
Abstract
Background:
Evasion of immune surveillance and suppression of the immune system are important hallmarks of tumour development in colon cancer. The goal of this study was to establish a tumour profile based on biomarkers that reflect a tumour’s immune susceptibility status and to determine their relation to patient outcome.
Methods:
The study population consisted of 285 stage I-IV colon cancer patients of which a tissue micro array (TMA) was available. Sections were immunohistochemically stained for the presence of Foxp3+ cells and tumour expression of HLA Class I (HLA-A, -B, -C) and non-classical HLA-E and HLA-G. All markers were combined for further analyses, resulting in three tumour immune phenotypes: strong immune system tumour recognition, intermediate immune system tumour recognition and poor immune system tumour recognition.
Results:
Loss of HLA class I expression was significantly related to a better OS (P-value 0.005) and DFS (P-value 0.008). Patients with tumours who showed neither HLA class I nor HLA-E or -G expression (phenotype a) had a significant better OS and DFS (P-value <0.001 and 0.001, respectively) compared with phenotype b (OS HR: 4.7, 95% CI: 1.2–19.0, P=0.001) or c (OS HR: 8.2, 95% CI: 2.0–34.2, P=0.0001). Further, the tumour immune phenotype was an independent predictor for OS and DFS (P-value 0.009 and 0.013, respectively).
Conclusion:
Tumours showing absence of HLA class I, HLA-E and HLA-G expressions were related to a better OS and DFS. By combining the expression status of several immune-related biomarkers, three tumour immune phenotypes were created that related to patient outcome. These immune phenotypes represented significant, independent, clinical prognostic profiles in colon cancer.
Similar content being viewed by others


Main
Historically, the immune system has been attributed an important role in controlling tumour growth and metastasis (Halvorsen and Seim, 1989a; Naito et al, 1998; Murphy et al, 2000a; Menon et al, 2004a). Evasion of immune surveillance and suppression of the immune system are two important traits cancer cells have to acquire during the process of tumorigenesis (Cavallo et al, 2011). Research of the last century has indicated that the influence of the immune system on tumour cells, both in the tumour micro-environment as well as during the process of tumour metastasis, also contributes to tumour progression (Schreiber et al, 2011). The cancer immune-editing hypothesis describes both the host-protective as well as the tumour-promoting actions the immune system might have on developing tumours, shaping tumour immunogenicity (Dunn et al, 2002, 2004a, 2004b, 2006; Smyth et al, 2006; Swann and Smyth, 2007; Vesely et al, 2011). Tumours are thought to be ‘edited’ through a Darwinian selection process into poorly immunogenic tumour cell variants invisible to the immune system and able to grow progressively. Immune editing might therefore have substantial effects on patient’s prognosis.
Several mechanisms taking place at the tumour cell level contribute to this process. The first mechanism is downregulation of human leukocyte antigen (HLA) class I expression. Downregulation of HLA class I minimises the level of tumour-associated antigen (TAA) expression by tumour cells and therefore their recognition and subsequently destruction by cytotoxic T cells (CTL) (Halvorsen and Seim, 1989b; Murphy et al, 2000b; Menon et al, 2004b; Cavallo et al, 2011). The second mechanism is the ability of tumour cells to regulate the expression of non-classical HLA class I molecules (HLA-E and HLA-G) on the cell surface. Expression of these markers has been found to inhibit natural killer (NK) cell recognition in the blood stream and therefore results in further tumour cell escape from immune surveillance (Khong and Restifo, 2002; Marin et al, 2003; Wischhusen et al, 2007; Zilberman et al, 2012). HLA-E is regularly expressed in various healthy tissues and correlated with HLA class I expression (Palmisano et al, 2005). In contrast, HLA-G is rarely found in healthy tissues but is frequently observed in tumours (Wischhusen et al, 2007). Third, tumour cell immune reactivity can become suppressed by the attraction of immunosuppressive regulatory T cells (Tregs) into the tumour micro-environment (Zitvogel et al, 2006; Nosho et al, 2010). Tregs are able to modulate the antitumour immune response, as they suppress the activity of CTL through direct cell-to-cell contact or via the release of cytokines like transforming growth factor-β (Needham et al, 2006; Zou, 2006; Curiel, 2007). Tregs and CTLs therefore show opposing actions in tumour immunity (Liu et al, 2011).
Previously, both the downregulation of HLA class I, presence of Tregs and HLA-E and -G expressions have been shown to be of clinical relevance in several types of cancers (Curiel et al, 2004; Hiraoka et al, 2006; Gao et al, 2007; de Kruijf et al, 2010b). In colorectal cancer (CRC), various studies have described the impact of the level of HLA class I tumour expression or the presence of Foxp3+ Treg cells on patients with varying results (Moller et al, 1991; Menon et al, 2002; Loddenkemper et al, 2006; Watson et al, 2006; Benevolo et al, 2007; Salama et al, 2009; Deng et al, 2010; Suzuki et al, 2010). In general, loss of HLA class I tumour expression seemed to result in a better prognosis (Sandel et al, 2005; Watson et al, 2006). The presence of high levels of Foxp3+ cells in CRC patients was related to a worse prognosis in some studies, although this relation could not always be established in CRC patients (Loddenkemper et al, 2006; Salama et al, 2009; Deng et al, 2010; Suzuki et al, 2010; Zeestraten et al, 2011). Studies on the prognostic value of HLA-E and HLA-G showed that expression of these molecules correlated with poor prognosis and tumour progression (Ye et al, 2007; Yie et al, 2007a, 2007b, 2007c).
Previous studies have shown a complex interaction between different immune markers, highlighting the need for combined marker analysis (de Kruijf et al, 2010a, 2010b; Zeestraten et al, 2011). The purpose of this study was to investigate the prognostic value of the immune-related biomarkers, HLA Class I, HLA-E and -G and Foxp3+, to establish distinct patterns that reflect a tumour’s immune-escape mechanism by combining these markers and to relate these patterns to clinical outcome.
Materials and methods
Study population
The patient population comprised a consecutive series of 470 colorectal cancer patients all treated with surgery for their primary tumour in the Leiden University Medical Center (LUMC) between 1991 and 2001. Of these patients’ tumour materials, clinico-pathological data and information on the follow-up were collected in retrospect. This research was performed according to the code of conduct for responsible use. Mucinous differentiation was defined as fully (>50%), partly (0–50%) or no mucinous differentiation. Tumour-node-metastasis (TNM) was defined by the Union for International Cancer Control (UICC) (Schiffmann et al, 2013). Tumour differentiation was defined as good, moderate or poor, as described in the pathology report. Patients with rectal cancer, patients with a history of cancer other than basal cell carcinoma or cervical carcinoma in situ, patients with more than one colon tumour at the same time and patients who received radio- or chemotherapy treatment before resection were excluded from the analysis (n=185 in total). The study cohort therefore consisted of 285 colon cancer patients.
Antibodies
The mouse monoclonal antibodies HCA2 and HC10 were used, which recognise the heavy chains of HLA Class I, and were kindly provided by Professor Dr. J. Neefjes. The reactivity spectrum of HCA2 comprises all HLA-A chains (except HLA-A24), as well as some HLA-B, HLA-C, HLA-E, HLA-F and HLA-G chains. HC10 reacts with HLA-B and HLA-C heavy chains and some HLA-A (HLA-A10, HLA-A28, HLA-A29, HLA-A30, HLA-A31, HLA-A32 and HLA-A33) (de Kruijf et al, 2010a). The mouse antibodies against human Foxp3 (ab20034 clone 236A/E7; Abcam) were used for Treg identification. The reactivity spectrum of Foxp3 is composed of regulatory T cells and may include small numbers of CD8+ cells but is generally considered to be the best single marker for Treg identification (Hill et al, 2007; Generali et al, 2009). For HLA-E and HLA-G identification, mouse monoclonal antibodies against HLA-E (ab2216 clone MEM-E/02: AbCam, UK) and HLA-G (4H84: Exbio, Czech Republic) were used. MEM-E/02 recognises denatured HLA-E (Menier et al, 2003; Lo et al, 2008), whereas 4H84 recognises denatured HLA-G molecules and also binds to free heavy chains of classical HLA class I molecules (Menier et al, 2003; Polakova et al, 2004; Zhao et al, 2012).
TMA production and immunohistochemistry
The histopathological characteristics of the tumour material from all patients included were determined by qualified pathologists according to the current standards. Of the formalin-fixed paraffin-embedded (FFPE) tumour blocks of the primary tumours, sections were cut for haematoxylin and eosin staining. On the basis of microscopic inspection of the slides, histopathologically representative bulk tumour regions from each tumour block were identified and punched for preparation of tumour tissue microarray (TMA) blocks. From each donor block, three 0.6 mm diameter tissue cores were punched from the identified tumour areas and transferred into a receiver paraffin block using a custom-made precision instrument. Immunohistochemical staining (IHC) for Foxp3+ cells, non-classical HLA-E and HLA-G and classical HLA class I tumour expression was performed on 4 μm-thick sections, which were cut from each receiver block and mounted on glass.
The sections were deparaffinised and rehydrated according to the standard procedures. Endogenous peroxidase was blocked for 20 min in 0.3% hydrogen peroxide in PBS. For antigen retrieval, slides were boiled in 0.01 M EDTA buffer (pH 8) for 10 min at maximum power in a microwave oven. Sections were incubated overnight with anti-Foxp3+, -HLA-E or -HLA-G antibodies at predetermined optimal dilution. After 30 min of incubation with Envision anti-mouse (K4001; DAKO Cytomation, Glostrup, Denmark), sections were visualised using diaminobenzidine solution (DAB+). Tissue sections were counterstained with haematoxylin, dehydrated and finally mounted in pertex. The IHC for HCA2 and HC10 was performed using the Autostainer Link 48 (DAKO). For antigen retrieval, Envision TM Target Retrieval Solution (DAKO), pH low, was used. The sections were incubated for 18 h with either HCA2 or HC10 antibodies at predetermined optimal dilution, followed by incubation with Envision FLEX/HRP (DAKO). Sections were visualised using DAB+ liquid solution (DAKO). Finally these slides were counterstained with haematoxylin as well, dehydrated and finally mounted in pertex. All slides were stained simultaneously to avoid interassay variation. For each patient, normal epithelium, stromal cells or lymphoid cells served as internal positive control for HLA class I antibody reactivity. Placenta tissue slides served as positive control for HLA-E and HLA-G staining. Slides from human tonsil tissue served as positive control for Foxp3+ staining. Negative controls were tissue slides that did undergo the whole immunohistochemical staining without primary antibody.
Evaluation of immunohistochemistry
Microscopic analysis of HCA2, HC10, HLA-E and HLA-G expressions and presence of Foxp3+cells was performed by two independent observers in a blinded manner (M.S.R.: 100% of the cohort, E.C.M.Z. 30% of the cohort). The Cohen’s Kappa was >0.75 for all stainings, indicating substantial agreement between the two observers. The scores of the three 0.6 mm punches were averaged. For HCA2 and HC10, the percentage of tumour cells with membranous staining was assessed. HLA class I expression status was determined according to the standard set by the International HLA and Immunogenetics Workshop (Chew et al, 2007). HCA2 and HC10 expression percentages were divided into two categories: 0–5% of the tumor cells show expression and 5–100% show expression. If <5% of the tumour cells showed expression for each of the two markers, this was determined to represent loss of HLA class I expression; if expression in <5% of the tumour cells of one of the two markers, this was determined as HLA class 1 downregulation; and if expression in >5% of the tumour cells for each of the two markers, this was denoted as HLA class I expression. For HLA-E and HLA-G, intensity of tumour staining (absent, weak, moderate or strong intensity) was determined. For HLA-E, an absence of staining and weak staining together vs moderate and strong staining together were used for the final analysis. For HLA-G, an absence of tumour staining was analysed vs weak, moderate and strong tumour staining together, because HLA-G is normally not expressed on healthy tissues in comparison with HLA-E (Palmisano et al, 2005; Wischhusen et al, 2007). Quantification of the number of Foxp3+ cells was microscopically assessed in the entire tumour punches of the TMA, and the absolute number of positive cells was used for the analysis.
Determination of microsatellite stability status
DNA was extracted from 2 mm tumour cores. Paraffin was dissolved in xylene, tissue was rehydrated in ethanol (100%/70%) and dried for 10 min at 37 °C. Nucleospin 96 Tissue kit (Machery-Nagel, Düren, Germany) was used for DNA extraction according to the manufacturer’s protocol.
MSS status was tested using the MSI Analysis System Version 1.2 (Promega, Mannheim, Germany) and interpreted by an experienced pathologist, as described previously (Zeestraten et al, 2012).
Statistical analysis
Statistical analyses were performed using the statistical package SPSS (version 17.0 for Windows; IBM, Armonk, NY, USA). The Student’s t-test and the χ2-test were used to evaluate associations between tumour expressions of HLA class I, and non-classical HLA-E and HLA-G and tumour infiltration of Foxp3+ cells and various clinico-pathological variables. Overall survival (OS) was defined as time of surgery until death and disease-free survival (DFS) as time of surgery until death or relapse of disease, whichever came first. The Kaplan–Meier method was used for calculation of survival probabilities and the log-rank test for comparison of survival curves between these three phenotypes. Cox regression was used for univariate and multivariate analysis for OS and DFS. Significant variables (P<0.05) in univariate analysis were included in multivariate analysis.
Results
HLA class I expression
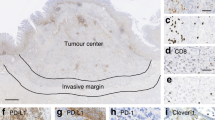
Microscopic quantification of HLA class I expression was performed on 242 patients as, due to staining artifacts and loss of material during the staining procedure, the IHC results of 43 cases could not be analysed. Representative images of HLA class 1 staining and frequencies of HLA class I expression in the different groups are shown in Figures 1 and 2. Patient characteristics and data on HLA class I expression are shown in Table 1. As HCA2 also reacts with some HLA-G chains (de Kruijf et al, 2010a), we examined the relationship between HCA-2 reactivity and HLA-G expression and found no correlation (P=0.348).

Representative images of HLA class I, HLA-E, HLA-G and Foxp3+ staining. Representative images of immuno-histochemical stainings for HLA Class I expression (HCA2 and HC10), HLA-E and HLA-G expression and presence of FOXP3+ on the left side with magnifications on the right side, performed according to standard protocols (details in Material and Methods). (A) HCA2-positive tumour (B) HC10-positive tumour (C) HC10-negative tumour with positive internal control (D) HLA-E-positive tumour (E) HLA-G- positive tumour and (F) Presence of Foxp3+ cells as indicated by the arrows.

Frequencies of HLA class I tumour expression, Foxp3+ tumour infiltration and HLA-E and -G tumour expression. Pie-charts indicating the frequencies of all stainings including missings due to staining artifacts and loss during the staining procedure. Details about group composition and scoring methods are written in Material and Methods. (A) Frequency of HLA class I tumour expression. (B) Frequency of Foxp3+ tumour cell infiltration. (C) Frequencies of HLA-E tumour expression. (D) Frequency of HLA-G tumour expression.
Patients whose tumours showed loss of HLA class I had a significantly better OS and DFS (logrank P-value 0.005 and 0.008) compared with patients with tumours with HLA class I downregulation or expression (Figure 3). The hazard ratios (HRs) for OS and DFS for HLA class I tumour expression are shown in Tables 2a and 2b.

Survival curves stratified for HLA class I tumour expression in colon cancer. (A) Kaplan–Meier curve for OS in the study population of 285 colon cancer patients stratified for HLA class I tumour expression status. (B) Kaplan–Meier curve for DFS in the study population of 285 CRC patients again stratified for HLA class I tumour expression.
Foxp3+ cells
The number of Foxp3+ cells could be evaluated only in 245 patients, because, due to staining artefacts and loss of material during the staining procedure, the IHC results of 40 cases could not be analysed. The mean number of positive cells per tumour punch was 19 with a median of 12.0. In 4.1% (n=10) of the patients, no Foxp3+ cells were present. Representative images of Foxp3+ staining are shown in Figure 1. Patients with expression of HLA class I showed significant higher levels of Foxp3+ cells at borderline significance in their tumour punches compared with HLA class I downregulation or loss: a mean of 21 Foxp3+ cells in the expression group vs a mean of 12 and 14 positive cells in the downregulation group and in the loss of HLA class I group, respectively, P=0.07. Patients with stage 1 tumours showed significantly higher levels of Foxp3+ cells compared with patients with stage 2, stage 3 and stage 4 tumours: mean level of Foxp3+ cells in stage 1 tumours was 38 compared with 13, 17 and 20 for the stage 2, 3 and 4 tumours, P=<0.001. For further analysis, Foxp3+ was categorised as below vs above median, based on the median due to the skewness in the spread of the data. Frequencies are shown in Figure 2. The presence of Foxp3+ cells in the tumour micro-environment was not related to OS (logrank P-value 0.114) or DFS (logrank P-value 0.155).
HLA-E and HLA-G
Representative images for HLA-E and HLA-G and frequencies in the different groups are shown in Figures 1 and 2. HLA-E and HLA-G were not related to OS and DFS (logrank P-values for OS 0.809 and 0.239, respectively, logrank P-values for DFS 0.876 and 0.117, respectively). None of the clinico-pathological characteristics were significantly related to tumour expression of HLA-E or HLA-G (data not shown).
A combined variable of HLA-E and HLA-G scores was created (cEG). Expression was considered positive when both HLA-E and HLA-G were expressed (HLA-E+/-G+ further denoted as cEG+) and negative when either HLA-E or HLA-G was not expressed (HLA-E+/-G- or HLA-E-/-G+ or HLA-E-/-G- further denoted as cEG-). Positive cEG was found in 14.7% (42 of 244) of tumours. Patient characteristics and data on the combined variable HLA-E and -G expression can be found in Table 1. None of the clinico-pathological variables shown in Table 1 were significantly related to tumour expression of cEG. cEG was not significantly related to OS (logrank P-value 0.245) and DFS (logrank P-value 0.100).
Multivariate analysis for single immune markers
Both for OS and DFS, a univariate analysis was performed for the following parameters: sex, age, TNM stage, HLA class I expression status, mucinous differentiation, tumour grade, adjuvant therapy and microsatellite status. In the univariate analysis for OS, age (P-value <0.001), TNM status (P-value <0.001) and HLA class I expression status (P-value 0.011) were significant predictors of survival. The same was true for the univariate analysis for DFS with a P-value of <0.001 for age and TNM status and a P-value of 0.02 for HLA class I expression. Therefore, all three were included in the multivariate analysis. In this analysis, age and TNM stage remained significant for both OS and DFS (OS and DFS P-values all <0.001): HLA class I was a borderline independent significant predictor for OS (P-value 0.08) (Tables 2a and 2b).
Analysis of tumour immune phenotypes
Except for HLA class I, none of the tumour immune markers showed a significant correlation with patients’ clinical outcome. The interaction between tumour cells and immune cells, however, is complex and multifaceted. Therefore, we hypothesised that analysis of combined tumour immune markers; describing a tumour’s immune phenotype may better reflect outcome of the interaction between tumour cells and the immune system. We combined all of the data into one combined variable. The Kaplan–Meier curves performed with this combined variable indeed revealed three distinct patterns in relation to patient outcome (Figures 4 and 5). The entire population could be divided into three phenotypes:
-
a)
Strong immune system tumour recognition: patients with tumours that showed loss of HLA class I expression, presence of Foxp3+ cells in the tumour micro-environment and negative cEG expression (n=11).
-
b)
Intermediate immune system tumour recognition: patients with tumours that showed downregulation of HLA class I expression and negative cEG expression but were found to have Foxp3+ cells in the tumour micro-environment or patients with tumours that showed normal HLA class I expression irrespective of cEG expression and the presence of Foxp3+ cells (n=184).
-
c)
Poor immune system tumour recognition: patients with tumours showing normal or downregulated HLA class I and no presence of Foxp3+ cells irrespective of their cEG expression (n=460).

Survival curves stratified for combined tumour expression of HLA class I, HLA-E, HLA-G and Foxp3+ in colon cancer. (A) Kaplan–Meier curve for OS in the study population of 285 colon cancer patients stratified for all the different combinations between tumour expression of HLA class I, combined expression of HLA-E and HLA-G (cEG) and the presence of Foxp3+ cells (Tregs) based on which three distinct patterns could be distinguished as shown in Figure 5 (B) Kaplan–Meier curve for DFS in the study population of 285 colon cancer patients stratified for all the different combinations between tumour expression of HLA class I, combined expression of HLA-E and HLA-G (cEG) and the presence of Foxp3+ cells (Tregs) based on which three distinct patterns could be distinguished as shown in Figure 5.

Survival curves stratified for immune phenotypes in colon cancer. (A) Kaplan–Meier curve for OS in the study population of 285 colon cancer patients stratified for all the different combinations between tumour expression of HLA class I, combined expression of HLA-E and HLA-G (cEG) and the presence of Foxp3+ cells (Tregs), based on which three immune phenotypes could be distinguished. See results section for explanation of the phenotypes. (B) Kaplan–Meier curve for DFS in the study population of 285 colon cancer patients stratified for all the different combinations between tumour expression of HLA class I, combined expression of HLA-E and HLA-G (cEG) and the presence of Foxp3+ cells (Tregs), based on which three immune phenotypes could be distinguished. See results section for explanation of the phenotypes.
These three phenotypes showed significant differences for OS (logrank P-value <0.001) and DFS (logrank P-value 0.001). The HRs of the three phenotypes for OS and DFS are shown in Tables 2a and 2b.
Multivariate analysis for tumour immune phenotypes
Again, both for OS and DFS, a univariate analysis was performed for the following parameters: sex, age, TNM stage, tumour immune phenotype, mucinous differentiation, tumour grade, adjuvant therapy and microsatellite status. In univariate analysis, next to age and TNM status, the tumour immune phenotype was a significant predictor for OS (P-value 0.001) and DFS (P-value 0.002). Therefore, all these three parameters were included in multivariate analysis. The tumour immune phenotype was an independent significant predictor for both OS (P-value 0.009) and DFS (P-value 0.013), and HRs are shown in Tables 2a and 2b.
Discussion
Tumour–immune interactions may be important for the prognosis of cancer patients (Khong and Restifo, 2002). In this study, we showed that, by combining the immune-related markers HLA class I, HLA-E, HLA-G and Foxp3+, we were able to determine three distinct patterns in survival, which might represent how immune surveillance controls tumour growth and metastasis.
The first marker of tumour immunogenicity used was the level of HLA class I expression of cancer cells. Our results are comparable with the results of other studies that were able to determine a prognostic effect of the HLA class I status in colon cancer (Menon et al, 2002; Watson et al, 2006). Watson et al (2006) showed that tumours with downregulation of HLA class I had a worse survival comparable with our results. In contrast, Menon et al (2002) showed a survival benefit in patients with downregulated HLA-A tumours. However, when HLA-A and HLA-B/C were combined, statistical significance was lost. Further, patients with expression of HLA class I were related to a better survival in the study by Watson et al (2006), whereas our study showed an improved survival in patients with loss of HLA class I expression. Possible explanations for these differences might be a different definition for HLA class I expression, differences in staining techniques and scoring or a different patient cohort, especially regarding the number of tumours showing microsatellite instability (MSI), which is associated with loss of HLA class I and a better prognosis (Dierssen et al, 2007; Mouradov et al, 2013). In our study, 33% of the tumours with loss of HLA class I showed the MSI phenotype, in comparison with 14% and 13% for HLA class I downregulation and expression. Results from Menon et al (2002) showed that 50% of the tumours with loss of HLA class I had the MSI phenotype. Unfortunately, Watson et al (2006) did not mention microsatellite status of their study cohort.
As hypothesised, loss of HLA class I expression in tumour cells could also be related to a better patient survival because such cells, once they metastasise to the bloodstream, are eliminated by NK cell attacks (Menon et al, 2002; Hokland and Kuppen, 2005; Watson et al, 2006). Tumours with loss of HLA class I have also shown to have significantly higher NK cell infiltration (Menon et al, 2004b). More interestingly, the tumours showing loss of HLA class I in our cohort were also the ones that showed to be negative for HLA-E and -G expression (phenotype a). Absence of the HLA-E and -G expression makes them even more susceptible to NK cell elimination (Khong and Restifo, 2002; Marin et al, 2003; Wischhusen et al, 2007; Zilberman et al, 2012). Further, this is also confirmed by CRC tumours with the loss of HLA class I expression, which do not metastasise to the liver (Tikidzhieva et al, 2012).
The presence of the third marker Foxp3+ is thought to represent the inhibition of host-protective antitumour responses. When stimulated, they inhibit the function of CTL (Schreiber et al, 2011). Although the exact mechanism by which these cells are drawn into the tumour micro-environment remains unexplained, their immunosuppressive effect has been proven with a high density of tumour-infiltrating Foxp3+ cells found to be associated with an unfavourable prognosis in a wide range of human carcinomas, including breast and lung cancer (Bates et al, 2006; Petersen et al, 2006). However, in colon cancer, different results are reported as well (Salama et al, 2009; Suzuki et al, 2010). One possible explanation for these opposite results might be a different micro-environment of colon cancer, which is colonised with many gastro-intestinal bacteria, triggering the production of pro-inflammatory cytokines causing tumour-enhancing effects. Instead of the specificity of infiltrating T cells for tumour antigens, T cells in colon cancer could be more specific for the microflora and suppress inflammation and immune responses from bacterial invasion, resulting in an antitumourigenic effect, which could explain the better prognosis of patients with tumours with a strong Foxp3+ infiltration (Ladoire et al, 2011). We were not able to demonstrate differences in disease outcome for Foxp3+ tumour infiltration supporting this latter hypothesis, but we did see differences in Foxp3+ infiltration if we combined them with HLA class I expression and with HLA-E and -G expression, especially in patients who have retained their HLA class I expression. Patients with normal HLA class I expression and absence of Foxp3+ cell infiltration showed a worse patient outcome. We hypothesise that the tumours of these patients have had a minimal CTL attack because the HLA class I expression is preserved, indicating no selective outgrowth of HLA class I-negative or downregulated tumours directed by CTL. As CTL and Foxp3+ cells show opposing actions (Liu et al, 2011) and CTLs are supposed to be absent in these tumours, Foxp3+ cell infiltration might not be necessary. These tumours could therefore progress aggressively as immune surveillance is poor. In contrary, tumours with HLA class I expression, which were able to attract Foxp3+ cells, showed a slightly better prognosis. In this case, Foxp3+ cell infiltration might indicate CTL activity, resulting in suppression of tumour growth.
Therefore, in our opinion, the clinical relevance of the studies by Watson et al (2006) and several others does not provide an optimal perspective on prognosis (Menon et al, 2002) because expression of a single immune marker is not sufficient for the selection of high-risk colon cancer patients or treatment allocation. As shown by our results and previous studies, immune markers are related to each other (Galon et al, 2006; de Kruijf et al, 2010a, 2010b; Angell and Galon, 2013).
When all markers were combined, patients showing the worst prognosis were patients with HLA class I downregulation, negative or positive cEG expression and absence of Foxp3+ cells denoted as phenotype c. We hypothesise that these poor immune system recognised tumours were able to elicit only a minimal CTL attack because they partly preserved HLA class I expression and subsequently attracted little to no Foxp3+ cells in their tumour micro-environment. Furthermore, these tumours showed a positive expression of HLA-E and -G, further escaping immune surveillance through inhibition of NK-cell recognition (Khong and Restifo, 2002; Marin et al, 2003; Wischhusen et al, 2007; Zilberman et al, 2012). These tumour cells can therefore quickly progress to the bloodstream and might eventually metastasise.
It is important to realise that what we are evaluating is just a ‘snapshot’ of the ongoing process of cancer immune editing in the patient’s primary tumour at time of resection. Still, from a clinical point of view, at the patient’s bedside this is usually the only data available based on which clinical decision making has to take place, and these data can actually be of clinical value to, for example, the allocation of adjuvant therapy as opposed by de Kruijf et al (2010b) in breast cancer and other studies (Ghiringhelli et al, 2004; Ladoire et al, 2008; ).
Our study does have a few limitations. Not all combinations between HLA class I, HLA-E and -G and Foxp3+ were present in our cohort. There was no representation of tumours with loss of HLA class I, which were HLA-E and -G positive. Therefore, we were not able to investigate the prognosis of these tumours, but we hypothesise that these tumours have a worse prognosis as these tumours might escape NK-cell attack. Although there is a physiological correlation between HLA-E and HLA class I molecules, this has been found to be disturbed in tumours, suggesting further escape from immune recognition through upregulation of HLA-E (de Palmisano et al, 2005; Kruijf et al, 2010a). To truly investigate these tumours, our study has to be validated in a bigger cohort. Second, the antibodies we used for HLA class I detection only detected the heavy chain but not the trimeric complex consisting of β2-microglobuline heavy chain and antigen (Perosa et al, 2003). Therefore, we should be careful using the term total loss of HLA class I. Third, we did not investigate the role of NK cells in patients with loss or downregulation of HLA class I, possibly explaining the positive prognostic effect of patients with loss of HLA class I expression. However, NK-cell infiltration at the tumour site is scarce, indicating that tumour staining for NK cells might be minimally informative (Sandel et al, 2005).
In conclusion we were able to identify local immune escape mechanisms of colon cancer, where the presence of Foxp3+ cell infiltration favors a better prognosis, indicating CTL activity. HLA-E and -G expressions might have a pivotal role in distant immune escape mechanisms; in case of loss or downregulation of HLA class I, HLA-E and -G expressions determine distant metastases and prognosis of colon cancer patients. Furthermore, we were able to determine three distinct survival patterns in colon cancer patients based on immune surveillance. In the future, these findings might contribute to better treatment allocation and maybe even the development of new cancer immuno-therapies.
Change history
21 January 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Angell H, Galon J (2013) From the immune contexture to the Immunoscore: the role of prognostic and predictive immune markers in cancer. Curr Opin Immunol 25 (2): 261–267.
Bates GJ, Fox SB, Han C, Leek RD, Garcia JF, Harris AL, Banham AH (2006) Quantification of regulatory T cells enables the identification of high-risk breast cancer patients and those at risk of late relapse. J Clin Oncol 24 (34): 5373–5380.
Benevolo M, Mottolese M, Piperno G, Sperduti I, Cione A, Sibilio L, Martayan A, Donnorso RP, Cosimelli M, Giacomini P (2007) HLA-A, -B, -C expression in colon carcinoma mimics that of the normal colonic mucosa and is prognostically relevant. Am J Surg Pathol 31 (1): 76–84.
Cavallo F, De GC, Nanni P, Forni G, Lollini PL (2011) 2011: the immune hallmarks of cancer. Cancer Immunol Immunother 60 (3): 319–326.
Chew SF, Kanaan C, Tait BD (2007) HLA expression and cancer—14th IHIWS immunohistochemistry quality control exercise exchange results. Tissue Antigens 69 (Suppl 1): 248–251.
Curiel TJ (2007) Tregs and rethinking cancer immunotherapy. J Clin Invest 117 (5): 1167–1174.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon-Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10 (9): 942–949.
de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ (2010a) HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol 185 (12): 7452–7459.
de Kruijf EM, van Nes JG, Sajet A, Tummers QR, Putter H, Osanto S, Speetjens FM, Smit VT, Liefers GJ, van de Velde CJ, Kuppen PJ (2010b) The predictive value of HLA class I tumor cell expression and presence of intratumoral Tregs for chemotherapy in patients with early breast cancer. Clin Cancer Res 16 (4): 1272–1280.
Deng L, Zhang H, Luan Y, Zhang J, Xing Q, Dong S, Wu X, Liu M, Wang S (2010) Accumulation of foxp3+ T regulatory cells in draining lymph nodes correlates with disease progression and immune suppression in colorectal cancer patients. Clin Cancer Res 16 (16): 4105–4112.
Dierssen JW, de Miranda NF, Ferrone S, Van Puijenbroek M, Cornelisse CJ, Fleuren GJ, van Wezel T, Morreau H (2007) HNPCC versus sporadic microsatellite-unstable colon cancers follow different routes toward loss of HLA class I expression. BMC Cancer 7: 33.
Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD (2002) Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol 3 (11): 991–998.
Dunn GP, Koebel CM, Schreiber RD (2006) Interferons, immunity and cancer immunoediting. Nat Rev Immunol 6 (11): 836–848.
Dunn GP, Old LJ, Schreiber RD (2004a) The immunobiology of cancer immunosurveillance and immunoediting. Immunity 21 (2): 137–148.
Dunn GP, Old LJ, Schreiber RD (2004b) The three Es of cancer immunoediting. Annu Rev Immunol 22: 329–360.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoue F, Bruneval P, Cugnenc PH, Trajanoski Z, Fridman WH, Pages F (2006) Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313 (5795): 1960–1964.
Gao Q, Qiu SJ, Fan J, Zhou J, Wang XY, Xiao YS, Xu Y, Li YW, Tang ZY (2007) Intratumoral balance of regulatory and cytotoxic T cells is associated with prognosis of hepatocellular carcinoma after resection. J Clin Oncol 25 (18): 2586–2593.
Generali D, Bates G, Berruti A, Brizzi MP, Campo L, Bonardi S, Bersiga A, Allevi G, Milani M, Aguggini S, Dogliotti L, Banham AH, Harris AL, Bottini A, Fox SB (2009) Immunomodulation of FOXP3+ regulatory T cells by the aromatase inhibitor letrozole in breast cancer patients. Clin Cancer Res 15 (3): 1046–1051.
Ghiringhelli F, Larmonier N, Schmitt E, Parcellier A, Cathelin D, Garrido C, Chauffert B, Solary E, Bonnotte B, Martin F (2004) CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 34 (2): 336–344.
Halvorsen TB, Seim E (1989a) Association between invasiveness, inflammatory reaction, desmoplasia and survival in colorectal cancer. J Clin Pathol 42 (2): 162–166.
Halvorsen TB, Seim E (1989b) Association between invasiveness, inflammatory reaction, desmoplasia and survival in colorectal cancer. J Clin Pathol 42 (2): 162–166.
Hill JA, Feuerer M, Tash K, Haxhinasto S, Perez J, Melamed R, Mathis D, Benoist C (2007) Foxp3 transcription-factor-dependent and -independent regulation of the regulatory T cell transcriptional signature. Immunity 27 (5): 786–800.
Hiraoka N, Onozato K, Kosuge T, Hirohashi S (2006) Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin Cancer Res 12 (18): 5423–5434.
Hokland M, Kuppen PJ (2005) Natural killer cells: from ‘disturbing’ background to central players of immune responses. Mol Immunol 42 (4): 381–383.
Khong HT, Restifo NP (2002) Natural selection of tumor variants in the generation of ‘tumor escape’ phenotypes. Nat Immunol 3 (11): 999–1005.
Ladoire S, Arnould L, Apetoh L, Coudert B, Martin F, Chauffert B, Fumoleau P, Ghiringhelli F (2008) Pathologic complete response to neoadjuvant chemotherapy of breast carcinoma is associated with the disappearance of tumor-infiltrating foxp3+ regulatory T cells. Clin Cancer Res 14 (8): 2413–2420.
Ladoire S, Martin F, Ghiringhelli F (2011) Prognostic role of FOXP3+ regulatory T cells infiltrating human carcinomas: the paradox of colorectal cancer. Cancer Immunol Immunother 60 (7): 909–918.
Liu F, Lang R, Zhao J, Zhang X, Pringle GA, Fan Y, Yin D, Gu F, Yao Z, Fu L (2011) CD8(+) cytotoxic T cell and FOXP3(+) regulatory T cell infiltration in relation to breast cancer survival and molecular subtypes. Breast Cancer Res Treat 130 (2): 645–655.
Lo ME, Sibilio L, Melucci E, Tremante E, Suchanek M, Horejsi V, Martayan A, Giacomini P (2008) HLA-E: strong association with beta2-microglobulin and surface expression in the absence of HLA class I signal sequence-derived peptides. J Immunol 181 (8): 5442–5450.
Loddenkemper C, Schernus M, Noutsias M, Stein H, Thiel E, Nagorsen D (2006) In situ analysis of FOXP3+ regulatory T cells in human colorectal cancer. J Transl Med 4: 52.
Marin R, Ruiz-Cabello F, Pedrinaci S, Mendez R, Jimenez P, Geraghty DE, Garrido F (2003) Analysis of HLA-E expression in human tumors. Immunogenetics 54 (11): 767–775.
Menier C, Saez B, Horejsi V, Martinozzi S, Krawice-Radanne I, Bruel S, Le DC, Reboul M, Hilgert I, Rabreau M, Larrad ML, Pla M, Carosella ED, Rouas-Freiss N (2003) Characterization of monoclonal antibodies recognizing HLA-G or HLA-E: new tools to analyze the expression of nonclassical HLA class I molecules. Hum Immunol 64 (3): 315–326.
Menon AG, Janssen-van Rhijn CM, Morreau H, Putter H, Tollenaar RA, van de Velde CJ, Fleuren GJ, Kuppen PJ (2004a) Immune system and prognosis in colorectal cancer: a detailed immunohistochemical analysis. Lab Invest 84 (4): 493–501.
Menon AG, Janssen-van Rhijn CM, Morreau H, Putter H, Tollenaar RA, van de Velde CJ, Fleuren GJ, Kuppen PJ (2004b) Immune system and prognosis in colorectal cancer: a detailed immunohistochemical analysis. Lab Invest 84 (4): 493–501.
Menon AG, Morreau H, Tollenaar RA, Alphenaar E, Van PM, Putter H, Janssen-van Rhijn CM, van de Velde CJ, Fleuren GJ, Kuppen PJ (2002) Down-regulation of HLA-A expression correlates with a better prognosis in colorectal cancer patients. Lab Invest 82 (12): 1725–1733.
Moller P, Momburg F, Koretz K, Moldenhauer G, Herfarth C, Otto HF, Hammerling GJ, Schlag P (1991) Influence of major histocompatibility complex class I and II antigens on survival in colorectal carcinoma. Cancer Res 51 (2): 729–736.
Mouradov D, Domingo E, Gibbs P, Jorissen RN, Li S, Soo PY, Lipton L, Desai J, Danielsen HE, Oukrif D, Novelli M, Yau C, Holmes CC, Jones IT, McLaughlin S, Molloy P, Hawkins NJ, Ward R, Midgely R, Kerr D, Tomlinson IP, Sieber OM (2013) Survival in stage II/III colorectal cancer is independently predicted by chromosomal and microsatellite instability, but not by specific driver mutations. Am J Gastroenterol 108 (11): 1785–1793.
Murphy J, O'Sullivan GC, Lee G, Madden M, Shanahan F, Collins JK, Talbot IC (2000a) The inflammatory response within Dukes' B colorectal cancers: implications for progression of micrometastases and patient survival. Am J Gastroenterol 95 (12): 3607–3614.
Murphy J, O'Sullivan GC, Lee G, Madden M, Shanahan F, Collins JK, Talbot IC (2000b) The inflammatory response within Dukes' B colorectal cancers: implications for progression of micrometastases and patient survival. Am J Gastroenterol 95 (12): 3607–3614.
Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, Ohtani H (1998) CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res 58 (16): 3491–3494.
Needham DJ, Lee JX, Beilharz MW (2006) Intra-tumoural regulatory T cells: a potential new target in cancer immunotherapy. Biochem Biophys Res Commun 343 (3): 684–691.
Nosho K, Baba Y, Tanaka N, Shima K, Hayashi M, Meyerhardt JA, Giovannucci E, Dranoff G, Fuchs CS, Ogino S (2010) Tumour-infiltrating T-cell subsets, molecular changes in colorectal cancer, and prognosis: cohort study and literature review. J Pathol 222 (4): 350–366.
Palmisano GL, Contardi E, Morabito A, Gargaglione V, Ferrara GB, Pistillo MP (2005) HLA-E surface expression is independent of the availability of HLA class I signal sequence-derived peptides in human tumor cell lines. Hum Immunol 66 (1): 1–12.
Perosa F, Luccarelli G, Prete M, Favoino E, Ferrone S, Dammacco F (2003) Beta 2-microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J Immunol 171 (4): 1918–1926.
Petersen RP, Campa MJ, Sperlazza J, Conlon D, Joshi MB, Harpole Jr DH, Patz Jr EF (2006) Tumor infiltrating Foxp3+ regulatory T-cells are associated with recurrence in pathologic stage I NSCLC patients. Cancer 107 (12): 2866–2872.
Polakova K, Kuba D, Russ G (2004) The 4H84 monoclonal antibody detecting beta2m free nonclassical HLA-G molecules also binds to free heavy chains of classical HLA class I antigens present on activated lymphocytes. Hum Immunol 65 (2): 157–162.
Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, Platell C, Iacopetta B (2009) Tumor-infiltrating FOXP3+ T regulatory cells show strong prognostic significance in colorectal cancer. J Clin Oncol 27 (2): 186–192.
Sandel MH, Speetjens FM, Menon AG, Albertsson PA, Basse PH, Hokland M, Nagelkerke JF, Tollenaar RA, van de Velde CJ, Kuppen PJ (2005) Natural killer cells infiltrating colorectal cancer and MHC class I expression. Mol Immunol 42 (4): 541–546.
Schiffmann L, Eiken AK, Gock M, Klar E (2013) Is the lymph node ratio superior to the Union for International Cancer Control (UICC) TNM system in prognosis of colon cancer? World J Surg Oncol 11: 79.
Schreiber RD, Old LJ, Smyth MJ (2011) Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 331 (6024): 1565–1570.
Smyth MJ, Dunn GP, Schreiber RD (2006) Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol 90: 1–50.
Suzuki H, Chikazawa N, Tasaka T, Wada J, Yamasaki A, Kitaura Y, Sozaki M, Tanaka M, Onishi H, Morisaki T, Katano M (2010) Intratumoral CD8(+) T/FOXP3 (+) cell ratio is a predictive marker for survival in patients with colorectal cancer. Cancer Immunol Immunother 59 (5): 653–661.
Swann JB, Smyth MJ (2007) Immune surveillance of tumors. J Clin Invest 117 (5): 1137–1146.
Tikidzhieva A, Benner A, Michel S, Formentini A, Link KH, Dippold W, von Knebel DM, Kornmann M, Kloor M (2012) Microsatellite instability and Beta2-Microglobulin mutations as prognostic markers in colon cancer: results of the FOGT-4 trial. Br J Cancer 106 (6): 1239–1245.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ (2011) Natural innate and adaptive immunity to cancer. Annu Rev Immunol 29: 235–271.
Watson NF, Ramage JM, Madjd Z, Spendlove I, Ellis IO, Scholefield JH, Durrant LG (2006) Immunosurveillance is active in colorectal cancer as downregulation but not complete loss of MHC class I expression correlates with a poor prognosis. Int J Cancer 118 (1): 6–10.
Wischhusen J, Waschbisch A, Wiendl H (2007) Immune-refractory cancers and their little helpers—an extended role for immunetolerogenic MHC molecules HLA-G and HLA-E? Semin Cancer Biol 17 (6): 459–468.
Ye SR, Yang H, Li K, Dong DD, Lin XM, Yie SM (2007) Human leukocyte antigen G expression: as a significant prognostic indicator for patients with colorectal cancer. Mod Pathol 20 (3): 375–383.
Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM (2007a) Expression of HLA-G is associated with prognosis in esophageal squamous cell carcinoma. Am J Clin Pathol 128 (6): 1002–1009.
Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM (2007b) Expression of human leucocyte antigen G (HLA-G) is associated with prognosis in non-small cell lung cancer. Lung Cancer 58 (2): 267–274.
Yie SM, Yang H, Ye SR, Li K, Dong DD, Lin XM (2007c) Expression of human leukocyte antigen G (HLA-G) correlates with poor prognosis in gastric carcinoma. Ann Surg Oncol 14 (10): 2721–2729.
Zeestraten EC, Maak M, Shibayama M, Schuster T, Nitsche U, Matsushima T, Nakayama S, Gohda K, Friess H, van de Velde CJ, Ishihara H, Rosenberg R, Kuppen PJ, Janssen KP (2012) Specific activity of cyclin-dependent kinase I is a new potential predictor of tumour recurrence in stage II colon cancer. Br J Cancer 106 (1): 133–140.
Zeestraten EC, Van Hoesel AQ, Speetjens FM, Menon AG, Putter H, van de Velde CJ, Kuppen PJ (2011) FoxP3- and CD8-positive infiltrating immune cells together determine clinical outcome in colorectal cancer. Cancer Microenviron 6 (1): 31–39.
Zhao L, Teklemariam T, Hantash BM (2012) Reassessment of HLA-G isoform specificity of MEM-G/9 and 4H84 monoclonal antibodies. Tissue Antigens 80 (3): 231–238.
Zilberman S, Schenowitz C, Agaugue S, Benoit F, Riteau B, Rouzier R, Carosella ED, Rouas-Freiss N, Menier C (2012) HLA-G1 and HLA-G5 active dimers are present in malignant cells and effusions: the influence of the tumor microenvironment. Eur J Immunol 42 (6): 1599–1608.
Zitvogel L, Tesniere A, Kroemer G (2006) Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol 6 (10): 715–727.
Zou W (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6 (4): 295–307.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Zeestraten, E., Reimers, M., Saadatmand, S. et al. Combined analysis of HLA class I, HLA-E and HLA-G predicts prognosis in colon cancer patients. Br J Cancer 110, 459–468 (2014). https://doi.org/10.1038/bjc.2013.696
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.696
Keywords
This article is cited by
-
Targeting MHC-I molecules for cancer: function, mechanism, and therapeutic prospects
Molecular Cancer (2023)
-
Prognostic significance of HLA-G in patients with colorectal cancer: a meta-analysis and bioinformatics analysis
BMC Cancer (2023)
-
A novel anoikis-related risk model predicts prognosis in patients with colorectal cancer and responses to different immunotherapy strategies
Journal of Cancer Research and Clinical Oncology (2023)
-
HLA and tumour immunology: immune escape, immunotherapy and immune-related adverse events
Journal of Cancer Research and Clinical Oncology (2023)
-
Immune-related lncRNA classification of head and neck squamous cell carcinoma
Cancer Cell International (2022)
