had 4, 9 (5%) had 5; while one (1%) patient had 6 concurrent mutations (Figure 1).

Spectrum and frequency of gene mutations in 175 Mayo clinic patients with WHO defined chronic myelomonocytic leukemia.
In a univariate survival analysis that included the aforementioned mutations, only the presence of ASXL1 mutations (P=0.01), absence of TET2 mutations (P=0.005) and presence of DNMT3A mutations (P=0.02) were associated with inferior survival. The number of concurrent mutations per patient did not affect outcome (P=0.3). In a multivariable analysis, the presence of ASXL1 (P=0.01) and the absence of TET2 (P=0.03) mutations retained their negative prognostic impact. In order to determine the prognostic interaction between these two mutations, patients were stratified into four mutational categories: ASXL1wt/TET2wt (n=56), ASXL1mut/TET2wt (n=31), ASXL1mut/TET2mut (n=50) and ASXL1wt/TET2mut (n=38). Survival data in these four groups showed significant difference in favor of ASXL1wt/TET2mut (median survival 38 months; P=0.016), compared with those with ASXL1wt/TET2wt (19 months), ASXL1mut/TET2wt (21 months) and ASXL1mut/TET2mut (16 months); there was no significant difference in survival among the latter three groups (P=0.3) (Figure 2).

Survival data for 175 patients with chronic myelomonocytic leukemia stratified by ASXL1 and TET2 mutational status.
In multivariable analysis, presence of ASXL1 (P=0.01) and absence of TET2 mutations (P=0.003) remained significant when risk factors used in the Mayo prognostic model (hemoglobin <10 gm/dl, absolute monocyte count >10x10(9)/L, platelet count <100x10(9)/L, presence of circulating immature myeloid cells) were added to the model;14 the same was true for ASXL1wt/TET2mut (P=0.036). In a separate multivariable analysis that included the Mayo prognostic model as a single variable along with presence of ASXL1 and absence of TET2 mutations or absence of ASXL1wt/TET2mut mutational status, the respective hazard ratios were 1.4 (95% CI 1.07–2.1; P=0.012), 1.5 (95% CI 1.07–2.1; P=0.03) and 1.8 (95% CI 1.2–2.7; P=0.001). On a univariate analysis, LFS was worse in ZRSR2-mutated cases (P=0.03). This relevance, however, was lost on a multivariable analysis that included circulating blasts (P=0.01) and high risk karyotype (P=0.03).
Discussion
Clonal cytogenetic abnormalities are seen in ~30%,13, 15 while gene mutations are seen in >90% of patients with CMML.1, 2, 16 These mutations can broadly be classified into the following categories: (i) mutations involving epigenetic regulator genes: TET2 (~60%), DNMT3A, IDH1, and IDH2 (IDH mutations <10%); (ii) mutations involving histone modification and chromatin regulation: ASXL1 (~40%) and EZH2 (<5%); (iii) mutations involving the splicing machinery: SF3B1, SRSF2 (~50%), U2AF1 and ZRSR2; (iv) mutations involving DNA damage response genes: Tp53 (~1%) and PHF6; (v) mutations in transcription factors and signal transduction pathways: JAK2, KRAS, NRAS (RAS~30%), CBL (~10–15%), FLT3, RUNX1(~15%) and mutations such as SETBP1 (~15%).1, 2, 16, 17, 18, 19 Of these, mutations involving TET2 (~60%), SRSF2 (~50%), ASXL1 (~40%) and the RAS pathway (~30%) are most frequent, with only frameshift and nonsense ASXL1 mutations independently impacting OS.1, 2
The ASXL1 (additional sex combs like 1) gene (chromosome 20q11) regulates chromatin by interacting with the polycomb-group repressive complex proteins (PRC1 and PRC2).20 Histone 2A lysine 119 (H2AK119Ub) and H3K27me3 play synergistic roles in PRC-mediated gene repression.11, 21 Abdel-Wahab et al.21 demonstrated that ASXL1 mutations resulted in loss of PRC2-mediated H3K27 tri-methylation, while Balasubramani et al.11 demonstrated that ASXL1 truncations conferred enhanced activity on the ASXL1–BAP1 complex. This complex results in global erasure of H2AK119Ub and depletes H327Kme3, promoting dysregulated transcription. The current study once again demonstrates the frequent occurrence of ASXL1 mutations (45%) in CMML and confirms the adverse prognostic impact imparted by frameshift and nonsense mutations on OS.
TET2 (ten-eleven translocation (TET) oncogene family member 2) is a member of the TET family of proteins.22 Although TET2 mutations are widely prevalent in CMML, thus far, they have not been shown to independently impact either OS or LFS.1 In the current study, TET2 mutations were seen in 46% of CMML patients and the absence of TET2 mutations negatively impacted OS. Additionally, the presence of clonal TET2 mutations, in the absence of clonal ASXL1 mutations (ASXL1wt/TET2mut), had a favorable impact on OS. The mechanism behind this association is unclear. In MDS and younger patients with CMML (age <65 years), the presence of clonal TET2 mutations, in the absence of clonal ASXL1 mutations, have been associated with response to hypomethylating agents (5-azacitidine and decitabine).5, 23 Treatment data on our cohort of patients were incomplete and it is currently unknown as to whether this favorable impact was an effect of better responses to hypomethylating agents or not.
Approximately, 80% of patients with MDS have one or more oncogenic driver mutations (SF3B1~24%, TET2~22%, SRSF2~15% and ASXL1~15%).4 In a large study (n=738), Papaemmanuil et al.4 demonstrated that driver mutations had an equivalent prognostic significance and LFS steadily declined as the number of driver mutations increased. 78% had at least one oncogenic mutation, while 43% had 2 or 3 and 10% had 4–8 mutations. Variants of unclear significance in oncogenic genes such as ASXL1 also adversely impacted outcomes. In the current study, 98% of the CMML patients had at least one mutation, 12% had 2, 14% had 3 and 17% had >3 mutations. The number of oncogenic mutations in CMML did not impact either the LFS or OS.
In summary, nearly all patients with CMML express one or more myeloid neoplasm-relevant mutations. Similar to prior studies, the three most frequent mutations include TET2, ASXL1 and SRSF2.1, 2 Unlike in MDS, survival outcomes in CMML were not affected by the number of concurrent driver mutations. We confirm the negative prognostic impact on OS imparted by ASXL1 mutations1, 2 and also suggest a favorable prognostic impact from TET2 mutations, unless accompanied by ASXL1 mutations. These findings need validation in a larger data set.
References
Itzykson R, Kosmider O, Renneville A, Gelsi-Boyer V, Meggendorfer M, Morabito M et al. Prognostic score including gene mutations in chronic myelomonocytic leukemia. J Clin Oncol 2013; 31: 2428–2436.
Patnaik MM, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA et al. ASXL1 and SETBP1 mutations and their prognostic contribution in chronic myelomonocytic leukemia: a two-center study of 466 patients. Leukemia 2014; 28: 2206–2212.
Patnaik MM, Parikh SA, Hanson CA, Tefferi A . Chronic myelomonocytic leukaemia: a concise clinical and pathophysiological review. Br J Haematol 2014; 165: 273–286.
Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013; 122: 3616–3627, quiz 3699.
Patnaik MM, Wassie EA, Padron E, Onida F, Itzykson R, Lasho TL et al. Chronic myelomonocytic leukemia in younger patients: molecular and cytogenetic predictors of survival and treatment outcome. Blood Cancer J 2015; 5: e280.
Tefferi A, Lim KH, Abdel-Wahab O, Lasho TL, Patel J, Patnaik MM et al. Detection of mutant TET2 in myeloid malignancies other than myeloproliferative neoplasms: CMML, MDS, MDS/MPN and AML. Leukemia 2009; 23: 1343–1345.
Itzykson R, Kosmider O, Renneville A, Morabito M, Preudhomme C, Berthon C et al. Clonal architecture of chronic myelomonocytic leukemias. Blood 2013; 121: 2186–2198.
Abdel-Wahab O, Levine RL . Mutations in epigenetic modifiers in the pathogenesis and therapy of acute myeloid leukemia. Blood 2013; 121: 3563–3572.
Kohlmann A, Grossmann V, Klein HU, Schindela S, Weiss T, Kazak B et al. Next-generation sequencing technology reveals a characteristic pattern of molecular mutations in 72.8% of chronic myelomonocytic leukemia by detecting frequent alterations in TET2, CBL, RAS, and RUNX1. J Clin Oncol 2010; 28: 3858–3865.
Kosmider O, Gelsi-Boyer V, Ciudad M, Racoeur C, Jooste V, Vey N et al. TET2 gene mutation is a frequent and adverse event in chronic myelomonocytic leukemia. Haematologica 2009; 94: 1676–1681.
Balasubramani A, Larjo A, Bassein JA, Chang X, Hastie RB, Togher SM et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat Commun 2015; 6: 7307.
Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 2009; 114: 937–951.
Wassie EA, Itzykson R, Lasho TL, Kosmider O, Finke CM, Hanson CA et al. Molecular and prognostic correlates of cytogenetic abnormalities in chronic myelomonocytic leukemia: a Mayo Clinic-French Consortium Study. Am J Hematol 2014; 89: 1111–1115.
Patnaik MM, Padron E, LaBorde RR, Lasho TL, Finke CM, Hanson CA et al. Mayo prognostic model for WHO-defined chronic myelomonocytic leukemia: ASXL1 and spliceosome component mutations and outcomes. Leukemia 2013; 27: 1504–1510.
Such E, Cervera J, Costa D, Sole F, Vallespi T, Luno E et al. Cytogenetic risk stratification in chronic myelomonocytic leukemia. Haematologica 2011; 96: 375–383.
McCullough KB, Patnaik MM . Chronic Myelomonocytic Leukemia: a Genetic and Clinical Update. Curr Hematol Malig Rep 2015; 10: 292–302.
Meggendorfer M, Roller A, Haferlach T, Eder C, Dicker F, Grossmann V et al. SRSF2 mutations in 275 cases with chronic myelomonocytic leukemia (CMML). Blood 2012; 120: 3080–3088.
Laborde RR, Patnaik MM, Lasho TL, Finke CM, Hanson CA, Knudson RA et al. SETBP1 mutations in 415 patients with primary myelofibrosis or chronic myelomonocytic leukemia: independent prognostic impact in CMML. Leukemia 2013; 27: 2100–2102.
Gelsi-Boyer V, Trouplin V, Adelaide J, Bonansea J, Cervera N, Carbuccia N et al. Mutations of polycomb-associated gene ASXL1 in myelodysplastic syndromes and chronic myelomonocytic leukaemia. Br J Haematol 2009; 145: 788–800.
Abdel-Wahab O, Pardanani A, Patel J, Wadleigh M, Lasho T, Heguy A et al. Concomitant analysis of EZH2 and ASXL1 mutations in myelofibrosis, chronic myelomonocytic leukemia and blast-phase myeloproliferative neoplasms. Leukemia 2011; 25: 1200–1202.
Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell 2012; 22: 180–193.
Yamazaki J, Taby R, Vasanthakumar A, Macrae T, Ostler KR, Shen L et al. Effects of TET2 mutations on DNA methylation in chronic myelomonocytic leukemia. Epigenetics 2012; 7: 201–207.
Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood 2014; 124: 2705–2712.
Acknowledgements
Current study is supported in part by grants from the ‘The Henry J Predolin Foundation for Research in Leukemia, Mayo Clinic, Rochester, MN, USA’. This publication was supported by CTSA Grant Number KL2 TR000136 from the National Center for Advancing Translational Science (NCATS). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Patnaik, M., Lasho, T., Vijayvargiya, P. et al. Prognostic interaction between ASXL1 and TET2 mutations in chronic myelomonocytic leukemia. Blood Cancer Journal 6, e385 (2016). https://doi.org/10.1038/bcj.2015.113
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bcj.2015.113
This article is cited by
-
Novel scheme for defining the clinical implications of TP53 mutations in myeloid neoplasia
Journal of Hematology & Oncology (2023)
-
Epigenetic regulation in hematopoiesis and its implications in the targeted therapy of hematologic malignancies
Signal Transduction and Targeted Therapy (2023)
-
Mutational landscape of chronic myelomonocytic leukemia and its potential clinical significance
International Journal of Hematology (2022)
-
Myelodysplastic/myeloproliferative neoplasms-unclassifiable with isolated isochromosome 17q represents a distinct clinico-biologic subset: a multi-institutional collaborative study from the Bone Marrow Pathology Group
Modern Pathology (2022)
-
CSF3R T618I mutant chronic myelomonocytic leukemia (CMML) defines a proliferative CMML subtype enriched in ASXL1 mutations with adverse outcomes
Blood Cancer Journal (2021)
