
Abstract
Background:
Tumours are responsive to temozolomide (TMZ) if they are deficient in O6-methylguanine-DNA methyltransferase (MGMT), and mismatch repair (MMR) proficient.
Methods:
The effect of TMZ on medulloblastoma (MB) cell killing was analysed with clonogenic survival assays. Expression of DNA repair genes and enzymes was investigated using microarrays, western blot, and immunohistochemistry. DNA sequencing and promoter methylation analysis were employed to investigate the cause of loss of the expression of MMR gene MLH1.
Results:
Temozolomide exhibited potent cytotoxic activity in D425Med (MGMT deficient, MLH1 proficient; IC50=1.7 μ M), moderate activity against D341Med (MGMT proficient, MLH1 deficient), and DAOY MB cells (MGMT proficient, MLH1 proficient). MGMT inhibitor O6-benzylguanine sensitised DAOY, but not D341Med cells to TMZ. Of 12 MB cell lines, D341Med, D283Med, and 1580WÜ cells exhibited MMR deficiency due to MLH1 promoter hypermethylation. DNA sequencing of these cells provided no evidence for somatic genetic alterations in MLH1. Expression analyses of MMR and MGMT in MB revealed that all patient specimens (n=74; expression array, n=61; immunostaining, n=13) are most likely MMR proficient, whereas some tumours had low MGMT expression levels (according to expression array) or were totally MGMT deficient (3 out of 13 according to immunohistochemistry).
Conclusion:
A subset of MB may respond to TMZ as some patient specimens are MGMT deficient, and tumours appear to be MMR proficient.
Similar content being viewed by others


Main
Medulloblastoma (MB) is the most common malignant paediatric brain tumour. Standard treatment includes surgery, chemotherapy, and irradiation (for older children and adolescents). The anti-cancer drug that has shown promise in the treatment of recurrent paediatric brain tumours is the methylating agent temozolomide (TMZ). Already a part of standard treatment of glioblastomas (Stupp et al, 2005), this orally administered drug causes only mild and tolerable side-effects (Broniscer et al, 2007; Nicholson et al, 2007). In an ongoing clinical trial for recurrent paediatric brain tumours (Phase II), the efficacy of TMZ (alone) is being tested against that of intravenous carboplatin/etoposide (HIT-REZ 2005; http://clinicaltrials.gov/ct2/show/NCT00749723). Another clinical trial compares the effectiveness of the well-tested combination of TMZ and irinotecan hydrochloride with or without bevacizumab (http://clinicaltrials.gov/ct2/show/NCT01217437) in recurrent MB.
TMZ spontaneously hydrolyses to MTIC (3-methyl-(triazen-1-yl)imidazole-4 carboxamide), which then degrades into the DNA-reactive methyldiazonium cation that forms methyl adducts at O-6 (G), N-7 (G), N-1 (A), and N-3 (A) positions (Marchesi et al, 2007). The O6-methylguanine (O6-meG)/cytosine (O6-meG:C) pair can result into the O6-meG:T mismatch in the progeny DNA after a first round of replication. The mismatch repair (MMR) system, a protein complex including MSH2, MSH6, MLH1, and PMS2 proteins attempts to repair the O6-meG:T mismatch by removing a patch of the newly synthesised strand containing thymine. However, if O6-meG remains in the template strand, thymine could still be misincorporated opposite O6-meG and a futile cycle of repair by the MMR system takes place during this S phase of the cell cycle. This process triggers collapsed replication fork and subsequent DNA double-strand break that then leads to G2/M arrest (Drablos et al, 2004). Another DNA repair protein that modulates TMZ activity is MGMT (O6-methylguanine-DNA methyltransferase), which can directly excise the methyl lesion at the O6 position of guanine (Pegg, 1990; Gerson, 2002). In theory, a defective MMR and/or presence of expression of MGMT can lead to the tumour’s tolerance to TMZ. Previous studies demonstrated that increased expression (Dunn et al, 2009; Karayan-Tapon et al, 2010) or reduced promoter methylation (Dunn et al, 2009; Karayan-Tapon et al, 2010) (which also increases expression level) of MGMT correlated with lower overall survival of adult glioblastoma (GBM) patients whose treatment regimen included TMZ. Cahill et al (2007) reported that among radiotherapy/TMZ-treated GBM patients, a subset of recurrent tumours had lost the expression of the MMR protein MSH6. As clinical studies involving the use of TMZ in paediatric MB have started only fairly recently, the association between MGMT/MMR status and clinical response and outcome among TMZ-treated MB patients are yet to be reported. In this study, we aimed to investigate the impact of MGMT/MMR status on the response of MB cell lines to TMZ. We then proceeded to analyse the expression of MMR and MGMT genes and proteins by expression array (61 MB and 12 MB cell lines) and immunostaining (13 MB) to examine the potential clinical relevance of the MMR system and MGMT in MB. The underlying cause for MMR deficiency in MB cells was further explored by sequencing and methylation analysis of the MLH1 gene.
Material and methods
Cell lines and culture conditions
We used the MB cell lines DAOY (from a 4-year-old male) (Jacobsen et al, 1985), D341Med (from a 3.5-year-old male) (Friedman et al, 1988), D425Med (from a 5-year-old male) (Bigner et al, 1990), D556Med (from a 7-year-old female) (Aldosari et al, 2002), D283Med (from ascitic fluid of a 6-year-old child with metastatic MB) (Friedman et al, 1985), MHH-MED-1 (from the cerebrospinal fluid of a 10-year-old male) (Pietsch et al, 1994), MHH-MED-3 (from a 3-year-old female) (Pietsch et al, 1994), MHH-MED-4 (from a 4-year-old male) (Pietsch et al, 1994), and UW228-2 (from a 9-year-old female) (Keles et al, 1995). Cell lines were cultured as previously described (Hartmann et al, 2005; Faoro et al, 2011). Three cell lines (1580WÜ, 5-year-old female; MEB-MED-8S, 1-year-old male; and MEB-MED-8A, 1-year-old male) were established by one of the authors (TP) and were used as described previously (Hartmann et al, 2005).
Tumour samples
Two MB series were analysed: (a) 61 primary MB samples whose expression array data (Affymetrix HG U133 Plus 2.0 arrays, Santa Barbara, CA, USA) (Kool et al, 2008, 2012) were reanalysed for the present study, and (b) 13 archival MB tissue samples from patients diagnosed at the University Medical Center Hamburg-Eppendorf, Germany. All these archival tissue samples were reviewed by an experienced neuropathologist (CH) and the diagnosis of MB was confirmed in all. Written informed consent was obtained from all patients and/or from the parents of all patients. All cases were made anonymous before the investigation.
Drugs
Temozolomide (Schering-Plough; Kenilworth, NJ, USA) and O6-benzylguanine (O6-BG) (Sigma Aldrich, Basel, Switzerland) were used as described elsewhere (Faoro et al, 2011).
Western blot analysis
MB cells were incubated with lysis buffer (50 mM Tris HCl, pH 7.8, 0.15 M NaCl, 1% NP-40, Roche (Basel, Switzerland) complete mini EDTA-free protease-inhibitor cocktail) on ice for 30 min. Lysis was checked under the microscope and the supernatant was isolated after centrifugation. Eighty microgram of protein extracts were mixed with an equal volume of 2 × SDS loading buffer (20% glycerol, 4% SDS, 0.2 M DTT, 0.01% bromophenol blue, 0.125 M Tris HCl, pH 6.8), electrophoresed in 12% Tris-Glycine SDS polyacrylamide gel, and transferred onto Biotrace PVDF membranes (Pall Co., Ann Arbor, MI, USA). The subsequent steps for western blotting were performed using the WesternBreeze Chemiluminescent Western Blot Immunodetection kit (Invitrogen, Carlsbad, CA, USA), with the alkaline phosphatase-conjugated anti-mouse secondary antibody and CDP-Star Chemiluminescent substrate. Mouse monoclonal antibodies used were specific for MLH1 (Oncogene, Cambridge, MA, USA), MSH2 (Oncogene), MSH6 (BD Biosciences, San Jose, CA, USA), PMS2 (Oncogene), and βIII tubulin (Millipore, Billerica, MA, USA). The expression of MGMT and β-actin were analysed by western blot analysis as described previously (Faoro et al, 2011).
Clonogenic survival assay
To assess clonogenic survival, the number of seeded cells was adjusted to obtain around 100 colonies per cell culture dish for a given treatment (von Bueren et al, 2009). Where required, the specific MGMT inhibitor O6-BG at a concentration of 25 μ M was added to the cell cultures 2 h before the addition of various concentrations of TMZ. The cultures were then maintained at 37°C in a humidified atmosphere containing 5% CO2, and allowed to grow for 9 (DAOY), 14 (D341Med), or 12 (D425Med) days, respectively. This was followed by the fixation of the cells in methanol/acetic acid (75% : 25%) and the staining with Giemsa dye. Colonies with more than 50 cells were counted. Clonogenic assays were repeated three times as independent experiments using triplicate cultures. The relative colony formation (percentage of clonogenic survival) was plotted against the drug concentrations.
MGMT and MLH1 sequencing
Genomic DNA was extracted from DAOY, D341Med, D283Med, and 1580WÜ cells using standard procedures. The DNA fragments encompassing four exons and adjacent intronic sequences of MGMT (OMIM 156569) gene were PCR amplified and directly sequenced using BigDye Terminator v1.1 Cycle Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and the ABI PRISM 3100 genetic analyser. For DAOY, D341Med, D283Med, and 1580WÜ, the genomic DNA regions covering 19 exons of MLH1 (OMIM 120436; GenBank accession number NM_0000249) were also PCR amplified and sequenced as described recently (Buerki et al, 2012).
MLH1 promoter hypermethylation analysis
The promoter methylation status of MLH1 in DAOY, D341Med, D283Med, and 1580WÜ cells was evaluated using the SALSA ME011-B1 MS-MLPA (methylation-specific, multiplex-ligation-dependent probe amplification) MMR Probemix Kit (MRC Holland, Amsterdam, The Netherlands) (Buerki et al, 2012). The kit included five MLH1-specific probes: MLH1 1 (237 bp; −659 bp distance to ATG start); MLH1 2 (265 bp; −383 bp distance to ATG start); MLH1 3 (189 bp; −246 bp distance to ATG start); MLH1 4 (166 bp; −13 bp distance to ATG start); and MLH1 5 (292 bp; +208 bp distance to ATG start) (Nygren et al, 2005; Gylling et al, 2009). All reactions were carried out according to the manufacturer’s protocol and fragment analysis was performed on an ABI PRISM 310 Genetic analyser (Buerki et al, 2012). Analyses were performed using three independent experiments.
Gene expression by microarray analysis
The gene expression data of MB tumours (n=61), which are available from NCBI’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/; accession number GSE10327) and published by Kool et al (2012), were reanalysed, whereas those of the 12 MB cell lines (D341Med, D425Med, D283Med, DAOY, MHH-MED-1, D556Med, 1580WÜ, MHH-MED-3, MEB-MED-8S, MHH-MED-4, MEB-MED-8A, and UW228-2) were newly generated. In both series, Affymetrix HG U133 Plus 2.0 arrays (Affymetrix) were used. Data analysis was performed using the R2 microarray analysis and visualisation platform (http://r2.amc.nl).
Immunohistochemistry
Thirteen additional MB specimens were examined immunohistochemically for MMR and MGMT protein expression using standard immunohistochemical techniques (Truninger et al, 2005; Menigatti et al, 2009). In brief, tumours were fixed in buffered formalin and embedded in paraffin. Four micrometre thick sections were mounted on glass slides coated with organosilane (DakoCytomation, Hamburg, Germany), deparaffinised, and rehydrated. Antigen retrieval was accomplished by heating the sections in a pressure cooker at 120°C for 2 min in 10 mM citrate-buffered solution (pH 6.0). DAKO (Hamburg, Germany) peroxidase blocking reagent and goat serum were sequentially used to suppress non-specific staining due to endogenous peroxidase activity and unspecific binding of antibodies, respectively. Incubations with primary monoclonal antibodies were performed as follows: anti-MSH2: 24 h at 4°C with Ab NA27 (Calbiochem, Darmstadt, Germany), 1 : 50; anti-MSH6: 24 h at 4°C with Ab 610919 (BD Biosciences), 1 : 65; anti-MLH1: 24 h at 4°C with Ab 551091 (BD Biosciences), 1 : 200; anti-PMS2: 24 h at 4°C with Ab 556415 (BD Biosciences), 1 : 85; and anti-MGMT: 24 h at 4°C with Ab MS-470-P1 (NeoMarker, Kalamazoo, MI, USA), 1 : 170. After washing, anti-mouse secondary antibodies conjugated to peroxidase labelled polymer (DAKO EnVision+ kit) were applied for 30 min at RT, and the peroxidase activity was developed by incubation with 3,3′diaminobenzidine chromogen solution (DAKO). The sections were then counterstained with hematoxylin. Immunoreactivity of the tissue was verified by labelling with antibodies against the Ki-67 antigen (anti-Ki-67: 24 h at 4°C with Ab M 7240, DAKO, 1 : 125). Slides were evaluated qualitatively for nuclear staining of tumour cells.
Statistical analysis
Whenever possible, data points are reported as mean±standard deviation (s.d.). (GraphPad Software, San Diego, CA, USA) software was used to calculate IC50 values and their 95% confidence intervals, and to statistically compare the fitted midpoints (log IC50) of the two curves (von Bueren et al, 2011).
Results
Temozolomide sensitivity depends on both the MMR status and on its MGMT expression
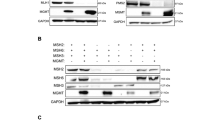
We investigated the effect of TMZ on the clonogenic survival with and without O6-BG pretreatment in DAOY, D425Med, and D341Med cells. Results from clonogenic survival assay (CFA) indicated that D425Med cells were highly sensitive to TMZ (Figure 1). The TMZ IC50 values and their 95% confidence intervals for D425Med (IC50: 1.705 μ M), DAOY (IC50: 383.8 μ M), and D341Med cells (IC50: 178.2 μ M) are shown in Figure 1. We have previously shown that the MGMT gene in D425Med cells is epigenetically silenced (Faoro et al, 2011), explaining the cell line’s sensitivity to TMZ. On the other hand, DAOY and D341Med cells expressed MGMT (Faoro et al, 2011), consistent with their tolerance to TMZ. Upon addition of the MGMT inhibitor O6-BG, the depletion of MGMT protein expression was evident for both DAOY and D341Med cells (data not shown). However, as indicated in Figure 1, O6-BG addition was able to potentiate the cytotoxic activity of TMZ in DAOY (TMZ IC50 value of 151.9 μ M, significantly lower than the basal IC50 of 383.8 μ M; P<0.001), the most TMZ-resistant MB cell line analysed in our study. In contrast, no significant increased cytotoxic activity of TMZ was evident in D425Med and D341Med cells upon O6-BG addition. This observation was expected for the MGMT-deficient D425Med cells. For the MGMT-proficient D341Med cells, these results were quite surprising. We first considered the possibility that the D341Med cells used in these experiments carried a mutated yet immuno-detectable MGMT protein, that is, a mutant protein resistant to inhibition by O6-BG, such as those variants previously reported in BCNU/O6-BG-resistant MB sublines (Bacolod et al, 2002, 2004). Sequencing did not reveal any evidence for somatic genetic alterations in MGMT, when assessing D341Med, DAOY and D425Med cells. However, when we analysed the MMR status by western blot (Figure 2), losses of MLH1 and PMS2 expression were found in D341Med cells, while all the MMR proteins investigated were normally expressed in DAOY.

Effect of temozolomide alone (straight lines) or temozolomide with a 2-h pretreatment with O6-BG (O6-benzylguanine; dashed lines) on the clonogenic survival of the medulloblastoma cell lines DAOY (A), D341Med (B), and D425Med (C). Single cells were seeded into culture dishes and treated with the denoted concentrations of temozolomide in absence or presence of O6-BG and then allowed to further grow for 9 (DAOY), 14 (D341Med), or 12 (D425Med) days, respectively. Data are presented as the mean±standard deviation (s.d.) of three independent experiments performed in triplicates. The IC50 values and their 95% confidence intervals were calculated from the regression curve and are indicated for each data set for temozolomide alone and temozolomide with a 2-h pretreatment with O6-BG for DAOY, D341Med, and D425Med cells. Differences between the two curves are represented by P-values (P<0.001, P<0.01, P<0.05; NS, not significant).

Analysis of protein expression of mismatch repair polypeptides MSH6, MSH2, PMS2, and MLH1 in D341Med, and DAOY cells by western blotting (βIII Tubulin expression used as loading control). Positive controls were protein extracts from HeLa cells. Representative blot (two independent experiments) is shown.
MB tumours are MMR proficient, some MB cells are MMR deficient, and the majority of MB tumours and MB cells are MGMT proficient
The loss of MMR function in one of three MB cell lines, prompted us to investigate the frequency of MMR deficiency in MB tumours. We analysed the expression levels of the four MMR genes among 61 primary MB tumours. The clinical, histological, and molecular subgroup (WNT, SHH, group 3 (GRP3) and group 4) characteristics of the 61 patients are described elsewhere (Kool et al, 2012). Included as reference were the expression data of nine normal cerebellum (CB; without information about age) samples which were assessed also by Affymetrix HG U133 Plus 2.0 array by Roth et al (2006), and data were available from NCBI’s Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/; accession number GSE3526). As shown in Figure 3 the expression levels of MLH1, MSH2, and MSH6 across 61 tumour samples (and all four molecular subtypes) were higher compared with the MMR gene expression assessed in the CB samples. The expression of PMS2 in primary MB tissues was within the range of the reference CB. Taken together, these data suggest that MMR defects associated with loss of expression of MMR genes are absent or rare in MB tumours. We then examined the expression level of MMR genes among 12 established MB cell lines; including the three cell lines used in the CFAs. Expression array analysis (Figure 4) indicated that of the 12 cell lines, 3 had undetectable expression of the MMR gene MLH1: D341Med, D283Med, and 1580WÜ. This confirmed what we found in D341Med cells by western blot: loss of the MLH1 protein expression (Figure 2). In D341Med cells, the non-detection of the PMS2 protein (even though the mRNA is expressed) can be explained by its degradation in the absence of the heterodimeric partner MLH1 (Figure 4) (Yao et al, 1999). Expression array analysis also showed that MGMT expression is detectable in all of the 61 MB tumours with transcription levels equal to or higher compared to those of CB samples (Figure 5A). Microarray analysis of the 12 MB cells revealed that only 2 cell lines (D425Med and Meb-Med-8a) did not express MGMT mRNA (Figure 5B). Unfortunately, tissue blocks of the 61 MB tumours used in the microarray study were not available for the analysis of the MMR protein expression. Therefore, we tested an additional 13 MB tumours (Table 1) by immunohistochemistry and found that all of them were positive for the presence of MSH2, MSH6, MLH1, and PMS2. A representative example is shown in Figure 6. Apparently, three of the 13 tumours (23%) showed complete loss of MGMT protein expression (Table 1).

The expression levels (Affymetrix HG U133 Plus 2.0 arrays) of the mismatch repair genes MLH1, MSH2, MSH6, and PMS2 in 61 medulloblastoma (Kool et al, 2012) grouped into the four molecular subtypes (Taylor et al, 2012) of medulloblastoma (WNT, SHH, Group 3 (GRP3) and Group 4 (GRP4)). Also included are the gene expression levels of mismatch repair genes for nine normal cerebellum (CB) samples published previously (Roth et al, 2006).

The expression levels (Affymetrix HG U133 Plus 2.0 arrays) of mismatch repair genes MLH1, MSH2, MSH6, and PMS2 in 12 medulloblastoma cell lines.

(A) The expression levels (Affymetrix HG U133 Plus 2.0 arrays) of MGMT in 61 medulloblastoma (Kool et al, 2012) grouped into the four molecular subtypes (Taylor et al, 2012) of medulloblastoma (WNT, SHH, Group 3 (GRP3), and Group 4 (GRP4)) compared with nine samples of normal cerebellum (CB), (B) MGMT expression levels of 12 medulloblastoma cell lines.

Immunohistochemistry of MMR proteins, MGMT, and Ki-67 antigen in 13 medulloblastoma tissues samples. Demonstration of nuclear expression of MLH1 (upper left), MSH2 (middle), MSH6 (upper right), PMS2 (lower left), MGMT (middle), and Ki-67 antigen (lower right) in a medulloblastoma from a 10-year-old girl (chromogen diaminobenzidine, scale bar applies to all pictures).
Mechanism of MLH1 silencing in D341Med, D283Med, and 1580WÜ cells
We then asked whether the lack of expression of MLH1 in D341Med, D283Med, and 1580WÜ cell lines can be explained by hypermethylation of the gene’s promoter region. Using SALSA ME011-B1 MMR assay, we were able to demonstrate that all three cell lines exhibited extensive CpG island methylation in at least four of five MLH1 promoter regions that were investigated (Table 2). This methylation pattern appeared to be sufficient for silencing of the MLH1 expression (Figure 4). In contrast, DAOY (which is positive for MLH1 expression) was methylated in only one CpG site (Table 2). DNA sequencing also indicated that none of the aforementioned cell lines (including DAOY) were mutated in MLH1.
Discussion
The two important molecular predictors of TMZ efficacy are MGMT expression and MMR status. Using MB cell line models, we were able to demonstrate that TMZ is most effective when the MB cells have a functioning MMR, in addition to low level of MGMT expression (whose correlation to TMZ sensitivity in MB has been previously demonstrated through in vitro viability assays (Daniel et al, 2010; Faoro et al, 2011) and xenograft studies (Daniel et al, 2010)). For cells with defective MMR, MGMT depletion (e.g., by O6-BG) will not potentiate TMZ activity, as we have shown in this study, and has been reported previously using colon cancer cell lines (Liu et al, 1996).
Thus far, there is limited information available regarding the prevalence and clinical relevance of MMR deficiency among MB. A recent report by Viana-Pereira et al (2009) indicated that 4 out of 36 (11%) MB tumours had microsatellite instability (the resulting phenotype of defective MMR). However, an earlier report found that MLH1, MSH2, and PMS2 (MSH6 was not probed) were expressed in all of 22 MB analysed by western blotting (Lee et al, 1998). In the present study, we demonstrated that all of the 74 MB tumours (61 analysed by expression profiling and by immunostaining) seem to be MMR proficient. In addition, MMR deficiency has been described in only a small fraction of malignant glioma in the paediatric age group (Vladimirova et al, 2008; Pollack et al, 2010). Furthermore, very rare cases of MMR deficiency originating from biallelic germline mutations of a MMR gene have been reported (Poley et al, 2007; Scott et al, 2007). Taken collectively, these studies point to the low prevalence of MMR deficiency in MB tumours. Indeed, the transcript levels of MMR genes MLH1, MSH2, and MSH6 were clearly higher in MB tumours when compared with CB samples. The transcript levels of PMS2 in MB tumours (expression levels were slightly higher when compared with CB tissues) need to be interpreted with caution as the probe seems to detect PMS2 and PMS2CL (the probe sets 209805_at and 221206_at can detect both PMS2 and PMS2CL mRNAs; noted in affymetrix.com).
This current study also showed that, in contrast to the primary MB tumours, 3 out of 12 MB cell lines had defective MMR system due to promoter hypermethylation of the MLH1 gene. As we have not been able to obtain primary tumour specimens from which the cell lines were established, we can not conclude whether this epigenetic event occurred in the primary tumours or during the establishment and propagation of the corresponding cell lines. There are possible explanations for the observation that 3 out of 12 MB cell lines are, due to MLH1 promoter hypermethylation are MMR deficient, while primary MB tumours are MMR proficient. First, the cancer cell lines may have been selected from a sub-population of cancer cells specifically adapted to growth in tissue culture. Some cells from the primary tumour may have possessed MLH1 promoter hypermethylation, thus MMR-deficiency. Although the great majority of the tumour cells were MMR proficient, those MMR-deficient cells might have been selected during the establishment of the tumour cell line. Another possible explanation is that at least part of the methylations might have been acquired in vitro during cell propagation.
In line with our observation regarding MB tumours and cell lines is the fact that colorectal cancer cell lines display higher rates of methylations (Suter et al, 2003) when compared with primary colorectal cancers (Lengauer et al, 1998) (e.g., MLH1 promoter methylation was detected in 30% of colorectal cancer cell lines (Suter et al, 2003)). Thus, it seems that MB tumours are, like the great majority of cancers rarely MMR deficient (Lengauer et al, 1998). Some MB cell lines are MMR deficient. So how does MMR deficiency confer an advantage on cells in vitro? In MMR-deficient cells, mutations in oncogenes and tumour suppressor accumulate more easily, thus resulting in clonal expansion. These these cells might be more conducive to transformation into culture. This inconsistency between tumours and cell lines highlight the limitations when using cancer cell lines as a model to study MB or other cancers. Differences between primary tumours and cancer cell lines have been reported for gene copy numbers, as several cancer-related genes show higher amplification and deletion frequencies in cell lines than tumours of their respective histology as demonstrated in MB for several genes including c-MYC gene amplification (Grotzer et al, 2001).
The expression of MGMT (detected at the mRNA level, the protein level, activity, or indirectly as promoter methylation) is detectable in many MB (Hongeng et al, 1997; Bobola et al, 2001; Rood et al, 2004; Faoro et al, 2011). An important issue is whether data from microarray analysis (which detects mRNA expression) and those results from immunohistochemical analysis (which detects protein expression) can be interpreted similarly. According to our gene expression analysis, MGMT expression seems to be present in primary MB, and expression values of ∼20% of the tumours are rather low (similar MGMT mRNA expression levels as CB samples). We would in general expect higher MGMT transcript levels – similar transcript levels as observed for MMR genes MLH1, MSH2, and MSH6 – in MGMT-proficient tumours when compared with CB samples. Silber et al (1993) showed a generally higher MGMT expression in brain tumours compared to adjacent normal brain tissues. Whether in those tumours with low MGMT mRNA expression the protein might be detectable in tumour cells by immunohistochemistry is questionable. Moreover, we need to consider that we can not exclude the possibility that the low MGMT expression assessed at the transcript level by microarray analysis could be the result from the contamination with tumour stroma cells or non-tumour cells expressing MGMT mRNA. Of note, a snap-frozen MB tumour sample was included in the microarray study when ⩾70% of the sample contained tumour cells (Kool et al, 2008, 2012). The fact that 2 out of 12 MB cell lines were MGMT deficient is providing further evidence that a small proportion of MB tumours might be MGMT deficient. In addition, there is marked intratumoral heterogenicity for MGMT immunoreactivity in the brain tumours including MB (Rood et al, 2004). Ideally, we would have analysed the two different cohorts by both techniques (microarray analysis and immunohistochemistry) and this is a limitation of our study. As we do not have the tissue blocks of the primary MB used in the microarray study, and as we have very limited amounts of tissues from the tumours analysed by immunohistochemistry, we were unable to assess MGMT/MMR proteins and mRNA expression in the two patient cohorts. Although one would expect concordance when comparing MGMT promoter methylation status, MGMT mRNA expression, and MGMT protein expression, discrepancies have been reported in several tumour entities including MB (Rood et al, 2004) and these results highlight the limitations of the methods used to date to assess MGMT protein/mRNA expression and promoter methylation. Given the low frequency of MMR deficiency in MB tumours (likely no one in our series (n=74)), MGMT expression level seems to be a more relevant factor mediating tolerance to TMZ in MB and could therefore have a role as predictive/prognostic factor. Studies of GBM patients suggest that the level of MGMT expression is a predictor of TMZ response and outcome (i.e., higher MGMT expression correlated to poorer overall survival rate) (Dunn et al, 2009; Karayan-Tapon et al, 2010). However, recent reports investigating a limited number of paediatric brain tumour patients (including MB) (Sardi et al, 2009; Faoro et al, 2011) point to the weakness of such an association (Sardi et al, 2009; Faoro et al, 2011). Several explanations are possible. First, multimodal therapy of MB patients includes not only alkylating agents but also other cytotoxic drugs and radiotherapy. Second, most of the reported MGMT expression levels on MB were performed in primary tumours before any chemotherapy. It is possible that MGMT expression is further upregulated during the course of treatment. This has been demonstrated in vitro, where continuous exposure of O6-BG alkylating drugs such as BCNU led to further upregulation of MGMT (and BCNU resistance) in MB cell lines (Bacolod et al, 2002, 2004). Zhang et al (2010) applied a similar approach to a glioma cell line (U373VR) (continuous exposure to TMZ) and also detected MGMT upregulation in the resulting TMZ-resistant line. Third, just like MGMT upregulation, MMR deficiency may arise during the course of TMZ treatment. In the aforementioned study by Zhang et al (2010), a glioma sub-line which acquired TMZ resistance also showed decreased expression of MSH6. Moreover, recent studies indicate that MMR gene abnormalities can be acquired in malignant glioma with initially intact MMR as a somatic mutation (MSH6) after treatment with alkylating agents (Pollack et al, 2010). Finally, another resistance mechanism may be involved. For instance, a third DNA repair mechanism, base excision repair (BER), has been shown to contribute to TMZ resistance in cancer cells (Marchesi et al, 2007). The rational for this finding is that TMZ also causes other DNA damages, such as N7-meG and the highly cytotoxic N3-meA, which are repaired by BER.
The depletion of MGMT by adding O6-BG before TMZ therapy proved to be effective in potentiating the anti-cancer toxicity against MB in preclinical studies (Friedman et al, 1995; Bobola et al, 2005). In a clinical trial investigating a combination of TMZ/O6-BG in paediatric patients with recurrent brain tumours (including MB) (Broniscer et al, 2007) showed modest anti-tumour activity of the drug combination (Broniscer et al, 2007). The use of O6-BG, however, bears the risk of serious complications since O6-BG can also potentiate TMZ in killing normal bone marrow cells. Owing to hematopoietic toxicity and lack of efficacy, the combination of O6-BG/BCNU was not further tested in clinical trials of paediatric CNS tumours (Adams et al, 2008). Moreover, O6-BG can elevate the mutagenicity of TMZ, as it inhibits the very protein (MGMT) that repairs its mutagenic adduct (O6-meG) (Cai et al, 2000). Accordingly, continuous exposure of MB cells to O6-BG and BCNU may result in the development of BCNU-resistant cells that have mutated MGMT. The mutant MGMT, it turned out, was inhibited by O6-BG much less effectively (Bacolod et al, 2002, 2004).
In summary, certain molecular factors such as MGMT and MMR expressions/activities may influence the efficacy of TMZ in MB. Our data demonstrate that a subset of MB may successfully respond to TMZ as some MB tumours are MGMT deficient, and MB tumours fortunately appear to be MMR proficient. However, as MMR deficiency is evident in 3 out of 12 MB cell lines (in all cases due to transcriptional silencing of MLH1 gene), we may speculate that this defect can arise during/after treatment of MB patients, a hypothesis that needs to be tested by investigating recurrent MB for these DNA repair proteins. Thus, it would be preferable to evaluate MGMT and MMR in tumour tissues obtained from surgery of the relapse before treatment with TMZ.
Accession codes
Change history
04 October 2012
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adams DM, Zhou T, Berg SL, Bernstein M, Neville K, Blaney SM (2008) Phase 1 trial of O6-benzylguanine and BCNU in children with CNS tumors: a Children’s Oncology Group study. Pediatr Blood Cancer 50 (3): 549–553
Aldosari N, Wiltshire RN, Dutra A, Schrock E, McLendon RE, Friedman HS, Bigner DD, Bigner SH (2002) Comprehensive molecular cytogenetic investigation of chromosomal abnormalities in human medulloblastoma cell lines and xenograft. Neuro-Oncol 4 (2): 75–85
Bacolod MD, Johnson SP, Ali-Osman F, Modrich P, Bullock NS, Colvin OM, Bigner DD, Friedman HS (2002) Mechanisms of resistance to 1,3-bis(2-chloroethyl)-1-nitrosourea in human medulloblastoma and rhabdomyosarcoma. Mol Cancer Ther 1 (9): 727–736
Bacolod MD, Johnson SP, Pegg AE, Dolan ME, Moschel RC, Bullock NS, Fang Q, Colvin OM, Modrich P, Bigner DD, Friedman HS (2004) Brain tumor cell lines resistant to O6-benzylguanine/1,3-bis(2-chloroethyl)-1-nitrosourea chemotherapy have O6-alkylguanine-DNA alkyltransferase mutations. Mol Cancer Ther 3 (9): 1127–1135
Bigner SH, Friedman HS, Vogelstein B, Oakes WJ, Bigner DD (1990) Amplification of the c-myc gene in human medulloblastoma cell lines and xenografts. Cancer Res 50: 2347–2350
Bobola MS, Berger MS, Ellenbogen RG, Roberts TS, Geyer JR, Silber JR (2001) O6-Methylguanine-DNA methyltransferase in pediatric primary brain tumors: relation to patient and tumor characteristics. Clin Cancer Res 7 (3): 613–619
Bobola MS, Silber JR, Ellenbogen RG, Geyer JR, Blank A, Goff RD (2005) O6-methylguanine-DNA methyltransferase, O6-benzylguanine, and resistance to clinical alkylators in pediatric primary brain tumor cell lines. Clin Cancer Res 11 (7): 2747–2755
Broniscer A, Gururangan S, MacDonald TJ, Goldman S, Packer RJ, Stewart CF, Wallace D, Danks MK, Friedman HS, Poussaint TY, Kun LE, Boyett JM, Gajjar A (2007) Phase I trial of single-dose temozolomide and continuous administration of o6-benzylguanine in children with brain tumors: a pediatric brain tumor consortium report. Clin Cancer Res 13 (22 Part 1): 6712–6718
Buerki N, Gautier L, Kovac M, Marra G, Buser M, Mueller H, Heinimann K (2012) Evidence for breast cancer as an integral part of lynch syndrome. Genes Chromosomes Cancer 51 (1): 83–91
Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, Batchelor TT, Futreal PA, Stratton MR, Curry WT, Iafrate AJ, Louis DN (2007) Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 13 (7): 2038–2045
Cai Y, Wu MH, Xu-Welliver M, Pegg AE, Ludeman SM, Dolan ME (2000) Effect of O6-benzylguanine on alkylating agent-induced toxicity and mutagenicity. In Chinese hamster ovary cells expressing wild-type and mutant O6-alkylguanine-DNA alkyltransferases. Cancer Res 60 (19): 5464–5469
Daniel RA, Rozanska AL, Mulligan EA, Drew Y, Thomas HD, Castelbuono DJ, Hostomsky Z, Plummer ER, Tweddle DA, Boddy AV, Clifford SC, Curtin NJ (2010) Central nervous system penetration and enhancement of temozolomide activity in childhood medulloblastoma models by poly(ADP-ribose) polymerase inhibitor AG-014699. Br J Cancer 103: 1588–1596
Drablos F, Feyzi E, Aas PA, Vaagbo CB, Kavli B, Bratlie MS, Pena-Diaz J, Otterlei M, Slupphaug G, Krokan HE (2004) Alkylation damage in DNA and RNA – repair mechanisms and medical significance. DNA Repair (Amst) 3 (11): 1389–1407
Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R, Crooks D, Husband D, Shenoy A, Brodbelt A, Wong H, Liloglou T, Haylock B, Walker C (2009) Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer 101 (1): 124–131
Faoro D, von Bueren AO, Shalaby T, Sciuscio D, Hurlimann ML, Arnold L, Gerber NU, Haybaeck J, Mittelbronn M, Rutkowski S, Hegi M, Grotzer MA (2011) Expression of O6-methylguanine-DNA methyltransferase in childhood medulloblastoma. J Neurooncol 103 (1): 59–69
Friedman HS, Burger PC, Bigner SH, Trojanowski JQ, Brodeur GM, He XM, Wikstrand CJ, Kurtzberg J, Berens ME, Halperin EC, Bigner DD (1988) Phenotypic and genotypic analysis of a human medulloblastoma cell line and transplantable xenograft (D341 Med) demonstrating amplification of c-myc. Am J Pathol 130: 472–484
Friedman HS, Burger PC, Bigner SH, Trojanowski JQ, Wikstrand CJ, Halperin EC, Bigner DD (1985) Establishment and characterization of the human medulloblastoma cell line and transplantable xenograft D283 Med. J Neuropathol Exp Neurol 44: 592–605
Friedman HS, Dolan ME, Pegg AE, Marcelli S, Keir S, Catino JJ, Bigner DD, Schold SC (1995) Activity of temozolomide in the treatment of central nervous system tumor xenografts. Cancer Res 55 (13): 2853–2857
Gerson SL (2002) Clinical relevance of MGMT in the treatment of cancer. J Clin Oncol 20 (9): 2388–2399
Grotzer MA, Hogarty MD, Janss AJ, Liu X, Zhao H, Eggert A, Sutton LN, Rorke LB, Brodeur GM, Phillips PC (2001) MYC messenger RNA expression predicts survival outcome in childhood primitive neuroectodermal tumor/medulloblastoma. Clin Cancer Res 7 (8): 2425–2433
Gylling A, Ridanpaa M, Vierimaa O, Aittomaki K, Avela K, Kaariainen H, Laivuori H, Poyhonen M, Sallinen SL, Wallgren-Pettersson C, Jarvinen HJ, Mecklin JP, Peltomaki P (2009) Large genomic rearrangements and germline epimutations in Lynch syndrome. Int J Cancer 124 (10): 2333–2340
Hartmann W, Koch A, Brune H, Waha A, Schuller U, Dani I, Denkhaus D, Langmann W, Bode U, Wiestler OD, Schilling K, Pietsch T (2005) Insulin-like growth factor II is involved in the proliferation control of medulloblastoma and its cerebellar precursor cells. Am J Pathol 166 (4): 1153–1162
Hongeng S, Brent TP, Sanford RA, Li H, Kun LE, Heideman RL (1997) O6-Methylguanine-DNA methyltransferase protein levels in pediatric brain tumors. Clin Cancer Res 3 (12 Part 1): 2459–2463
Jacobsen PF, Jenkyn DJ, Papadimitriou JM (1985) Establishment of a human medulloblastoma cell line and its heterotransplantation into nude mice. J Neuropathol Exp Neurol 44: 472–485
Karayan-Tapon L, Quillien V, Guilhot J, Wager M, Fromont G, Saikali S, Etcheverry A, Hamlat A, Loussouarn D, Campion L, Campone M, Vallette FM, Gratas-Rabbia-Re C (2010) Prognostic value of O6-methylguanine-DNA methyltransferase status in glioblastoma patients, assessed by five different methods. J Neurooncol 97 (3): 311–322
Keles GE, Berger MS, Srinivasan J, Kolstoe DD, Bobola MS, Silber JR (1995) Establishment and characterization of four human medulloblastoma-derived cell lines. Oncol Res 7: 493–503
Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, Clifford SC, Pietsch T, von Bueren AO, Rutkowski S, McCabe M, Collins VP, Backlund ML, Haberler C, Bourdeaut F, Delattre O, Doz F, Ellison DW, Gilbertson RJ, Pomeroy SL, Taylor MD, Lichter P, Pfister SM (2012) Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol 123 (4): 473–484
Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, Mrsic A, Ylstra B, Grajkowska W, Hartmann W, Pietsch T, Ellison D, Clifford SC, Versteeg R (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3 (8): e3088
Lee SE, Johnson SP, Hale LP, Li J, Bullock N, Fuchs H, Friedman A, McLendon R, Bigner DD, Modrich P, Friedman HS (1998) Analysis of DNA mismatch repair proteins in human medulloblastoma. Clin Cancer Res 4: 1415–1419
Lengauer C, Kinzler KW, Vogelstein B (1998) Genetic instabilities in human cancers. Nature 396 (6712): 643–649
Liu L, Markowitz S, Gerson SL (1996) Mismatch repair mutations override alkyltransferase in conferring resistance to temozolomide but not to 1,3-bis(2-chloroethyl)nitrosourea. Cancer Res 56 (23): 5375–5379
Marchesi F, Turriziani M, Tortorelli G, Avvisati G, Torino F, De Vecchis L (2007) Triazene compounds: mechanism of action and related DNA repair systems. Pharmacol Res 56 (4): 275–287
Menigatti M, Truninger K, Gebbers JO, Marbet U, Marra G, Schar P (2009) Normal colorectal mucosa exhibits sex- and segment-specific susceptibility to DNA methylation at the hMLH1 and MGMT promoters. Oncogene 28 (6): 899–909
Nicholson HS, Kretschmar CS, Krailo M, Bernstein M, Kadota R, Fort D, Friedman H, Harris MB, Tedeschi-Blok N, Mazewski C, Sato J, Reaman GH (2007) Phase 2 study of temozolomide in children and adolescents with recurrent central nervous system tumors: a report from the Children’s Oncology Group. Cancer 110 (7): 1542–1550
Nygren AO, Ameziane N, Duarte HM, Vijzelaar RN, Waisfisz Q, Hess CJ, Schouten JP, Errami A (2005) Methylation-specific MLPA (MS-MLPA): simultaneous detection of CpG methylation and copy number changes of up to 40 sequences. Nucleic Acids Res 33 (14): e128
Pegg AE (1990) Mammalian O6-alkylguanine-DNA alkyltransferase: regulation and importance in response to alkylating carcinogenic and therapeutic agents. Cancer Res 50 (19): 6119–6129
Pietsch T, Scharmann T, Fonatsch C, Schmidt D, Oeckler R, Freihoff D, Albrecht S, Wiestler AD, Zeltzer P, Riehm H (1994) Characterization of five new cell lines derived from human primitive neuroectodermal tumors of the central nervous system. Cancer Res 54: 3278–3287
Poley JW, Wagner A, Hoogmans MM, Menko FH, Tops C, Kros JM, Reddingius RE, Meijers-Heijboer H, Kuipers EJ, Dinjens WN (2007) Biallelic germline mutations of mismatch-repair genes: a possible cause for multiple pediatric malignancies. Cancer 109 (11): 2349–2356
Pollack IF, Hamilton RL, Sobol RW, Nikiforova MN, Nikiforov YE, Lyons-Weiler MA, LaFramboise WA, Burger PC, Brat DJ, Rosenblum MK, Gilles FH, Yates AJ, Zhou T, Cohen KJ, Finlay JL, Jakacki RI (2010) Mismatch repair deficiency is an uncommon mechanism of alkylator resistance in pediatric malignant gliomas: a report from the Children’s Oncology Group. Pediatr Blood Cancer 55 (6): 1066–1071
Rood BR, Zhang H, Cogen PH (2004) Intercellular heterogeneity of expression of the MGMT DNA repair gene in pediatric medulloblastoma. Neuro-Oncol 6 (3): 200–207
Roth RB, Hevezi P, Lee J, Willhite D, Lechner SM, Foster AC, Zlotnik A (2006) Gene expression analyses reveal molecular relationships among 20 regions of the human CNS. Neurogenetics 7 (2): 67–80
Sardi I, Cetica V, Massimino M, Buccoliero AM, Giunti L, Genitori L, Arico M (2009) Promoter methylation and expression analysis of MGMT in advanced pediatric brain tumors. Oncol Rep 22 (4): 773–779
Scott RH, Mansour S, Pritchard-Jones K, Kumar D, MacSweeney F, Rahman N (2007) Medulloblastoma, acute myelocytic leukemia and colonic carcinomas in a child with biallelic MSH6 mutations. Nat Clin Pract Oncol 4 (2): 130–134
Silber JR, Mueller BA, Ewers TG, Berger MS (1993) Comparison of O6-methylguanine-DNA methyltransferase activity in brain tumors and adjacent normal brain. Cancer Res 53 (14): 3416–3420
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352 (10): 987–996
Suter CM, Norrie M, Ku SL, Cheong KF, Tomlinson I, Ward RL (2003) CpG island methylation is a common finding in colorectal cancer cell lines. Br J Cancer 88 (3): 413–419
Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, Eberhart CG, Parsons DW, Rutkowski S, Gajjar A, Ellison DW, Lichter P, Gilbertson RJ, Pomeroy SL, Kool M, Pfister SM (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123 (4): 465–472
Truninger K, Menigatti M, Luz J, Russell A, Haider R, Gebbers JO, Bannwart F, Yurtsever H, Neuweiler J, Riehle HM, Cattaruzza MS, Heinimann K, Schar P, Jiricny J, Marra G (2005) Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology 128 (5): 1160–1171
Viana-Pereira M, Almeida I, Sousa S, Mahler-Araujo B, Seruca R, Pimentel J, Reis RM (2009) Analysis of microsatellite instability in medulloblastoma. Neuro Oncol 11 (5): 458–467
Vladimirova V, Denkhaus D, Soerensen N, Wagner S, Wolff JE, Pietsch T (2008) Low level of microsatellite instability in paediatric malignant astrocytomas. Neuropathol Appl Neurobiol 34 (5): 547–554
von Bueren AO, Oehler C, Shalaby T, von Hoff K, Pruschy M, Seifert B, Gerber NU, Warmuth-Metz M, Stearns D, Eberhart CG, Kortmann RD, Rutkowski S, Grotzer MA (2011) c-MYC expression sensitizes medulloblastoma cells to radio- and chemotherapy and has no impact on response in medulloblastoma patients. BMC Cancer 11: 74
von Bueren AO, Shalaby T, Oehler-Janne C, Arnold L, Stearns D, Eberhart CG, Arcaro A, Pruschy M, Grotzer MA (2009) RNA interference-mediated c-MYC inhibition prevents cell growth and decreases sensitivity to radio- and chemotherapy in childhood medulloblastoma cells. BMC Cancer 9: 10
Yao X, Buermeyer AB, Narayanan L, Tran D, Baker SM, Prolla TA, Glazer PM, Liskay RM, Arnheim N (1999) Different mutator phenotypes in Mlh1- versus Pms2-deficient mice. Proc Natl Acad Sci USA 96 (12): 6850–6855
Zhang J, Stevens MF, Laughton CA, Madhusudan S, Bradshaw TD (2010) Acquired resistance to temozolomide in glioma cell lines: molecular mechanisms and potential translational applications. Oncology 78 (2): 103–114
Acknowledgements
We would like to thank Dr Bert Vogelstein, Dr Josef Jiricny, and Dr Stefan M Pfister for helpful comments. We thank Dr Darell D Bigner for kindly providing D283Med cells. We are grateful to Dr Udo zur Stadt and Guido Looft for some assistance in sequencing experiments. Temozolomide was a generous gift of Schering-Plough (Kenilworth, NJ). We acknowledge the following sources of funding: The Swiss Research Foundation Child and Cancer (AOVB, MAG), German Children’s Cancer Foundation/Deutsche Kinderkrebsstiftung (AOVB, SR, TP), Krebsliga Zürich (GM), Krebsliga beider Basel (KH), Oncosuisse (KH), German Research Foundation (TP) and Fördergemeinschaft Kinderkrebszentrum Hamburg e. V. (AOVB, SR).
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
von Bueren, A., Bacolod, M., Hagel, C. et al. Mismatch repair deficiency: a temozolomide resistance factor in medulloblastoma cell lines that is uncommon in primary medulloblastoma tumours. Br J Cancer 107, 1399–1408 (2012). https://doi.org/10.1038/bjc.2012.403
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.403
Keywords
This article is cited by
-
NUSAP1 potentiates chemoresistance in glioblastoma through its SAP domain to stabilize ATR
Signal Transduction and Targeted Therapy (2020)
-
Medulloblastoma epigenetics and the path to clinical innovation
Journal of Neuro-Oncology (2020)
-
Casein kinase 2 inhibition sensitizes medulloblastoma to temozolomide
Oncogene (2019)
-
The germline variants in DNA repair genes in pediatric medulloblastoma: a challenge for current therapeutic strategies
BMC Cancer (2017)
-
Overcoming multiple drug resistance mechanisms in medulloblastoma
Acta Neuropathologica Communications (2014)
