
Abstract
Background:
We evaluated week-on/week-off axitinib dosing plus chemotherapy in patients with gastrointestinal tumours, including tumour thymidine uptake by fluorine-18 3′-deoxy-3′-fluorothymidine positron emission tomography (18FLT-PET).
Methods:
During a lead-in period, patients received twice daily (b.i.d.) axitinib 7 mg (n=3) or 10 mg (n=18) for 7 days followed by a 7-day dosing interruption; serial 18FLT-PET scans were performed before day 1 and on days 7, 10, and 14. Axitinib plus FOLFIRI or FOLFOX was then administered in 2-week cycles; axitinib was interrupted on day 10 of each cycle for 7 days.
Results:
The maximum tolerated dose of axitinib was 10 mg b.i.d., in a week-on/week-off schedule, combined with FOLFIRI or FOLFOX. Common all-causality grade 3 adverse events were neutropenia (38%), hypertension (33%), and fatigue (29%). Of 21 patients, 2 (10%) had a partial response and 12 (57%) had stable disease. Following 7 days of continuous axitinib dosing, tumour 18FLT uptake decreased –49% from baseline and recovered to –28% and –17% from baseline, respectively, after 3 and 7 days of axitinib interruption.
Conclusion:
Axitinib administered in a week-on/week-off schedule combined with FOLFIRI or FOLFOX is supported by 18FLT-PET data and was well tolerated in patients with gastrointestinal tumours.
Similar content being viewed by others

Main
In patients with metastatic colorectal cancer (mCRC), improved outcomes have been observed in patients treated with bevacizumab (humanised monoclonal antibody to vascular endothelial growth factor (VEGF)) plus chemotherapy compared with chemotherapy alone (Hurwitz et al, 2004; Saltz et al, 2008). Combinations of chemotherapy plus other targeted agents with different mechanisms of action have been explored. However, to date, addition of tyrosine kinase inhibitors (TKIs) of VEGF receptors (VEGFRs) to chemotherapy regimens has failed to improve efficacy in patients with mCRC compared with placebo (Hecht et al, 2011; Van Cutsem et al, 2011) or bevacizumab (Hecht et al, 2010; Schmoll et al, 2012; Cunningham et al, 2013) in randomised phase II or phase III trials.
Preclinical studies have shown that agents that bind VEGF may transiently normalise tumour vasculature and increase uptake of cytotoxic drugs (Goel et al, 2011). However, more potent antiangiogenic agents, such as TKIs that target VEGFRs, may actually induce hypoxia and decrease blood flow to the tumour, slowing tumour cell division and impeding delivery of cytotoxic agents, which may antagonise the efficacy of chemotherapeutic agents (Ma and Waxman, 2009). Because of the potential chemotherapy-antagonising effects, intermittent dosing of a potent antiangiogenic agent with a short half-life may be the optimal strategy when administered in combination with chemotherapy.
Axitinib is a potent and selective second-generation inhibitor of VEGFRs 1, 2, and 3 (Hu-Lowe et al, 2008) that showed single-agent efficacy in phase II studies of various tumour types (Rixe et al, 2007; Cohen et al, 2008; Rini et al, 2009; Schiller et al, 2009; Fruehauf et al, 2011). Axitinib was approved in the United States (Pfizer Inc, 2012), Japan, the European Union, and other countries for the second-line treatment of advanced renal cell carcinoma. A phase I study showed that axitinib may be combined with full-dose 5-fluorouracil (5-FU)/leucovorin/irinotecan (FOLFIRI) or 5-FU/leucovorin/oxaliplatin (FOLFOX) without intolerable toxicities and without affecting the plasma concentration of these drugs in patients with mCRC or other solid tumours (Sharma et al, 2010). However, improvements in efficacy have not been evident with continuous axitinib plus chemotherapy compared with bevacizumab plus chemotherapy when used as first- or second-line mCRC treatment (Bendell et al, 2013; Infante et al, 2013).
In this study, we explored the safety and efficacy of axitinib dose interruptions before administration of FOLFIRI or FOLFOX in patients with mCRC or other advanced gastrointestinal cancers. Serial fluorine-18 3′-deoxy-3′-fluorothymidine positron emission tomography (18FLT-PET) scans (Salskov et al, 2007) were performed to explore the effect of 7-day axitinib exposure and subsequent interruption in thymidine uptake and tumour cell proliferation, to aid in determining the optimal administration of axitinib when combined with chemotherapy.
Materials and methods
Study design
This was a phase I portion of a randomised phase II study evaluating axitinib in combination with chemotherapy and bevacizumab in mCRC (Sharma et al, 2010; Infante et al, 2013). The objectives of this portion were to determine the maximum tolerated dose of axitinib administered in an interrupted-dosing schedule, estimate the optimal timing of FOLFIRI or FOLFOX administration following interruption of axitinib dosing based on 18FLT-PET scans, determine whether addition of axitinib administered in an interrupted-dosing schedule to FOLFOX or FOLFIRI can reverse pre-existing drug resistance among patients refractory to previous FOLFIRI or FOLFOX regimens, and assess tumour response and safety.
This study was conducted in accordance with the Declaration of Helsinki, the International Conference on Harmonisation Guidelines on Good Clinical Practice, the study protocol and applicable local regulatory requirements and laws. All participants provided written informed consent and agreed to comply with the study protocol. The protocol and informed consent forms were approved by an institutional review board/independent ethics committee. This trial is registered on ClinicalTrials.gov (NCT00460603).
Patients
All eligible adult patients had gastrointestinal cancer, measurable disease per Response Evaluation Criteria in Solid Tumours (RECIST, version 1.0) (Therasse et al, 2000), and appropriate lesions for 18FLT-PET/computed tomography (CT) analysis (outside a previous radiation field and a diameter >3 cm for a liver lesion, >1.5 cm for a pulmonary lesion and >2 cm for all other lesions). Patients had an Eastern Cooperative Oncology Group performance status of 0 or 1, adequate bone marrow, renal and hepatic function, and baseline blood pressure (BP) <130/80 mm Hg with antihypertensive medications permitted, and were eligible to receive FOLFIRI or FOLFOX. Key exclusion criteria were major surgery or investigational agent within 4 weeks before enrolment or minor surgery within 2 weeks before enrolment, clinically significant gastrointestinal abnormalities, proteinuria (⩾2+protein by dipstick or ⩾500 mg per 24 h), grade ⩾2 sensory neuropathy, serious or non-healing wound ulcer or bone fracture, current congestive heart failure, or significant bleeding episodes or arterial thrombotic events within 6 months before enrolment.
Study treatments
During a lead-in period, patients received an oral dose of axitinib 7 mg twice daily (b.i.d.) (cohort 1) or 10 mg b.i.d. (cohort 2) for 7 days, followed by a 7-day interruption of axitinib treatment (week-on/week-off schedule). Cohort 1 initially enrolled three patients, and cohort 2 was opened if none of the three patients in cohort 1 experienced dose-limiting toxicities from the lead-in axitinib dose. Dose-limiting toxicities were defined as grade 4 neutropenia or thrombocytopenia for ⩾8 days, or grade 4 febrile neutropenia; proteinuria ⩾2 g per 24 h; uncontrolled grade⩾3 non-haematologic toxicity for ⩾14 days; or inability to resume axitinib, FOLFOX or FOLFIRI within 14 days after stopping due to treatment-related toxicity. If one or more of the three patients in cohort 1 experienced dose-limiting toxicities, three or more additional patients would be enrolled to assess the safety of the axitinib interrupted-dosing schedule. Patients in cohort 1 who tolerated the 7 mg b.i.d. lead-in dose of axitinib could have their dose titrated to 10 mg b.i.d. in a week-on/week-off schedule.
Following the lead-in period, axitinib and chemotherapy (FOLFIRI or FOLFOX) were administered in 2-week cycles. On day 1 of each cycle, patients received irinotecan 180 mg m−2 intravenous infusion over 90 min (or oxaliplatin 85 mg m−2 over 120 min) concurrently with leucovorin 400 mg m−2 over 2 h via separate infusion lines, followed by 5-FU 400 mg m−2 bolus, then 5-FU 2400 mg m−2 infusion over 46–48 h. The choice of chemotherapy was at the discretion of the investigator according to the standard of care and individual patient tolerability. Chemotherapy doses were specifically adjusted for neutropenia, thrombocytopenia, nausea, vomiting, diarrhoea, and peripheral sensory neuropathy and for grade ⩾3 toxicities possibly related to chemotherapy.
On day 3 of each cycle, axitinib b.i.d. dosing was started following completion of the 5-FU infusion. Axitinib dosing was interrupted on day 10 of each cycle for 7 days. The axitinib dose was reduced by one dose level in patients who developed grade 3 non-haematologic adverse events or two readings of systolic BP >150 mm Hg or diastolic BP >100 mm Hg while receiving maximal antihypertensive therapy. The axitinib dose was interrupted in patients with grade 4 axitinib-related adverse events, two readings of systolic BP >160 mm Hg or diastolic BP >105 mm Hg, or ⩾2 g protein per 24 h, and resumed at one lower dose level when adverse events improved to grade ⩽2, BP decreased to <150/100 mm Hg, or<2 g protein per 24 h was present. Patients were treated until disease progression or intolerable toxicity.
Assessments
Patients were evaluated by physical examination at screening, day 1 of each cycle and at end of treatment. Tumours were radiologically assessed at screening and every 6 weeks following study initiation, according to RECIST (Therasse et al, 2000). Safety was monitored throughout the study period, and adverse events were graded according to the Common Terminology Criteria for Adverse Events (CTCAE, version 3.0) (Trotti et al, 2003). BP was measured at each clinic visit and b.i.d. by patients.
18FLT-PET/CT imaging
During the lead-in period, 18FLT-PET/CT scans were performed before day 1, on day 7 of continuous axitinib dosing and on days 10 and 14 (i.e., third and seventh day of axitinib dosing interruption). A CT scan was obtained at the beginning of each imaging session using a combined PET/CT scanner (General Electric Discovery STE and Discovery VCT; General Electric Healthcare, Waukesha, WI, USA). Continuous dynamic PET scanning in two-dimensional mode was then performed for ∼30 min immediately following administration of a 185–259 MBq (5–7 mCi) bolus of 18FLT (Siemens Molecular Imaging, Knoxville, TN, USA). After dynamic imaging, static whole-body scans in three-dimensional mode were obtained starting at 60 min using 5-min acquisitions per bed position. Images were reconstructed using ordered-subset expectation maximisation. Metastatic lesions were identified using static whole-body imaging data. A maximum standardised uptake value (SUV) was obtained from a region of interest drawn over the largest area of a lesion. For a lesion located within a high background organ (e.g., the liver), mean SUV in the region of interest was used. Kinetic analyses were performed using the dynamic image data, and Patlak influx constant (Kpat) was calculated (Schiepers et al, 2007). The reported SUV or Kpat values are the measurements from the most intense lesion identified on the patient’s first FLT-PET scan.
Statistical analysis
Descriptive statistics were used for efficacy and safety data. For the FLT-PET data, a reference arterial input time activity curve (TAC) was extracted from a dynamic PET study using independent component analysis (Hoh et al, 2010). This TAC is known to be contaminated by a metabolic fraction (mf), which was modelled as mf=C*(1.0−exp(−0.029*T) where C is a coefficient ranging from 0.4 to 0.8 and T is time in minutes (Schiepers et al, 2007). To minimise the effects of metabolites in determining the net uptake rate of FLT calculated with Patlak graphical analysis, only an early time interval was used (3–14 min).
Results
Patients and treatment
A total of 21 patients were enrolled between cohort 1 (n=3) and cohort 2 (n=18) (Table 1). Median duration of axitinib therapy was 527 days (range 55–663) in cohort 1 and 191 days (range 6–586) in cohort 2. Duration of axitinib therapy in individual patients is shown in Figure 1. Median daily dose of axitinib was 13.8 mg (range 12.4–14.0) in cohort 1 and 18.2 mg (range 9.3–20.0) in cohort 2. Fourteen patients received FOLFIRI and six received FOLFOX. Of 16 patients who had received previous chemotherapy, 9 received the same chemotherapy regimen in this study that they received previously (FOLFIRI in all cases). One patient with mCRC in cohort 2 did not receive chemotherapy. Median number of cycles of chemotherapy was 38 (range 4–48) in cohort 1 and 16 (range 2–42) in cohort 2. Twenty patients discontinued the study due to insufficient clinical response (n=13), withdrawal of consent (n=2), death due to disease progression (n=1) and adverse events (peripheral neuropathy, bacterial sepsis, elevated aspartate aminotransferase, and superior vena cava syndrome; n=1 each).

Duration of axitinib treatment.
Safety
There were no dose-limiting toxicities from the lead-in axitinib dose in the first six patients enrolled in the study, and the maximum tolerated dose of axitinib was determined to be 10 mg b.i.d., in a week-on/week-off schedule, in combination with either FOLFIRI or FOLFOX. The most common grade ⩾3 adverse events, occurring in seven patients during the dose-limiting toxicity observation period, were hypertension (n=3), fatigue (n=2), and polyuria, Escherichia urinary tract infection, ascites, abdominal pain, hyponatremia, nausea, decreased appetite, and hypovolemia (n=1 each). Following the dose-limiting toxicity observation period, two patients receiving axitinib 10 mg b.i.d. required dose reductions or interruptions due to grade 3 hypertension during their first seven days of treatment with axitinib plus chemotherapy. The most frequently reported all-causality adverse events were fatigue (81%), nausea (76%), diarrhoea (67%), neutropenia (52%), and hypertension (48%) (Table 2). The most common grade 3 adverse events were neutropenia (38%), hypertension (33%), and fatigue (29%) (Table 2). Four grade 4 adverse events (aspartate aminotransferase increased, myocardial infarction, pulmonary embolism, and superior vena cava syndrome; n=1 each) and one grade 5 adverse event (disease progression) were reported. The axitinib dose was reduced in 6 (29%) patients and interrupted in 11 (52%) patients due to treatment-related adverse events.
Efficacy
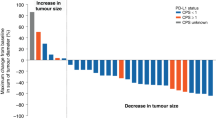
Two (10%) patients with CRC demonstrated a partial response, and twelve (57%) patients had stable disease. Maximum percentage change in tumour size by patient is shown in the Supplementary Figure. In the 17 patients with CRC, median progression-free survival (PFS) was 9.0 months (95% CI: 2.6–10.4) (Figure 2). One patient who discontinued chemotherapy after cycle 1 due to neutropenia continued to experience clinical benefit with single-agent axitinib (week-on/week-off schedule) and remains on therapy as of the data cutoff date.

Median progression-free survival for patients with colorectal cancer ( n =17). Abbreviations: CI=confidence interval; mPFS=median progression-free survival.
18FLT-PET/CT imaging
Complete sets of 18FLT-PET/CT data were available for 18 patients with lung (n=6), liver (n=5), lymph node (n=3) and abdominal wall and pelvic (n=2 each) lesions (Figure 3A and B). In one patient, no measurable lesion could be identified on either the dynamic or multi-bed images. Following an initial 7 days of continuous axitinib dosing, tumour 18FLT uptake (SUV) decreased –49% (s.e.m. 7%) from baseline (Figure 3C) in patients with liver lesions. Subsequently, following 3 and 7 days of axitinib interruption, tumour 18FLT uptake recovered to –28% (s.e.m. 7%) and –17% (s.e.m. 10%) from baseline, respectively. Maximum Kpat decreased –37% (s.e.m. 10%) from baseline after 7 days continuous axitinib dosing, and recovered to −27% (s.e.m. 14%) and +0.5% (s.e.m. 13%) from baseline, respectively, following 3 and 7 days of axitinib interruption (Figure 3D). Changes in 18FLT uptake by a target lesion in a representative patient are shown in Figure 4.

Patient level (A) SUVmax scores ( n =18) and (B) maximum Kpat values ( n =13; 5 patients were missing baseline or follow-up measurements); percentage change from baseline in (C) SUVmax scores ( n =18) and (D) maximum Kpat values ( n =12). aSUVmean scores and average Kpat values shown for the five patients with liver lesions. Abbreviations: Kpat=Patlak influx constant; SUV=standardised uptake value.

Changes in 18FLT uptake during and after axitinib treatment in a 3.0 × 3.1 cm2 right ilio-sacral region mass in a sample patient. Arrows denote site of target lesion at (A) baseline; (B) day 7 axitinib dosing; (C) day 10 (3 days after stopping axitinib dose); (D) day 14 (7 days after stopping axitinib); and (E) computed tomography slide of corresponding region of reference. Abbreviations: 18FLT= [18F] fluorothymidine; Kpat=Patlak influx constant; SUVmax=maximum uptake value.
Discussion
Enhanced efficacy with VEGF-binding agents, such as bevacizumab (Hurwitz et al, 2004; Saltz et al, 2008) or aflibercept (Van Cutsem et al, 2012), in combination with chemotherapy is supported by the ‘vascular normalisation’ hypothesis, which postulates that these agents normalise abnormal tumour vasculature and improve delivery of other anticancer therapies (Jain, 2001). This hypothesis has been substantiated in numerous preclinical and clinical studies (Goel et al, 2011). In contrast, to date none of the antiangiogenic TKIs have demonstrated improved efficacy in combination with chemotherapies in randomised phase II or phase III trials in mCRC (Hecht et al, 2010, 2011; Van Cutsem et al, 2011; Schmoll et al, 2012; Cunningham et al, 2013); results from an ongoing, randomised phase II trial (NCT01478594) evaluating the VEGFR TKI tivozanib plus FOLFOX vs bevacizumab plus FOLFOX are still awaited. This may be explained by the transient nature of vascular normalisation with TKIs, beyond which marked vascular pruning or regression may actually result in decreased delivery of chemotherapeutic agents to tumours (Goel et al, 2011). In addition, antiangiogenic TKIs may render tumours relatively hypoxic due to diminished blood flow and stop DNA synthesis, antagonising the cytotoxic effects of chemotherapies (Ma and Waxman, 2009). Taken together, this suggests that an interrupted schedule of antiangiogenic TKIs with a short half-life may be helpful in reversing the untoward effect on tumour vasculature and may optimise the intratumoural delivery of chemotherapeutic agents and restore tumour sensitivity. Intermittent dosing schedules may permit the use of antiangiogenic TKIs with reduced systemic toxicity and equivalent therapeutic benefit. The short half-life of axitinib (2–5 h) (Rugo et al, 2005) provided an opportunity to explore the impact of intermittent dosing of an antiangiogenic TKI on tumour blood flow, fluorodeoxythymidine uptake and antitumour efficacy.
In this study, axitinib administered in a week-on/week-off schedule in combination with FOLFIRI or FOLFOX was found to be well tolerated and demonstrated efficacy in patients with mCRC or other gastrointestinal tumours – 76% of whom had received previous chemotherapy. Common adverse events reported here were fatigue, nausea, diarrhoea, hypertension, and neutropenia, which were anticipated based on results from phase II studies of single-agent axitinib treatment of different tumours (Rixe et al, 2007; Cohen et al, 2008; Rini et al, 2009; Schiller et al, 2009; Fruehauf et al, 2011), as well as a phase I study of axitinib plus chemotherapy in patients with mCRC (Sharma et al, 2010). Noting that the majority of patients had received at least one previous chemotherapy regimen, our data suggest that interruption of axitinib dosing before administration of chemotherapy may improve clinical outcomes compared with continuous dosing. Although there is a risk of tumour progression when axitinib dosing is interrupted due to angiogenesis rebound, this effect was not observed in the present study. Further analysis of the efficacy and toxicity profiles of axitinib administered in an intermittent versus continuous dosing schedule in combination with chemotherapy would require further testing in a randomised clinical trial.
Imaging technology was proposed as a tool to quantify changes in tumour vasculature during antiangiogenic therapy and help optimise the dose and schedule of these agents in combination with chemotherapy (Jain, 2001). 18FLT-PET imaging has already been explored as a non-invasive method for diagnosis, tumour staging, and response to therapy (Bading and Shields, 2008). The present study used 18FLT-PET imaging to evaluate tumour cell proliferation following axitinib exposure and subsequent dosing interruption to help establish optimal administration of axitinib when combined with chemotherapy. Our 18FLT-PET results, based on data from both static (SUV) and dynamic (Kpat) scans, suggest that tumour cell proliferation decreases during the period of treatment with axitinib followed by a rebound in tumour metabolic activity during the withdrawal phase. Since cytotoxic agents preferentially target proliferating tumour cells, the continuous concurrent dosing of axitinib in combination with cytotoxic agents may inhibit the activity of the cytotoxic agents. This effect may partially account for the lack of improved outcomes with continuous administration of axitinib combined with chemotherapy in patients with mCRC (Bendell et al, 2013; Infante et al, 2013).
This study results suggest that axitinib administered in a week-on/week-off schedule in combination with chemotherapy may be an effective and well-tolerated treatment for CRC. This dosing strategy warrants further investigation.
Change history
18 February 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Bading JR, Shields AF (2008) Imaging of cell proliferation: status and prospects. J Nucl Med 49 (Suppl 2): 64S–80S.
Bendell JC, Tournigand C, Swieboda-Sadlej A, Barone C, Wainberg Z, Kim JG, Pericay C, Pastorelli D, Tarazi J, Rosbrook B, Bloom J, Ricart AD, Kim S, Sobrero AF (2013) Axitinib or bevacizumab plus FOLFIRI or modified FOLFOX-6 after failure of first-line therapy for metastatic colorectal cancer: a randomized phase II study. Clin Colorectal Cancer 12: 239–247.
Cohen EE, Rosen LS, Vokes EE, Kies MS, Forastiere AA, Worden FP, Kane MA, Sherman E, Kim S, Bycott P, Tortorici M, Shalinsky DR, Liau KF, Cohen RB (2008) Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol 26: 4708–4713.
Cunningham D, Wong RP, D'Haens G, Douillard JY, Robertson J, Stone AM, Van Cutsem E (2013) Cediranib with mFOLFOX6 vs bevacizumab with mFOLFOX6 in previously treated metastatic colorectal cancer. Br J Cancer 108: 493–502.
Fruehauf JP, Lutzky J, McDermott DF, Brown CK, Meric JB, Rosbrook B, Shalinsky DR, Liau KF, Niethammer AG, Kim S, Rixe O (2011) Multicenter, phase II study of axitinib, a selective second-generation inhibitor of vascular endothelial growth factor receptors 1, 2, 3, in patients with metastatic melanoma. Clin Cancer Res 17: 462–469.
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91: 1071–1121.
Hecht JR, Trarbach T, Hainsworth JD, Major P, Jager E, Wolff RA, Lloyd-Salvant K, Bodoky G, Pendergrass K, Berg W, Chen BL, Jalava T, Meinhardt G, Laurent D, Lebwohl D, Kerr D (2011) Randomized, placebo-controlled, phase III study of first-line oxaliplatin-based chemotherapy plus PTK787/ZK 222584, an oral vascular endothelial growth factor receptor inhibitor, in patients with metastatic colorectal adenocarcinoma. J Clin Oncol 29: 1997–2003.
Hecht JR, Yoshino T, Mitchell EP, Dees III MS, Countouriotis AM, Maneval EC, Kretzschmar A (2010) A randomized, phase IIb study of sunitinib plus 5-fluorouracil, leucovorin, and oxaliplatin (mFOLFOX6) versus bevacizumab plus mFOLFOX6 as first-line treatment for metastatic colorectal cancer (mCRC): interim results. J Clin Oncol 28 (15_suppl): Abstract 3532.
Hoh C, Brunken R, Saxena S, Buchsbaum M, Vera D (2010) Extraction of physiologic signals with independent component analysis (ICA). J Nucl Med 51 (Supplement 2): Abstract 1320.
Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu EY, McTigue MA, Murray BW, Kania RS, O'Connor P, Shalinsky DR, Bender SL (2008) Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res 14: 7272–7283.
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350: 2335–2342.
Infante JR, Reid TR, Cohn AL, Edenfield WJ, Cescon TP, Hamm JT, Malik IA, Rado TA, McGee PJ, Richards DA, Tarazi J, Rosbrook B, Kim S, Cartwright TH (2013) Axitinib and/or bevacizumab with modified FOLFOX-6 as first-line therapy for metastatic colorectal cancer: a randomized phase 2 study. Cancer 119: 2555–2563.
Jain RK (2001) Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 7: 987–989.
Ma J, Waxman DJ (2009) Dominant effect of antiangiogenesis in combination therapy involving cyclophosphamide and axitinib. Clin Cancer Res 15: 578–588.
Pfizer Inc. (2012) Inlyta® (axitinib) prescribing information Available at http://labeling.pfizer.com/ShowLabeling.aspx?id=759 (accessed 23 May 2013).
Rini BI, Wilding G, Hudes G, Stadler WM, Kim S, Tarazi J, Rosbrook B, Trask PC, Wood L, Dutcher JP (2009) Phase II study of axitinib in sorafenib-refractory metastatic renal cell carcinoma. J Clin Oncol 27: 4462–4468.
Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, Motzer RJ, Bycott P, Liau KF, Freddo J, Trask PC, Kim S, Rini BI (2007) Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol 8: 975–984.
Rugo HS, Herbst RS, Liu G, Park JW, Kies MS, Steinfeldt HM, Pithavala YK, Reich SD, Freddo JL, Wilding G (2005) Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol 23: 5474–5483.
Salskov A, Tammisetti VS, Grierson J, Vesselle H (2007) FLT: measuring tumor cell proliferation in vivo with positron emission tomography and 3'-deoxy-3'-[18F]fluorothymidine. Semin Nucl Med 37: 429–439.
Saltz LB, Clarke S, Diaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Lichinitser M, Yang TS, Rivera F, Couture F, Sirzen F, Cassidy J (2008) Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol 26: 2013–2019.
Schiepers C, Chen W, Dahlbom M, Cloughesy T, Hoh CK, Huang SC (2007) 18F-fluorothymidine kinetics of malignant brain tumors. Eur J Nucl Med Mol Imaging 34: 1003–1011.
Schiller JH, Larson T, Ou SH, Limentani S, Sandler A, Vokes E, Kim S, Liau K, Bycott P, Olszanski AJ, von Pawel J (2009) Efficacy and safety of axitinib in patients with advanced non-small-cell lung cancer: results from a phase II study. J Clin Oncol 27: 3836–3841.
Schmoll HJ, Cunningham D, Sobrero A, Karapetis CS, Rougier P, Koski SL, Kocakova I, Bondarenko I, Bodoky G, Mainwaring P, Salazar R, Barker P, Mookerjee B, Robertson J, Van Cutsem E (2012) Cediranib with mFOLFOX6 versus bevacizumab with mFOLFOX6 as first-line treatment for patients with advanced colorectal cancer: a double-blind, randomized phase III study (HORIZON III). J Clin Oncol 30: 3588–3595.
Sharma S, Abhyankar V, Burgess RE, Infante J, Trowbridge RC, Tarazi J, Kim S, Tortorici M, Chen Y, Robles RL (2010) A phase I study of axitinib (AG-013736) in combination with bevacizumab plus chemotherapy or chemotherapy alone in patients with metastatic colorectal cancer and other solid tumors. Ann Oncol 21: 297–304.
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van Glabbeke M, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92: 205–216.
Trotti A, Colevas AD, Setser A, Rusch V, Jaques D, Budach V, Langer C, Murphy B, Cumberlin R, Coleman CN, Rubin P (2003) CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol 13: 176–181.
Van Cutsem E, Bajetta E, Valle J, Köhne CH, Hecht JR, Moore M, Germond C, Berg W, Chen BL, Jalava T, Lebwohl D, Meinhardt G, Laurent D, Lin E (2011) Randomized, placebo-controlled, phase III study of oxaliplatin, fluorouracil, and leucovorin with or without PTK787/ZK 222584 in patients with previously treated metastatic colorectal adenocarcinoma. J Clin Oncol 29: 2004–2010.
Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, Ruff P, van Hazel GA, Moiseyenko V, Ferry D, McKendrick J, Polikoff J, Tellier A, Castan R, Allegra C (2012) Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol 30: 3499–3506.
Acknowledgements
This study was sponsored by Pfizer Inc. Medical writing support was provided by Joanna Bloom, PhD, of Engage Scientific Solutions and was funded by Pfizer Inc.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
CK Hoh has acted as a consultant for and received research funding from Pfizer Inc. J Tarazi and B Rosbrook are employees of Pfizer Inc and own Pfizer stock. S Kim, employed at Pfizer Inc at the time of the study described here and during preparation of this manuscript, is currently employed by Mirna Therapeutics, and owns stock in Pfizer Inc and Mirna Therapeutics. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Hoh, C., Burris, H., Bendell, J. et al. Intermittent dosing of axitinib combined with chemotherapy is supported by 18FLT-PET in gastrointestinal tumours. Br J Cancer 110, 875–881 (2014). https://doi.org/10.1038/bjc.2013.806
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2013.806
Keywords
This article is cited by
-
Antiangiogenic tyrosine kinase inhibitors in colorectal cancer: is there a path to making them more effective?
Cancer Chemotherapy and Pharmacology (2017)
-
18F-FLT PET/CT imaging in patients with advanced solid malignancies treated with axitinib on an intermittent dosing regimen
Cancer Chemotherapy and Pharmacology (2016)

{kind=link}