
Abstract
Background:
Bcl-2-like members have been found to be inherently overexpressed in many types of haematologic malignancies. The small-molecule S1 is a BH3 mimetic and a triple inhibitor of Bcl-2, Mcl-1 and Bcl-XL.
Methods:
The lethal dose 50 (LD50) values of S1 in five leukaemic cell lines and 41 newly diagnosed leukaemia samples were tested. The levels of Bcl-2 family members and phosphorylated Bcl-2 were semiquantitatively measured by western blotting. The interactions between Bcl-2 family members were tested by co-immunoprecipitation. The correlation between the LD50 and expression levels of Bcl-2 family members, alone or in combination, was analysed.
Results:
S1 exhibited variable sensitivity with LD50 values ranging >2 logs in both established and primary leukaemic cells. The ratio of pBcl-2/(Bcl-2+Mcl-1) could predict the S1 response. Furthermore, we demonstrated that pBcl-2 antagonised S1 by sequestering the Bak and Bim proteins that were released from Mcl-1, andpBcl-2/Bak, pBcl-2/Bax and pBcl-2/Bim complexes cannot be disrupted by S1.
Conclusion:
A predictive index was obtained for the novel BH3 mimetic S1. The shift of proapoptotic proteins from being complexed with Mcl-1 to being complexed with pBcl-2 was revealed for the first time, which is the mechanism underlying the index value described herein.
Similar content being viewed by others


Main
The Bcl-2 protein family includes antiapoptotic as well as proapoptotic members (Youle and Strasser, 2008). The antiapoptotic family members, including Bcl-2, Bcl-XL and Mcl-1, which are termed as Bcl-2-like proteins, counterbalance the proapoptotic members Bax and Bak via a shared BH3 domain (Adams and Cory, 1998; Wei et al, 2001; Letai et al, 2002; Adams and Cory, 2007).
Bcl-2-like members are overexpressed in many haematological malignancies including acute lymphoblastic leukaemia (ALL), acute myeloid leukaemia (AML), chronic lymphoblastic leukaemia (CLL) and chronic myeloid leukaemia (CML) (Kitada et al, 1998; Andreeff et al, 1999; Salomons et al, 1999; Jaiswal et al, 2003; Packham and Stevenson, 2005; Fonseca et al, 2009; Stam et al, 2010). In addition, Bcl-2 expression levels alone cannot fully explain its antiapoptotic activity; it is well known that the Bcl-2 protein undergoes multisite phosphorylation at the loop region and that phosphorylation at serine 70 is necessary for its full antiapoptotic function and may regulate its interaction with both multidomain and BH3-only proapoptotic members (Deng et al, 2000, 2004; Konopleva et al, 2006). Therefore, small molecules that target several Bcl-2-like proteins by mimicking a BH3 domain are favoured in antileukaemic strategies. For example, the orally available analogue ABT-263 is in clinical trials for lymphoid malignancies (Roberts et al, 2012). However, Mcl-1 and Bfl-1confer resistance to ABT-737 because of its off-target effects (Lei et al, 2006; Vogler et al, 2009). Other clinical therapeutic candidates, including AT101 and obatoclax, induce apoptosis but not through the Bax/Bak pathway (Lei et al, 2006; Mott et al, 2008). Their nonspecific activities may limit their therapeutic efficiency and potentially provoke undesirable side effects.
We previously identified a small-molecule BH3 mimetic and pan-Bcl-2 inhibitor named S1 that exhibits nanomolar to submicromolar affinity towards Mcl-1 and Bcl-2. It has been shown that S1 induces Bax/Bak-dependent apoptosis in multiple cell lines (Zhang et al, 2007, 2010, 2011; Ting et al., 2012). Its specificity and pan-inhibition give S1 advantages over other drug candidates. Currently, S1 is being developed for preclinical trials.
In this study, we tested S1 in five different leukaemic cell lines and 41 newly diagnosed ALL, AML, CLL and CML samples. We tried to establish an indicator for the S1 response in leukaemic cells to guide the future clinical development of S1. As expected, S1 inhibited Bcl-2, Bcl-XL and Mcl-1, and these individual protein levels cannot predict the S1 response. Surprisingly, an index of pBcl-2/(Bcl-2+Mcl-1) predicts S1 sensitivity best. The newly demonstrated pBcl-2/Bak dimer, both in vitro and in vivo, provides a useful understanding for how pBcl-2 prevents apoptosis. This study could contribute to the future clinical development of S1 and rationalise the use of other pan-Bcl-2 inhibitors alone or in combination with kinase inhibitor-based strategies.
Materials and methods
Patients
Cells from 41 patients diagnosed of leukaemia according to the World Health Organisation classification, who had not received treatment for the past 3 months, were studied. Tumour cells were obtained from peripheral blood. The characteristics of these patients are listed in Supplementary Table S2. In all cases, an informed consent was obtained in accordance with the guidelines and the approval of the Second Affiliated Hospital of Dalian Medical University (Dalian, Liaoning, China) and the Declaration of Helsinki.
Isolation and culture of primary cells
Mononuclear cells from peripheral blood samples were isolated by Ficoll-Hypaque sedimentation (Sigma Chemical Co., St Louis, MO, USA). Contaminating red cells were lysed in 0.8% ammonium chloride solution for 10 min. Cells were either used directly or cryopreserved in liquid nitrogen in the presence of 10% dimethyl sulphoxide and 60% heat-inactivated fetal calf serum (FCS; Gibco BRL, Grand Island, NY, USA). Cells from leukaemia patients were cultured in RPMI 1640 culture medium (Thermo Scientific HyClone, Beijing, China), supplemented with 10% FCS and 50 μg ml−1 penicillin–streptomycin. For CML chronic phase samples, CD34+ cells from patients were selected by binding to immunomagnetic beads (MiniMACS; Miltenyi Biotec, Auburn, CA, USA) from resuscitation mononuclear cells, according to the manufacturer’s instructions.
Cell lines, plasmids and reagents
Five leukaemic cell lines were used in the study: 1T cell-derived ALL (Jurkat), 1 CML (K562), 2 AML (HL60 and U937) and 1 CLL (CLL-AAT). These cell lines contain all the four disease types in the primary samples. All the cells were purchased from Cell Bank of Chinese Academy of Sciences and used within 6 months from resuscitation. Wild type (WT) and Bax−/−Bak−/− (DKO) mouse embryo fibroblasts (MEFs) (Lindsten et al, 2000) were provided by Andreas Strasser (The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia). All the cells were maintained in minimum essential medium supplemented with 10% FBS and 100 U ml−1 streptomycin and penicillin.
Human Bcl-2 cDNA was cloned in pUC19 plasmid. Nucleotides corresponding to 70, 87 serine (S) or 69 threonine (T) residue were substituted to create a conservative alteration to alanine (A) or glutamic acid (E) with a site-directed mutagenesis kit (Clontech, Beijing, China) and then modified by addition of the HA tag sequence at its NH2 terminus. Each single mutant was cloned into pET28b (+) and pCIneo mammalian expression vector (Promega Corp., Madison, WI, USA). To generate the HA-Bcl-2-AAA and HA-Bcl-2-EEE cell lines, K562 cells were transfected with the pCIneo vectors encoding HA-tagged Bcl-2 mutant. Transfection of K562 cell line was performed with Lipofectamine according to the manufacturer’s instructions. Under our condition, 20–30% of cells are routinely transfected. Then, the stably transfected cells were selected by addition of Geneticin (G418), purchased from Invitrogen (Grand Island, NY, USA), to the medium at a concentration of 800 μg ml−1. After 3 weeks, stably transfected selected cells were further cultured with G418 at a concentration of 400 μg ml−1. Five different expression clones were selected for other experiments.
Plasmid encoding BakΔTM was a kind gift from Qian Liu and Kalle Gehring (McGill University, Montreal, QC, Canada) (Moldoveanu et al, 2006). Proteins were expressed in Escherichia coli (BL21) and then purified as reported earlier (Dai et al, 2009). S1 was synthesised as described previously (Zhang et al, 2007), and dissolved in DMSO (10 mM). PD98059 was purchased from Sigma. Annexin V-FITC and Lipofectamine 2000 Transfection Reagent were from Invitrogen. ABT-737 was obtained from Selleck Chemicals (Houston, TX, USA).
Immunoblotting
Cells (5 × 106) were resuspended in 150 μl of 3-((3-cholamidopropyl) dimethylammonio)-1-propanesulphonate (CHAPS) buffer (1% CHAPS (w v−1), 100 mM NaCl, 5 mM Na3PO4, 2.5 mM EDTA, 1 μg ml−1 leupeptin, 1 μg ml−1 aprotinin and 1 μ M PMSF). After 40 min on ice, the lysates were cleared by centrifugation at 12 000 g for 30 min at 4 °C. Total protein (100 μg) were separated by SDS–PAGE, electrotransferred to polyvinylidene difluoride membranes and analysed following standard procedures. Signals were detected using SuperSignal West Femto (no. 34096; Thermo Fisher Scientific Inc., Barrington, IL, USA). The following antibodies were used: Bcl-2 (sc-130307), pBcl-2 (Ser70, sc-293129), Mcl-1 (sc-819), Bim (sc-11425), Bcl-XL (sc-8392), Bax (sc-493) and actin (sc-8432) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and Bak (ab32371) was from Abcam (Cambridge, MA, USA). For detection of phosphorylated Bcl-2 and Bim by western blotting, 1 : 100 dilutions were used. For detection of Bcl-2, Mcl-1, Bax and Bcl-XL, 1 : 200 dilutions were used. In all, 1 : 500 dilutions were used for Bak and actin. Specificity of the pBcl-2 (Ser70) antibody was tested in HL60 cells (Supplementary Figure S1).
Immunoprecipitation
Cells (25 × 106) were lysed in 1% CHAPS buffer. Whole-cell lysates were obtained, 500 μg total proteins were precleared with protein A-Sepharose and incubated overnight with 5 μg of the specific antibody. Immunocomplexes were captured with either protein A-Sepharose or protein G-Agarose (Sigma Chemical Co.). The presence of immunocomplexes was determined by western blot analysis.
Analysis of apoptosis
Phosphatidylserine exposure was quantified by surface Annexin V-FITC staining as described previously (Zhang et al, 2011). A total of 10 000 cells per sample were acquired in a FACSCalibur flow cytometry system using the CellQuest software (BD Biosciences, Becton Drive, Franklin Lakes, NJ, USA). The lethal dose 50 (LD50) was defined as the concentration of the drug required to reduce cell viability by 50%. For the cell viability assay, we plated 5000 cells per well before addition of different chemicals and developed the plates in reaction with MTT (Sigma Chemical Co.).
Isothermal titration calorimetry
Calorimetric experiments were performed using an iTC200 system (GE Healthcare, South Burlington, VT, USA). Thermodynamic constants were determined by the least-squares fitting using Origin version 7.0. Samples were dialysed into 20 mM Tris, pH 7.8, 150 mM NaCl and 0.5 mM Tris (2-carboxyethyphosphine) hydrochloride. The cell was loaded with 20 μ M EEE-Bcl-2 samples, and the injection syringe was loaded with 300 μ M Bak proteins. Typically titrations consisted of 16 injections of 10 μl, with 240-s equilibrations between injections. Immediately before use, all proteins were dialysed in PBS and centrifuged at 100 000 g for 10 min.
Statistical analysis
The expression of Bcl-2 family members, individually or in combination, was compared among the three groups using the Kruskal–Wallis test. Pairwise comparisons between groups were performed using the Wilcoxon’s rank-sum test. A Bonferroni correction was used for pairwise comparisons. Pretreatment to post-treatment changes in Bcl-2 family transcripts were assessed using the Wilcoxon’s signed rank test. All statistical tests were two-sided. For pairwise comparisons, P<0.017 was used to indicate statistical significance. For all other comparisons, P<0.05 was used to indicate statistical significance. Synergism, additive effects and antagonism were assessed using the Calcusyn software v.2.0 (Biosoft, Ferguson, MO, USA). The interaction among drugs was found synergistic when CI values were <1. Relative protein quantification was carried out with Kodak Carestream Molecular Imaging software (Carestream Health, Inc., New Haven, CT, USA). Results are expressed as mean±standard deviation (s.d.) of three independent experiments. Spearman’s correlation was used to assess the strength and direction of association between post-treatment cell viability and each Bcl-2 family member individually or in combination. The Cohen interpretation of correlation (r) indicates that when r<0.3, it represents weak correlation, 0.3–0.5 is moderate and >0.5 is strong.
Results
Leukaemic cell lines display differential sensitivity to S1
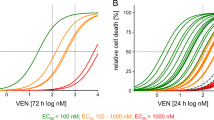
On the basis of previous reports (Zhang et al, 2011) and this study, S1 is a targeted, high-affinity small molecule that can occupy the BH3-binding groove of Bcl-2, Mcl-1 and Bcl-XL (Ki=58 nM for Mcl-1, 310 nM for Bcl-2 (Zhang et al, 2011) and 230 nM for Bcl-XL (Supplementary Figure S2A)). We assessed the apoptosis-inducing effect of S1 in five leukaemic cell lines. Cells were treated with S1 for 24 h at doses ranging from 5 to 40 μ M, and the LD50 value was measured by Annexin V labelling (Supplementary Table S1, LD50 of ABT-737 in these five cell lines were also analysed). S1 exerted a dose-dependent cytotoxic effect and differential sensitivity in the five cell lines (Figure 1A).

Individual Bcl-2 family protein expression levels did not predict the sensitivity to S1 -induced apoptosis in leukaemic cell lines. (A) Leukaemic cell lines were cultured with increasing doses of S1 for 24 h, and then stained with Annexin V for flow cytometry analysis. The means of three independent experiments are shown. Data are the mean±s.e.m. (n=3). (B) S1 and ABT-737 induce apoptosis in WT but not Bax–/–Bak–/– (DKO) MEFs. Wild-type or Bax–/–Bak–/– MEFs were incubated for 48 h with S1 or ABT-737, and cell viability assessed by the 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Data are the mean±s.e.m. (n=3). (C) Immunoblot analysis of five leukaemia cell line lysates (100 μg of protein) with the indicated antibodies. Actin was used as a loading control. (D) pBcl-2 level did not predict LD50 of S1 well in leukaemia cell lines. Protein expression levels were defined by immunoblot analysis using the Kodak Carestream Molecular Imaging software and plotted against the LD50. The Spearman’s rank correlation coefficient is indicated. Spearman’s correlation (r) and P-values are also shown.
Caspase-3/9 activation and Bax/Bak-dependent cytotoxicity further indicated that S1 induced apoptosis in the leukaemia cells via the intrinsic pathway (Supplementary Figure S2B) and acted as a BH3 mimetic (Supplementary Figure S3). To determine whether S1 triggers mitochondrial apoptosis, we studied the drug’s effects on Bax–/–Bak–/– (DKO) MEFs, which exhibit resistance to a broad range of apoptotic stimuli (Lindsten et al, 2000). Like that of ABT-737, S1’s activity was restricted to DKO MEFs (Figure 1B). S1 was cytotoxic to WT but not DKO MEFs even at high concentrations. Furthermore, S1 induced apoptosis even at the high concentrations in leukaemic cells and is not harmful to non-tumour cells (Supplementary Figure S4). ABT-737 induced early apoptosis within 3 h. For S1, significant apoptosis occurred at 12 h. The delay of apoptosis induction of S1 than ABT-737 may be due to the physicochemical properties and weaker affinity of S1 than ABT-737.
Expressions of individual Bcl-2 family members are not predictive of the S1 response
Given the variation in S1 sensitivity and the importance of the relative expression levels of Bcl-2 family proteins in predicting BH3 mimetics sensitivity (Deng et al, 2007), we investigated whether the expression levels of Bcl-2 family members contribute to the S1 response. The expression levels of Bcl-2 family members were assessed by immunoblotting with subsequent densitometric analysis in the aforementioned five leukaemic cell lines (Figure 1C). The data were analysed using the Kodak Carestream Molecular Imaging software. The expression of each individual family member in the five cell lines was plotted against the LD50 of S1. Among the seven Bcl-2 family members (Bcl-2, Bcl-XL, Mcl-1, Noxa, Bim, Bax, Bak) that we examined, no significant correlation was found between the expression levels of any single protein and the LD50 values (r<0.3, P>0.05; Supplementary Figures S5A–G). Subsequently, we analysed the expression of phosphorylated Bcl-2 at the serine 70 residue because it has been shown that the phosphorylation of Bcl-2 at this position is necessary for its full antiapoptotic function (May et al, 1994; Ito et al, 1997; Ruvolo et al, 2001). A phospho-specific (S70) Bcl-2 antibody was used. Specificity of the pBcl-2 antibody was tested in HL60 cells (Supplementary Figure S1). A moderate correlation was found between pBcl-2 levels and S1 sensitivity (r=0.47; Figure 1D), suggesting that pBcl-2 is not a good indicator of S1 sensitivity.
The pBcl-2/(Bcl-2+Mcl-1) ratio is predictive of the S1 response in both primary and established leukaemic cells
To determine the sensitivity of clinical leukaemic samples to S1, freshly isolated primary leukaemic cells derived from the peripheral blood of patients (ALL, AML, CLL and CML) were examined for cell death by flow cytometry using Annexin V staining. The majority of primary samples underwent apoptosis after treatment with S1; however, there was a wide range of sensitivity (LD50 at 40 h ranging from 0.24 to 80.4 μ M), which is in agreement with the results from the cell lines. The lethal dose 50 for ABT-737 was used for comparison with S1 in 11 primary samples (Supplementary Figure S6). After 20 h of treatment, the mean LD50 was 33.8 μ M and decreased to 23.7 μ M after longer exposure (40 h) to this inhibitor (Supplementary Table S2). Based on their responses, 41 primary samples could be classified into three groups: (I) a sensitive group (17 of 41 cases, 41.5%) with an LD50 <15 μ M, (II) an intermediate group (14 of 41, 34.1%) with an LD50 of 15–30 μ M and (III) a resistant group (10 of 41, 24.3%) with an LD50 >30 μ M (Figure 2A).

pBcl-2/(Bcl-2+Mcl-1) provides the most significant linear correlation for the sensitivity to S1 , both in primary and established cells. (A) Primary leukaemic cells derived from 41 patients were incubated with S1 (0–50 μ M). Cell survival was assessed by Annexin V binding, as determined by flow cytometric analysis. The lethal dose 50 values were calculated, and each data pillar represents one patient sample. Based on cell LD50 data, the response to S1 was classified as: resistant (>30 μ M), intermediate (15–30 μ M) and sensitive (<15 μ M). (B) pBcl-2/(Bcl-2+Mcl-1) ratios in function of S1 sensitivity (LD50) of 41 primary leukaemia samples. Spearman’s correlation (r) and P-values are shown. Relative protein quantification was carried out with Kodak Carestream Molecular Imaging software. (C) pBcl-2/(Bcl-2+Mcl-1) radio in resistant, intermediate and sensitive group. (D) Analysis of pBcl-2/(Bcl-2+Mcl-1) ratio as a function of S1 sensitivity (LD50) of leukaemia cell lines. Spearman’s correlation (r) and P-values are shown. *** indicates P<0.001, extremely significant.
Next, we determined the expression levels of Bcl-2 family members in these samples (Supplementary Figure S7). Similar to the results observed in the cell lines, pBcl-2 showed moderate linear correlation (r=0.48, P<0.001; Supplementary Figure S8A). We then plotted the expression of several combinations of Bcl-2 family members, including pBcl-2, against cell viability. The relative ratio of pBcl-2/(Bcl-2+Mcl-1) protein levels provided the best linear correlation (r=0.69, P<0.001; Figure 2B). The relative ratios of pBcl-2/Bcl-2 and pBcl-2/Mcl-1 did not show a linear correlation: r=0.24, P=0.01 and r=0.24, P=0.01, respectively (Supplementary Figures S8B and C). The inclusion of Bcl-XL into the pBcl-2/(Bcl-2+Mcl-1) model had no significant effect on the correlation (Supplementary Figure S8D). The pBcl-2/(Bcl-2+Mcl-1) ratio was calculated in the resistant, intermediate and sensitive groups. The lowest ratio (P<0.001) was found in the sensitive group, while the highest one was observed in the resistant group (Figure 2C). These data indicate that the pBcl-2/(Bcl-2+Mcl-1) ratio is a predictive ratio for the response to S1.
To further address the applicability of our predictive model in leukaemic cells, the pBcl-2/(Bcl-2+Mcl-1) ratio was determined in the aforementioned five cell lines. The pBcl-2/(Bcl-2+Mcl-1) ratio also provided the highest predictive value for response to S1 in the five cell lines (r=0.76, P=0.05; Figure 2D). Among the five cell lines tested, the most resistant cells (CLL-AAT cells) showed the highest ratio of 0.586, while the most sensitive cells (Jurkat cells) showed a ratio of 0.015 (Supplementary Table S1). Taken together, the pBcl-2/(Bcl-2+Mcl-1) ratio is predictive for the S1 response in a broad range of primary and established leukaemic tumour cells.
pBcl-2 levels modulate the sensitivity of leukaemic cells to S1
To further demonstrate whether the pBcl-2/(Bcl-2+Mcl-1) ratio predicts S1 sensitivity, we tested whether modulated Bcl-2 phosphorylation status can affect the sensitivity to S1. A non-phospho-mimetic mutant, HA-AAA-Bcl-2, and a phospho-mimetic mutant, HA-EEE-Bcl-2, were applied as described previously (Konopleva et al, 2006). Bcl-2 mutants and pCIneo control expression vectors were then transfected into K562 cells that express low levels of endogenous Bcl-2 (Weerasinghe et al, 2001). Then, G418 was used for selection and subsequent cloning. Independent clones were obtained from K562 cells transfected with different vectors. We have chosen the clones expressing relatively low levels of mutant Bcl-2 proteins and designed them as K562/AAA-Low and K562/EEE-Low. The clones expressing relatively high levels of mutant Bcl-2 proteins were named as K562/AAA-High and K562/EEE-High. K562/Vector was used as a control (Figure 3A). The expressions of other Bcl-2 family members were not changed in the transfected K562 cells. The control cells, and K562/AAA-Low and K562/AAA-High cells displayed similar sensitivity to S1. However, the K562/EEE-Low cells were resistant to S1 and K562/EEE-High cells displayed higher resistance, suggesting that Bcl-2 phosphorylation opposes the proapoptotic action of S1. Consistent with a mechanism whereby increased Bcl-2 phosphorylation impedes S1 suppression of Bcl-2 dimerisation with Bax, S1 potently blocked Bcl-2/Bax association in cells expressing exogenous AAA mutant, but not EEE mutant Bcl-2 (Supplementary Figure S9). These results suggest that increases in the pBcl-2 levels but not non-phosphorylated-Bcl-2 (npBcl-2) contribute to S1 resistance.

Bcl-2 phosphorylation modulates sensitivity to S1 in leukaemic cells. (A) K562-Vector cells and K562 expressing different levels of mutant Bcl-2 cells were treated with S1 for 12 or 24 h and Annexin V+ cells were analysed. Indicated protein levels were examined by immunoblotting. Apoptosis was quantified by Annexin V staining and determined by flow cytometric analysis. Results represented the mean of three independent experiments. (B) HL60 cells were treated for 24 h with 20 μ M PD98059 and total protein extracts were analysed by western blot for the expression of pERK1, pBcl-2(Ser70), Bcl-2 and actin. (C) HL60 cells were treated with the indicated doses of S1 alone or together with 20 μ M PD98059 for 24 h. Viability was assessed by 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Combination indexes (CIs) were calculated using the Calcusyn software and indicated inside the plot for each combination. (D) Cells from four resistant leukaemia patients were cotreated with 20 μ M PD98059 and 20 μ M S1 for 40 h and viability was assessed by MTT assay. Combination indexes were calculated as above.
As ERK has been identified as a direct kinase of Bcl-2 (Konopleva et al, 2006; Pérez-Galán et al, 2008), we addressed whether the MEK/ERK inhibitor PD98059 was able to enhance the cytotoxic effect of S1 by inhibiting Bcl-2 phosphorylation. Treatment of HL60 cells with 20 μ M PD98059 for 6 h blocked the constitutive activation of ERK1 (pERK1) and significantly reduced pBcl-2 (Ser70) levels while having no effect on the total Bcl-2 protein levels (Figure 3B). As expected, PD98059 significantly enhanced the toxicity of S1 in HL60 cells (CI=0.63; Figure 3C). Consequently, we chose four samples (three are ALL, one is CLL) from patients who belonged to the resistant group. The combination of PD98059 with S1 was highly synergistic in these cells (Figure 3D). Taken together, these results further support the idea that pBcl-2 have a central role in the ratio of pBcl-2/(Bcl-2+Mcl-1) and that decreasing pBcl-2 levels can increase S1 sensitivity.
Proapoptotic proteins are displaced from Mcl-1 by S1 in a resistant cell line but are sequestered by pBcl-2
To evaluate the molecular mechanism that determines S1 sensitivity, we tested the interference of S1 on Bcl-2 family protein heterodimers. First, the relative levels of endogenous Bcl-2 family member complexes in leukaemic cell lines were measured by the immunoprecipitation of antiapoptotic proteins in K562, HL60 and U937 cells (Figure 4A). Immunoprecipitation assays showed that in addition to the well-known Bcl-2 family member complexes, the new complexes pBcl-2/Bak, pBcl-2/Bim and pBcl-2/Bax were found for the first time. To validate existence of these novel complexes, we determined the direct binding of pBcl-2/Bak using isothermal titration calorimetry. The Bak protein was titrated into a solution of EEE-Bcl-2 protein that was heterologously expressed in E. coli. Consistent with the co-immunoprecipitation (co-IP) assays, Bak demonstrated apparent binding to the phospho-mimetic Bcl-2 mutants (Figure 4B).

Bak displaced from Mcl-1 associates with pBcl-2 in resistant cell lines. (A) K562, HL60 and U937 cells were lysed. Mcl-1, Bcl-2 and pBcl-2 immunoprecipitations were performed, and immunoprecipitated fractions were analysed by western blotting for the indicated proteins. (B) Isothermal calorimetric titration of the Bcl-2 phosphorylation mutant protein, EEE-Bcl-2, with the Bak protein. The EEE-Bcl-2 concentration was 30 μ M, and the Bak concentration was 300 μ M. Injection volumes were 20 μl. (C) Immunoprecipitation of K562 cell lysates. Cells were treated in the presence or absence of 5 μ M S1 for 8, 18 and 24 h. Bim, Bax, Bak, Bcl-2 and Mcl-1 levels were analysed by western blotting. (D) Immunoprecipitation of U937 cell lysates using the indicated antibodies. (Left panel) The immunoprecipitates were analysed for the presence of the indicated proteins by immunoblotting. Cells were treated in the presence or absence of 20 μ M S1 for 8, 18 and 24 h. (Middle panel) U937 cells were treated in the presence or absence of 20 μ M S1 for 24 h. Supernatants of the Mcl-1 immunoprecipitation were used for the pBcl-2 immunoprecipitation. Immunoblotting was performed as described previously. (Right panel) U937 cell lysates were detected by immunoblotting using the indicated antibodies. Ab, antibody.
To determine which proapoptotic proteins were displaced from antiapoptotic proteins by S1, K562 and U937 cells were treated with S1 for different times. The dynamics of the heterodimers were analysed using immunoprecipitation. An examination of the heterodimers containing Bcl-2 and Mcl-1 in K562 cells with 5 μ M S1 treatment showed a gradual decrease in the levels of Bcl-2/Bax, Bcl-2/Bim, Mcl-1/Bak and Mcl-1/Bim complexes (Figure 4C). These data are consistent with those of apoptosis levels shown in Supplementary Figure S2B.
In U937 cells, 10 μ M S1 can disrupt Mcl-1/Bak and Mcl-1/Bim complexes, as similarly demonstrated in K562 cells (Figure 4D, left panel), indicating that S1 also reached its target Mcl-1 in the resistant cell line. However, we observed an increase in the levels of Bcl-2/Bak and Bcl-2/Bim complexes. U937 cells displayed higher levels of Bcl-2 phosphorylation than those of HL60 and K562 cells (Figure 1C). We questioned whether S1 disrupted npBcl-2 complexes or pBcl-2 complexes. The results obtained in K562 (Figure 4B) and HL60 cells (Supplementary Figure S10) showed that S1 could disrupt npBcl-2/Bax, npBcl-2/Bak and npBcl-2/Bim complexes. However, the results (Figure 4D, left panel) demonstrated that S1 could not disrupt the heterodimers containing pBcl-2 by using a specific pBcl-2 antibody in the co-IP. We hypothesised that pBcl-2, not npBcl-2, recaptures the released Bak and Bim from Mcl-1 and thus prevents apoptosis. Moreover, we observed a shift of Bak and Bim from being complexed with Mcl-1 to being complexed with pBcl-2, while the pBcl-2/Bax complex remained unchanged (Figure 4D, left panel). To further study the changing composition of Bak and Bim complexes, we analysed pBcl-2/Bak, pBcl-2/Bax and pBcl-2/Bim complexes in the supernatant of Mcl-1 immunoprecipitations (Figure 4D, middle panel). Although the pBcl-2 levels remained constant during S1 treatment, we observed an increase in the level of pBcl-2/Bim and pBcl-2/Bak aggregated, indicating that pBcl-2 was able to sequester proapoptotic proteins released from Mcl-1. These results demonstrate that pBcl-2 was able to sequester the Bak and Bim proteins released from Mcl-1 and that the movement of proapoptotic proteins from Mcl-1 complexes to pBcl-2 complexes is the mechanism underlying the predictive pBcl-2/(Bcl-2+Mcl-1) ratio. Similar results were found in K562/EEE-High cells. Bak aggregated towards the phospho-mimetic HA-EEE-Bcl-2, while S1 released Bak from Mcl-1 (Supplementary Figure S11).
Taken together, our data explain why the pBcl-2/(Bcl-2+Mcl-1) ratio can predict the sensitivity to S1 treatment. It is because S1 disrupts the heterodimers containing Mcl-1 and npBcl-2 but not pBcl-2 dimers. As such, Bim and Bak shift between Mcl-1 and Bcl-2; pBcl-2/(Bcl-2+Mcl-1) determines whether proapoptotic proteins can be released by S1.
CML primary samples show greater S1 sensitivity when compared with other leukaemic cells
Further analysis of the pBcl-2/(Bcl-2+Mcl-1) ratios in the different types of leukaemic samples revealed that CML samples are the most sensitive to S1, with a mean LD50 value of 8.15 μ M. These data are in accordance with the lowest pBcl-2/(Bcl-2+Mcl-1) ratio of 0.18 (Figures 5A and B).

Chronic myeloid leukaemia was the most sensitive subset to S1 in the four types of primary leukaemia samples. Comparisons of S1 LD50 (A) and pBcl-2/(Bcl-2+Mcl-1) ratio (B) in the different type of primary leukaemic samples. **0.001<P<0.01, very significant; ***P<0.001, extremely significant.
Discussion
S1 is an authentic BH3 mimetic and a triple inhibitor of Bcl-2, Mcl-1 (Zhang et al, 2011) and Bcl-XL. It exhibits variable sensitivity in 5 established and 41 primary leukaemic cell lines with LD50 values ranging >2 logs (0.24–80.4 μ M). Through a semiquantitative analysis of western blot bands using the imaging software, we identified the pBcl-2/(Bcl-2+Mcl-1) ratio as the best predictive index for S1 sensitivity in both primary and established cells. This index may guide the preclinical studies of S1 for leukaemia in the future. Similar to previous findings that the relative ratio of (Mcl-1+BFL-1)/Bcl-2 provides a significant linear correlation with ABT-737 sensitivity (Al-Harbi et al, 2011), a combination of individual antiapoptotic proteins can predict the response to S1. This index thus reflects the complex interactions and crosstalk between Bcl-2 family members.
The predictive index in this study suggests an antiapoptotic function of pBcl-2 against S1. We have applied both the kinase inhibitor PD98059 and a phospho-mimetic mutant EEE-Bcl-2 plasmid to alter the levels of pBcl-2. The downregulation of pBcl-2 could improve the S1 response in resistant cell lines and primary samples, while the upregulation of pBcl-2 can enhance S1 resistance. In previous reports, pBcl-2 was found to reduce the activity of ABT-737 and obatoclax (Konopleva et al, 2006; Pérez-Galán et al, 2008). We propose a general mechanism for how Bcl-2 protein prevents apoptosis through its phosphorylation status. The demonstration of an antiapoptotic mechanism of pBcl-2 is crucial. Interestingly, although pBcl-2 was more abundant in resistant leukaemia cells than that in sensitive ones (Supplementary Figure S12), pBcl-2 itself cannot predict the sensitivity to S1 better than the pBcl-2/(Bcl-2+Mcl-1) ratio. This finding suggests that pBcl-2 has an antiapoptotic role in the network of the Bcl-2 family. S1 is a unique tool to investigate the exact functional role of pBcl-2 with other Bcl-2 family members. (I) It is the BH3 mimetic that acts through the Bcl-2-regulated apoptotic pathway only (Zhang et al, 2011; Ting et al., 2012). By contrast, obatoclax can kill cells without Bax/Bak (Lei et al, 2006). (II) It is the pan-inhibitor that can antagonise Mcl-1, unlike ABT-737 (Konopleva et al, 2006). As such, S1 can reflect the protein–protein interactions (PPIs) of Bcl-2 family members accurately and comprehensively.
We investigated the PPIs related to pBcl-2 in the S1-resistant U937 cell line. Surprisingly, we demonstrated a dynamic displacement of Bim and Bak. When Bim and Bak are released by S1 from Mcl-1, they are transferred to pBcl-2. Moreover, S1 can only disrupt npBcl-2/Bak, npBcl-2/Bim and npBcl-2/Bax but not pBcl-2/Bak, pBcl-2/Bim and pBcl-2/Bax complexes. This finding suggests that either the affinity of pBcl-2 to proapoptotic members is higher than that of npBcl-2 or that the binding pocket is so divergent with a common BH3 groove that it cannot be occupied by S1 or other pan-Bcl-2 inhibitors, such as obatoclax. The availability of pBcl-2/Bak, pBcl-2/Bax and pBcl-2/Bim (which S1 failed to interfere with) and the transfer of Bak and Bim from Mcl-1 to pBcl-2 reveal that the pBcl-2/(Bcl-2+Mcl-1) ratio rather than pBcl-2 itself is a better indicator of the S1 response in leukaemic cells. Nevertheless, pBcl-2 is a therapeutic target for leukaemia, and the combination of BH3 mimetics and kinase inhibitors could be a potent therapy.
In addition, our study suggests that different types of leukaemia exhibit variable basal pBcl-2 levels. CML is the most sensitive leukaemia to S1 due to its relatively low pBcl-2/(Bcl-2+Mcl-1) ratio. S1 may be a novel single agent therapeutic approach for CML.
A parallel analysis of the LD50 values of ABT-737 in 11 primary samples was shown in Supplementary Figure S6. The samples with high pBcl-2 levels were resistant to both ABT-737 and S1. By contrast, two samples with low pBcl-2/(Bcl-2+Mcl-1) ratio were sensitive to S1 but resistant to ABT-737 because of it high Mcl-1 expression level. For CLL samples, three samples with low Mcl-1 levels and high Bcl-2 levels were sensitive to ABT-737. Other samples with high levels of either pBcl-2 or Mcl-1 were resistant to ABT-737. We noted that CLL samples are not particularly sensitive to S1. This may be attributed to the fact that S1 is characterised as having pan-antiapoptotic Bcl-2 family activity with Ki’s low for Mcl-1, and 4- to 6-fold higher for Bcl-2 and Bcl-XL. The CLL samples practically expressed high levels of Bcl-2 or Bcl-XL, which S1 is only partially effective in antagonising.
In conclusion, S1 can efficiently kill different types of leukaemic cells by inhibiting Bcl-2, Mcl-1, and Bcl-XL. However, pBcl-2 reduces the S1 cytotoxicity by sequestering Bak and Bim, which are released from Mcl-1 upon S1 exposure. As such, it is the pBcl-2/(Bcl-2+Mcl-1) ratio that provides the predictive power for S1. CML is the most sensitive type of leukaemia to S1 due to its low pBcl-2/(Bcl-2+Mcl-1) ratio.
Change history
14 May 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adams JM, Cory S (1998) The Bcl-2 protein family: arbiters of cell survival. Science 281: 1322–1326
Adams JM, Cory S (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26: 1324–1337
Al-Harbi S, Hill BT, Mazumder S, Singh K, Devecchio J, Choudhary G, Rybicki LA, Kalaycio M, Maciejewski JP, Houghton JA, Almasan A (2011) An antiapoptotic BCL-2 family expression index predicts the response of chronic lymphocytic leukaemia toABT-737. Blood 118: 3579–3590
Andreeff M, Jiang S, Zhang X, Konopleva M, Estrov Z, Snell VE, Xie Z, Okcu MF, Sanchez-Williams G, Dong J, Estey EH, Champlin RC, Kornblau SM, Reed JC, Zhao S (1999) Expression of Bcl-2-related genes in normal and AML progenitors: changes induced by chemotherapy and retinoic acid. Leukaemia 13: 1881–1892
Dai H, Meng XW, Lee S-H, Schneider PA, Kaufmann SH (2009) Context-dependent Bcl-2/Bak interactions regulate lymphoid cell apoptosis. J Biol Chem 284: 18311–18322
Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A (2007) BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell 12: 171–185
Deng X, Gao F, Flagg T, May Jr WS (2004) Mono- and multisite phosphorylation enhances Bcl2’s antiapoptotic function and inhibition of cell cycle entry functions. Proc Natl Acad Sci USA 101: 153–158
Deng X, Kornblau SM, Ruvolo PP, May WS Jr (2000) Regulation of Bcl2 phosphorylation and potential significance for leukemic cell chemoresistance. J Natl Cancer Inst Monogr 28: 30–37
Fonseca R, Bergsagel PL, Drach J, Shaughnessy J, Gutierrez N, Stewart AK, Morgan G, Van Ness B, Chesi M, Minvielle S, Neri A, Barlogie B, Kuehl WM, Liebisch P, Davies F, Chen-Kiang S, Durie BG, Carrasco R, Sezer O, Reiman T, Pilarski L, Avet-Loiseau H International Myeloma Working Group (2009) International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukaemia 23: 2210–2221
Ito T, Deng X, Carr B, May WS (1997) Bcl-2 phosphorylation required for anti-apoptosis function. J Biol Chem 272: 11671–11673
Jaiswal S, Traver D, Miyamoto T, Akashi K, Lagasse E, Weissman IL (2003) Expression of BCR/ABL and BCL-2 in myeloid progenitors leads to myeloid leukaemias. Proc Natl Acad Sci USA 100: 10002–10007
Kitada S, Andersen J, Akar S, Zapata JM, Takayama S, Krajewski S, Wang HG, Zhang X, Bullrich F, Croce CM, Rai K, Hines J, Reed JC (1998) Expression of apoptosis-regulating proteins in chronic lymphocytic leukaemia: correlations with in vitro and in vivo chemoresponses. Blood 91: 3379–3389
Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi YX, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, May WS, Reed JC, Andreeff M (2006) Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukaemia. Cancer Cell 10: 375–388
Lei X, Chen Y, Du G, Yu W, Wang X, Qu H, Xia B, He H, Mao J, Zong W, Liao X, Mehrpour M, Hao X, Chen Q (2006) Gossypol induces Bax/Bak-independent activation of apoptosis and cytochrome c release via a conformational change in Bcl-2. FASEB J 20: 2147–2149
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192
Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB (2000) The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell 6: 1389–1399
May WS, Tyler PG, Ito T, Armstrong DK, Qatsha KA, Davidson NE (1994) Interleukin-3 and bryostatin-1 mediate hyperphosphorylation of BCL2 alpha in association with suppression of apoptosis. J Biol Chem 269: 26865–26870
Moldoveanu T, Liu Q, Tocilj A, Watson M, Shore G, Gehring K (2006) The X-ray structure of a BAK homodimer reveals an inhibitory zinc binding site. Mol Cell 24: 677–688
Mott JL, Bronk SF, Mesa RA, Kaufmann SH, Gores GJ (2008) BH3-only protein mimetic obatoclax sensitizes cholangiocarcinoma cells to Apo2L/TRAIL-induced apoptosis. Mol Cancer Ther 7: 2339–2347
Packham G, Stevenson FK (2005) Bodyguards and assassins: Bcl-2 family proteins and apoptosis control in chronic lymphocytic leukaemia. Immunology 114: 441–449
Pérez-Galán P, Roué G, López-Guerra M, Nguyen M, Villamor N, Montserrat E, Shore GC, Campo E, Colomer D (2008) BCL-2 phosphorylation modulates sensitivity to the BH3 mimetic GX15-070 (Obatoclax) and reduces its synergistic interaction with bortezomib in chronic lymphocytic leukaemia cells. Leukaemia 22: 1712–1720
Roberts AW, Seymour JF, Brown JR, Wierda WG, Kipps TJ, Khaw SL, Carney DA, He SZ, Huang DC, Xiong H, Cui Y, Busman TA, McKeegan EM, Krivoshik AP, Enschede SH, Humerickhouse R (2012) Substantial susceptibility of chronic lymphocytic leukaemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol 30: 488–496
Ruvolo PP, Deng X, May WS (2001) Phosphorylation of Bcl2 and regulation of apoptosis. Leukaemia 15: 515–522
Salomons GS, Smets LA, Verwijs-Janssen M, Hart AA, Haarman EG, Kaspers GJ, Wering EV, Der Does-Van Den Berg AV, Kamps WA (1999) Bcl-2 family members in childhood acute lymphoblastic leukaemia: relationships with features at presentation, in vitro and in vivo drug response and long-term clinical outcome. Leukaemia 13: 1574–1580
Stam RW, Den Boer ML, Schneider P, de Boer J, Hagelstein J, Valsecchi MG, de Lorenzo P, Sallan SE, Brady HJ, Armstrong SA, R Pieters (2010) Association of high-level MCL-1 expression with in vitro and in vivo prednisone resistance in MLL-rearranged infant acute lymphoblastic leukaemia. Blood 115: 1018–1025
Ting Song Xilong, Chang Zhichao, Zhang Yubo, Liu, Shen Xiaoyun (2012) S1, a novel pan-BH3 mimetic, induces apoptosis in Mcl-1-overexpressing cells through Bak. J Pharmacol Sci 119: 330–340
Vogler M, Butterworth M, Majid A, Walewska RJ, Sun XM, Dyer MJ, Cohen GM (2009) Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 113: 4403–4413
Weerasinghe P, Hallock S, Tang SC, Liepins A (2001) Role of Bcl-2 family proteins and caspase-3 in sanguinarine-induced bimodal cell death. Cell Biol Toxicol 17: 371–381
Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730
Youle RJ, Strasser A (2008) The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59
Zhang Z, Jin L, Qian X, Wei M, Wang Y, Wang J, Yang Y, Xu Q, Xu Y, Liu F (2007) Novel Bcl-2 inhibitors: discovery and mechanism study of small organic apoptosis-inducing agents. ChemBioChem 8: 113–121
Zhang Z, Song T, Zhang T, Gao J, Wu G, An L, Du G (2011) A novel BH3 mimetic S1 potently induces Bax/Bak-dependent apoptosis by targeting both Bcl-2 and Mcl-1. Int J Cancer 128: 1724–1735
Zhang Z, Wu G, Gao J, Song T (2010) Inclusion complex of a Bcl-2 inhibitor with cyclodextrin: characterization, cellular accumulation, and in vivo antitumor activity. Mol Pharms 7: 1348–1354
Zhang Z, Wu G, Xie F, Song T, Chang X (2011) 3-Thiomorpholin-8-oxo-8H-acenaphtho[1,2-b]pyrrole-9-carbonitrile (S1) based molecules as potent, dual inhibitors of B-cell lymphoma 2 (Bcl-2) and myeloid cell leukaemia sequence 1 (Mcl-1): structure-based design and structure–activity relationship studies. J Med Chem 54: 1101–1105
Acknowledgements
We would like to gratefully acknowledge support from National Natural Science Foundation of China (81272876). We also thank Kalle Gehring (McGill University, Montreal, QC, Canada) for providing plasmids. WT and DKO MEF cells were a gift from Dr Andreas Strasser (The Walter and Eliza Hall Institute of Medical Research, Melbourne, VIC, Australia).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zhang, Z., Liu, Y., Song, T. et al. An antiapoptotic Bcl-2 family protein index predicts the response of leukaemic cells to the pan-Bcl-2 inhibitor S1. Br J Cancer 108, 1870–1878 (2013). https://doi.org/10.1038/bjc.2013.152
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.152
Keywords
This article is cited by
-
Rationally derived drug combinations with the novel Mcl-1 inhibitor EU-5346 in breast cancer
Breast Cancer Research and Treatment (2019)
-
Myeloid cell leukemia-1 is an important apoptotic survival factor in triple-negative breast cancer
Cell Death & Differentiation (2015)
