
Abstract
Background:
We completed a phase I clinical trial to test the safety and toxicity of combined treatment with cixutumumab (anti-IGF-1R antibody) and selumetinib (MEK 1/2 inhibitor).
Methods:
Patients with advanced solid tumours, refractory to standard therapy received selumetinib hydrogen sulphate capsules orally twice daily, and cixutumumab intravenously on days 1 and 15 of each 28-day cycle. The study used a 3+3 design, with a dose-finding cohort followed by an expansion cohort at the maximally tolerated dose that included pharmacokinetic and pharmacodynamic correlative studies.
Results:
Thirty patients were enrolled, with 16 in the dose-finding cohort and 14 in the expansion cohort. Grade 3 or greater toxicities included nausea and vomiting, anaemia, CVA, hypertension, hyperglycaemia, and ophthalmic symptoms. The maximally tolerated combination dose was 50 mg twice daily of selumetinib and 12 mg kg−1 every 2 weeks of cixutumumab. Two patients achieved a partial response (one unconfirmed), including a patient with BRAF wild-type thyroid carcinoma, and a patient with squamous cell carcinoma of the tongue, and six patients achieved time to progression of >6 months, including patients with thyroid carcinoma, colorectal carcinoma, and basal cell carcinoma. Comparison of pre- and on-treatment biopsies showed significant suppression of pERK and pS6 activity with treatment.
Conclusions:
Our study of anti-IGF-1R antibody cixutumumab and MEK 1/2 inhibitor selumetinib showed that the combination is safe and well-tolerated at these doses, with preliminary evidence of clinical benefit and pharmacodynamic evidence of target inhibition.
Similar content being viewed by others

Main
Insulin-like growth factor-1 receptor (IGF-1R) is a central activator of key prosurvival signalling pathways including RAS/RAF/MEK/ERK and PI3K/Akt/mTOR. After binding IGF ligand, IGF-1R undergoes autophosphorylation, inducing various substrate activations and downstream activity. In particular, activated RAS triggers activation of RAF kinase which then phosphorylates MEK1 and MEK2. Activated MEK phosphorylates its known targets, ERK1 and ERK2, leading to dimerisation, nuclear translocation, and induction of target genes (Khokhlatchev et al, 1998; Chang and Karin, 2001). Insulin-like growth factor-1 receptor-mediated signalling drives numerous cellular processes, including proliferation, survival, apoptosis, differentiation, metabolism, and motility (Baserga, 1999). Activating mutations of pathway components, particularly KRAS and BRAF, are found in numerous cancers, and recent drug development efforts have focused on inhibition of these driver components (Rusconi et al, 2012). In addition, IGF-1 is a potent mitogen for cancer cell growth and expressed in most cancers, although very few activating mutations or gene amplifications have been identified for the IGF-1 receptor (Baserga, 1999).
Disappointingly, trials of single-agent inhibitors of IGF-1R and downstream targets have generally shown only modest clinical responses (Jin et al, 2013). The emergence of clinical resistance is suspected to arise from compensatory upregulation and crosstalk within the RAS/RAF/MEK/ERK and PI3K/AKT/mTOR pathways (O'reilly et al, 2006; Poulikakos and Solit, 2011; Turke et al, 2012; Britten, 2013). In light of this, many clinical studies are exploring toxicity and efficacy of combined approaches, such as PI3K and MEK inhibition in basal-type breast cancer, or dual BRAF-MEK inhibition in BRAF-mutant melanoma (Britten, 2013). Numerous in vitro studies have demonstrated greater apoptosis and growth inhibition with simultaneous inhibition of multiple IGF-1 pathway targets (Shelton et al, 2004; Bertrand et al, 2006; Yanochko and Eckhart, 2006; Ji, 2007; Buck et al, 2008; Roberts et al, 2012; Flanigan et al, 2013; Molina-Arcas et al, 2013; Renshaw et al, 2013), suggesting this approach might lessen compensatory crosstalk and upregulation within the pathways. In particular, combined inhibition of IGF-1 and MAP/ERK kinase led to increased apoptosis in models of KRAS-mutant lung cancer, melanoma, and colorectal cancer (Villanueva et al, 2010; Chen and Sweet-Cordero, 2013; Flanigan et al, 2013; Molina-Arcas et al, 2013).
Cixutumumab is a recombinant human IgG1/λ monoclonal antibody that blocks interaction of IGF-1R and ligands IGF-1 and IGF-2, leading to internalisation/degradation of IGF-1R. Selumetinib is a highly selective MEK 1/2 inhibitor that exhibits potent inhibition of phosphorylated ERK. Both drugs demonstrated safety and tolerability in single-agent phase I and II clinical trials (Imclone Systems I, 2006; Rothenberg et al, 2007; Rowinsky et al, 2007; Adjei et al, 2008; Mckian and Haluska, 2009; Banerji et al, 2010; Schöffski et al, 2013). To test the hypothesis that vertical inhibition of the IGF pathway would be safe and tolerable, we designed an open label, phase I dose-escalation clinical trial of combined therapy with cixutumumab and selumetinib in patients with advanced solid tumours, including analysis of pharmacokinetic (PK) profiling and pharmacodynamic (PD) inhibition of downstream targets in an expansion cohort.
Patients and methods
Study design
We undertook this study after approval of our institutional review board, and informed consent was obtained from all patients. This study was registered with www.ClinicalTrials.gov, with the identifier NCT01061749. The primary objective of the study was to determine the safety, toxicity, and recommended maximally tolerated dose (MTD) of the combination of selumetinib and cixutumumab in patients with advanced/metastatic solid tumours. The study consisted of an initial dose-escalation cohort, and a second expansion cohort of patients treated at MTD with additional correlative PK and PD studies. The dose-escalation portion used a standard 3+3 design with a targeted dose-limiting toxicity (DLT) rate at maximum of 20% (Storer, 1989). A minimum of three patients were treated at each dose level and monitored for toxicity during the initial 6 weeks of the treatment prior to dose escalation to the next level. If none of three patients experienced DLT, the dose was escalated to the next level; if one of three patients experienced DLT, the dose level was expanded to six patients. If one or more of the additional patients exhibited DLT, the previous dose level would be considered MTD and used for the following expansion cohort. The expansion cohort was designed to enrol an additional 10–15 patients treated at MTD to obtain further safety/toxicity data and also PK and PD correlative studies.
Eligibility criteria
Patients were required to have advanced or metastatic solid tumours refractory to any number of previous standard therapies with measurable disease of ⩾1 cm, and at least a 4-week washout period from prior chemotherapy or radiation and resolution of all related toxicities to grade 1 or less by CTCAE version 4.0. Patients with previous exposure to IGF-1R or RAF/MEK inhibitors were not enrolled on the study. Patients were required to be 18 years of age or older, with Eastern Cooperative Oncology Group performance status of 0–1, and possess a life expectancy of >3 months. Adequate haematologic, renal, hepatic, and cardiac function were required. In addition, patients underwent baseline ophthalmologic examination prior to beginning therapy, owing to previously reported ophthalmic toxicity with MEK inhibitors. Patients were excluded for uncontrolled medical comorbidities including hypertension, NYHA Class II heart failure, uncontrolled infection, chronic liver disease including hepatitis B and C infection, pregnancy or unwillingness to use contraception, active CNS metastases or primary tumours, HIV positive patients on HAART therapy, psychiatric disturbances, poorly controlled diabetes mellitus (HgbA1c ⩾7.5) or history of disorders/supplementation of growth hormone, and any serious intraocular or retinal pathology except for controlled glaucoma and cataracts. Coadministration of study drugs with inhibitors or inducers of CYP1A2 was prohibited.
Drug administration
Selumetinib (AZD6244, ARRY-142886) was provided by the NCI under a collaborative agreement between AstraZeneca Pharmaceuticals and the Division of Cancer Treatment and Diagnosis (DCTD). Cixutumumab (IMC-A12) was supplied by Imclone Systems and Eli Lilly Pharmaceuticals with distribution by NCI/DCTD. Patients received selumetinib hydrogen sulphate capsules orally twice daily continuously on a 28-day cycle, administered on an empty stomach based on prior favourable PK data with fasting (Leijen et al, 2011). Patients received cixutumumab intravenously on days 1 and 15 of each cycle. Dosing for the study was chosen with discussion and input from the National Cancer Institute Cancer Therapy and Evaluation Program. For IMC-A12, a starting dose of 12 mg kg−1 every 2 weeks was chosen based on prior favourable PK/PD data at 6 mg kg−1 weekly, and demonstrated safety at 15 mg kg−1 every 2 weeks (Higano et al, 2007), whereas 50 mg twice daily was chosen of selumetinib owing to the previous single-agent PD profile (Banerji et al, 2010). Dosing of selumetinib was initiated at 50 mg twice daily, less than the previous single-agent MTD of 75 mg twice daily, and cixutumumab at 12 mg kg−1 every 2 weeks, with an escalation schema as listed including a de-escalation option in case of early toxicity (Table 1).
Dose-limiting toxicities
Patients were evaluable for toxicity if they received at least one dose of study drugs. Patients who came off study for any reason other than toxicity during the first 6 weeks were not included for dose-escalation determinations. Dose-limiting toxicities for the combination were defined to occur during the first 6 weeks of treatment (owing to cixutumumab half-life), possess attribution status of possible, probable, or definitely related to study drugs, and meet one of the following criteria: any grade 4 toxicity, grade 3 nausea, and vomiting refractory to maximal medical management for >3 days, grade 3 hyperglycaemia refractory to maximal medical management for >7 days, grade 3 rash refractory to maximal medical management for >3 days, grade 3 lipase elevation with clinical signs of pancreatitis, grade 3 neutropenia with fever or duration >7 days, or any other grade 3 toxicity. Anaemia due to blood loss was not considered dose limiting.
Measurement of effect
A secondary objective of our study was to assess preliminary evidence of efficacy for combination therapy using RECIST response criteria version 1.0. Patients must have received two cycles of therapy to be evaluable for radiographic response. Patients were required to undergo imaging every 8 weeks or more frequently if clinically indicated. Patients meeting criteria for partial response were required to have a confirmatory scan 4 weeks later.
Pharmacokinetic and PD analysis
Patients enrolling in the expansion cohort consented to PK and PD studies. Selumetinib PKs were assessed after a single dose on day 1. Serial sampling of venous blood was performed before treatment and at 0.5, 1, 1.5, 2, 4, and 8 h after treatment. Samples were collected in EDTA tubes. After centrifugation, the resultant plasma was frozen at −70 °C until the time of analysis. Plasma concentrations of selumetinib and its main metabolite (N-desmethyl selumetinib) were determined using a validated liquid chromatographic-mass spectrometric assay over the concentration range of 2.00–2000 ng ml−1 (selumetinib) and 2.00–500 ng ml−1 (N-desmethyl selumetinib) by Quotient Bio Analytical Sciences, an LGC business (Fordham, UK). The assay exhibited acceptable performance (selumetinib: accuracy −2.7 to 0.0%; precision<5.3% CV; N-desmethyl selumetinib: accuracy −2.3 to 0.6%; precision <13.3% CV). Pharmacokinetic assessment of cixutumumab was not performed in this study, owing to the low likelihood of interaction of a monoclonal antibody with selumetinib.
Patients underwent two mandatory tumour biopsies for PD analysis, the first prior to treatment and the second after the first cycle of treatment. Multiple core biopsy samples obtained under ultrasound or CT guidance were formalin-fixed and embedded in paraffin blocks. Blinded quantification of immunohistochemical markers including total and phosphorylated ERK and S6 were performed by AstraZeneca R&D Laboratories (Cheshire, UK) according to established protocols. Antibodies for phospho-ERK (#4376, Thr202/Tyr204) and phospho-S6 (#4857, Ser235/236) were obtained from Cell-Signaling Technology, Inc. (Danvers, MA, USA). H-scores were calculated based on percentage of cells expressing the target and the intensity of the staining in least three separate samples for each tumour.
Statistical considerations
This study was a single institution, unblinded dose-finding trial using a standard 3+3 design. The dose was escalated in a stepwise manner with a total of six patients at the putative MTD. The targeted DLT rate was ⩽20%. Patients with no reported DLTs who were withdrawn from the study prior to 6 weeks of evaluation were not considered evaluable for safety. Proportions of patients with toxicities were summarised using descriptive statistics. Tumour responses were determined by RECIST criteria v. 1.0. Time to progression was defined from the starting treatment date to the date of disease progression, including radiographic progression, or clinical progression requiring discontinuation from the study. PK variables were calculated by standard noncompartmental methods using WinNonlin profession (version 6.3) as previously described (Adjei et al, 2008; Gabrielsson and Weiner, 2012). For exploratory analysis of PD correlates, Pearson correlations, Mann–Whitney tests, and unpaired t-tests were performed to screen for associations between PD target expression and PK exposure best overall response, and time to response. All P values are reported as two-sided, with the a priori level of significance set at <0.05.
Results
Patient characteristics
Thirty patients with advanced solid tumours were enrolled in the study between 8 January 2010 and 24 January 2013, receiving at least one dose of both agents. A variety of tumour types were included in this study, including 13 patients with gastrointestinal tumours (colorectal, pancreatic, and biliary) and 4 patients with thyroid cancers (Table 2). The majority of patients had received at least three prior chemotherapy treatments for their disease (median 3; range 0–12). Nineteen of the 30 patients remained on study for at least 8 weeks and were evaluable for disease response by radiographic imaging. Of the patients who came off study before completing two cycles, four patients did so due to a disease-related significant adverse event, three patients due to clinical progression or deterioration, and four patients due to drug-related toxicities (one patient on dose level 1 in the expansion cohort, and three on dose level 2).
Toxicities and adverse events
The combination of drugs was well-tolerated in most patients at the tested doses. None of the first three patients enrolled at dose level 1 exhibited DLT, and therefore patient four was escalated to dose level 2 (Table 1). We eventually enrolled a total of 10 patients to dose level 2. Three of these patients required replacement for DLT rate determinations owing to withdrawal from study before completing a cycle for nontoxicity reasons. Our first DLT occurred in the sixth evaluable patient, and we opted to enrol an additional three patients to confirm a DLT rate of ⩽33%. However, another DLT occurred in these additional patients, thus we de-escalated to dose level 1, enrolling an additional three patients to confirm the MTD. In summary, the rate of DLT at dose level 1 was one out of six patients, defining the MTD for the study at 50 mg twice daily selumetinib with cixutumumab 12 mg kg−1 every 2 weeks.
All DLTs in the study were ophthalmic symptoms. The first patient developing DLT on dose level 2 noted abrupt onset of scotoma and flashes obscuring her vision in both eyes on the second day of selumetinib treatment. Ophthalmologic evaluation and MRI of the brain revealed no abnormalities. The visual abnormalities resolved over 1–2 weeks after discontinuation of the study drugs. The second patient on dose level 2 awoke with blind spots in his right visual field, on day 2 of selumetinib therapy. Ophthalmic evaluation revealed marked decrease in visual acuity on the right with pigment epithelial abnormalities in the retina and macula. Follow-up exam 4 months later showed resolution of these changes, and the patient reported improvement in vision although not complete resolution. The third patient with ophthalmic DLT developed black dots in her field of vision after 1 day of treatment at dose level 1. Ophthalmic evaluation showed no change from baseline examination. In total, 40% of all patients in the study reported some degree of ophthalmic toxicity, more commonly blurry vision, floaters, or flashing lights. Only 10% of all patients developed dose-limiting ophthalmic toxicity. In addition to the previously described DLTs, an additional two patients had selumetinib doses held for a 2-week period for floaters/blurry vision without ophthalmic exam findings.
Additional adverse effects deemed at least possibly related to one or both of the study drugs in >5% of patients are summarised in Table 3. The most common study drug-related side effects experienced by patients included dermatologic rashes and irritation (77%), mucosal ulcers or irritation (53%), nausea and vomiting (50%), diarrhoea (43%), ophthalmic symptoms as previously discussed (40%), and poor appetite/weight loss (37%). Grade 3 or greater toxicities included nausea and vomiting (one patient), anaemia (one patient), CVA (one patient), hypertension (two patients), hyperglycaemia (four patients), and ophthalmic symptoms (two patients).
Two patients in the study experienced strokes. The first patient developed a seizure 2 days after starting the study drug and was found to have a thrombotic stroke. However, imaging was consistent with a subacute event and his symptoms had preceded study drug administration with detailed questioning. With the help of neurology consultation, it was felt that the event most likely occurred prior to starting the study drugs. However, the patient was discontinued from the study at that time. The second patient presented after 2 weeks of treatment with altered mental status and confusion, and was found to have multiple ischaemic infarcts on MRI evaluation. He was noted to have splenic infarcts on imaging prior to enrolling on study, and again with neurology consultation, we determined that most likely these were chronic embolic events preceding study entry. The patients did not have treatment-emergent hypertension.
Dose modifications
Two patients required dose reduction of both cixutumumab and selumetinib, one for significant muscle fatigue and reduced neck range of motion, and one for atrial fibrillation. An additional patient underwent dose reduction of cixutumumab for grade 3 hyperglycaemia. Doses were held for anaemia and fatigue (both selumetinib and cixutumumab held), dermatologic issues (selumetinib, three patients), GERD (selumetinib, one patient), and minor ophthalmic symptoms (selumetinib, two patients). One additional patient had study drugs held while completing a course of radiation for brain lesions. One patient experienced a burning sensation with cixutumumab infusion that did not require treatment.
Efficacy
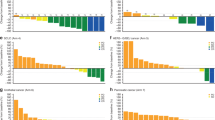
Nineteen patients underwent follow-up imaging and were evaluable for response. Median time to progression for all patients was 2.5 months (range 1.4–14.9, Figure 1A). As no patients died while actively receiving study drugs, death was not considered a progression event in our analysis.

(A) Time to progression for all patients, including patients who came off study for events not related to disease progression (censored). Median time to progression was 2.5 months. (B) Time to progression for evaluable patients by tumour type and dose level. Coloured bars indicate best radiographic response (red— progressive disease, blue—stable disease, and green—confirmed partial response). *Dose level 1, **Dose level 2. (C) Waterfall plot of best radiographic response by RECIST for evaluable patients by tumour type. Coloured bars indicate best clinical outcome (red—progressive disease, blue- stable disease, and green—partial response). Note that several patients came off study for clinical progression of disease despite meeting radiologic criteria for stable disease. Dotted lines indicate criteria for progression or partial response. A full color version of this figure is available at the British Journal of Cancer online.
A subset of patients remained on study for >6 months, including three patients with thyroid cancer (two BRAF WT, one mutant), two with colon cancer (one BRAF mutant, one unknown), and a patient with basal cell carcinoma (BRAF unknown). (Figure 1B, Supplementary Table 1). Regarding best responses in target lesions, two patients met RECIST criteria for partial response (>30% reduction in target lesions), but only one patient had a confirmatory scan 4 weeks later (Figure 1C). The other patient developed a new lesion and came off study for progressive disease.
Nine patients had prior BRAF mutation testing available, and two of the three patients with BRAF mutated tumours remained on study for >6 months (Supplementary Table S1).
Pharmacokinetics
Thirteen patients were evaluable for selumetinib PK analysis in the expansion cohort (Table 4). Consistent with previous reports, total selumetinib exhibited ∼20% variability in exposure with a plasma concentration–time profile exhibiting rapid absorption and elimination (Adjei et al, 2008). N-desmethyl metabolite levels ranged from 2.6–15% of selumetinib.
Pharmacodynamic analysis
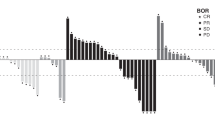
Paired on-treatment biopsies were obtained in five patients and immunohistochemical analysis was performed to measure expression of key downstream targets of IGF-1/MEK. One patient for each marker was excluded owing to inconclusive staining. Immunohistochemistry H-scores for pERK and pS6 were decreased after treatment in all evaluable patients for pS6, and three of the four patients for pERK, suggesting treatment was inhibiting downstream target activity (Figure 2A). Suppression of pERK and pS6 expression was found to significantly correlate with PK Cmax (pERK r=0.99, P=0.0013; pS6 r=0.92, P=0.025). Although our results are consistent with previous reports, owing to the small sample size the statistical significance should not be overinterpreted. Baseline pERK expression was also analysed against time to progression and best tumour response (Figure 2B). Patients with higher baseline pERK expression tended to have a shorter time on study (2.3 months vs 5.7 months) and increased tumour growth, (mean increase of 20% vs decrease of 2.4%), but small sample size limits the interpretation of this data.

(A) Pharmacodynamic target assessment was measured in several patients who underwent paired tumour biopsies, pre- and post-treatment. These were analysed by immunohistochemistry for expression of downstream targets including phospho-ERK, total ERK, phospho-S6, and total S6. Most patients had a decrease in the ratios of phosphorylated-to-total ERK and S6 after treatment. (B) Baseline ratio of phosphorylated-to-total ERK was compared with time to progression and % change in target lesions by RECIST. Patients with higher ratios at baseline tended to have a shorter time to progression and worse tumour response. Data are mean±SEM, compared using unpaired t-test.
Discussion
In the era of targeted therapy for solid tumours, the development of resistance remains an ongoing clinical problem and limits progression-free survival. Thus, trials utilising combination therapy are currently being extensively explored, in hopes of avoiding the interpathway crosstalk and compensatory upregulation that is thought to lead to resistance. In our study, we evaluated vertical inhibition of the IGF-1R pathway based on in vitro evidence that simultaneous blockade of upstream and downstream targets increased cell death. We have identified a recommended combined phase II dose at selumetinib 50 mg twice daily, and cixutumumab 12 mg kg−1 every 2 weeks.
In single-agent studies of selumetinib, the maximally tolerated dose was 75 mg twice daily, with dose-limiting toxicities including grade 3 acneiform rash and pleural effusion. Ophthalmic toxicities occurred in 26% of patients treated at the 75 mg dose and no CVAs were reported in this study (Banerji et al, 2010). Cixutumumab is a well-tolerated drug with no MTD reached in the initial clinical studies, with demonstrated safety up to 15 mg kg-1 every 2 weeks and a recommended phase II dose of 10 mg kg−1 every 2 weeks, as well as a favourable PK profile reached at 6 mg kg−1 weekly (Rothenberg et al, 2007; Mckian and Haluska, 2009). Dose-limiting toxicity-included hyperglycaemia which was generally manageable with oral antihyperglycemic agents, and other adverse events included dermatologic reactions, fatigue, and anaemia (Imclone Systems Inc, 2006).
In our combination study, the most common adverse effects were rash and gastrointestinal symptoms, and these did not occur any more frequently or more severely than previously described. We did note adverse events that were not previously reported, including mucosal irritation in over 50% of patients and hypertension in 10% of patients. Two patients in our study experienced CVA, and although these events were likely unrelated to the study drugs and neither patients experienced hypertension, the incidence of hypertension in our study raises the question of whether this combination may have off-target effects impacting vascular endothelium. Most importantly, we noted a higher incidence of ophthalmic toxicity in 40% of patients, including two grade 3 adverse events and DLT in 10% of patients. Owing to the small number of patients, it is unclear whether the use of combination therapy increased the risk of ophthalmic toxicity, but this should be a focus in future randomised trials. Many of the ophthalmic symptoms were nonspecific and inconsistent without a clear pattern, including blurry vision, scotoma, and floaters. Only one patient had confirmed abnormalities on ophthalmic examination, making objective assessment difficult. However, these symptoms appeared to resolve with holding of study drugs in two patients, thus the incidence is worthy of further investigation.
Pharmacokinetics of the oral hydrogen sulphate formulation of selumetinib have been previously described (Adjei et al, 2008; Banerji et al, 2010; Leijen et al, 2011; O'neil et al, 2011). Compared with previous reports at a dose of 50 mg twice daily, our profiling showed a similar Cmax and Tmax. In terms of PD effects, selumetinib demonstrates potent downstream inhibition of ERK1 and ERK2 phosphorylation (Yeh et al, 2007). We demonstrated similar treatment effect in our paired pre- and post-treatment biopsies, with inhibition of not only pERK but also pS6 in most patients. This suggests inhibition of the PI3K/AKT pathway as well, likely via inhibition of IGF-1R signalling. Previous studies in patient peripheral blood mononuclear cells showed a correlation between decrease in TPA-induced pERK and plasma selumetinib concentration (Banerji et al, 2010). In our small sampling of paired tumour biopsies, we also showed a significant correlation between plasma selumetinib Cmax and decrease in both pERK and pS6.
In this study we have demonstrated that combined inhibition of IGF-1R and MEK is feasible with regards to toxicity. The next step would require a randomised clinical trial including selumetinib alone vs the combination therapy in the most promising subsets of patients, to confirm synergistic activity with the combination. Our results do support the strategy of vertical inhibition while attempting to inhibit a particular pathway as well as further exploration of this specific combination.
This strategy produced meaningful clinical benefit for some patients, including stabilisation of tumour growth for >6 months or tumour shrinkage. In terms of clinical response, results in previous single-agent studies of both selumetinib and cixutumumab have been modest. In the initial, 16 patients enrolled in the dose-finding study of cixutumumab, there were no objective responses and only half of the patients achieved stable disease for >6 weeks (Higano et al, 2007; Mckian and Haluska, 2009). Two of these patients, one with hepatocellular carcinoma and one with male breast cancer achieved SD for >9 months. Selumetinib has shown sporadic complete responses in patients with BRAF-mutant melanoma, but only 50% of unselected patients were able to achieve stable disease for >6 weeks (Banerji et al, 2010).
We explored several possible biomarkers to characterise this subset of patients deriving clinical benefit within our study. The most significant clinical benefit was observed in patients with thyroid, colon, and head and neck squamous cell carcinoma (HNSCC). Prior in vitro data provide a strong rationale for efficacy in thyroid and colon cancers (Ji, 2007; Liu and Xing, 2008; Flanigan et al, 2013). In HNSCC, several studies report increased IGF-1R expression, and the role of EGFR-mediated MAPK signalling is well-established, thus the observed activity of our combination is quite plausible (Slomiany et al, 2007). MEK inhibition has been shown to have activity in patients with BRAF-mutant melanoma, but interestingly has not proved effective in other BRAF-mutant tumours such as colorectal and lung cancer. In our study, although two patients with BRAF-mutant tumours showed prolonged PFS, several patients with BRAF wild-type disease also achieved significant clinical benefit, suggesting that BRAF is not likely to be an isolated therapeutic biomarker for this combination. Of interest, high ERK phosphorylation was recently shown to be predictive of resistance to IGF-1R inhibition in small cell lung cancer (Zinn et al, 2013). In our study, we also observed that patients with a relatively higher baseline pERK/tERK ratio had a shorter time to progression and increased tumour growth by RECIST. Given the small number of patients, these observations are hypothesis-generating only, and more complete characterisation of pathway activity in future clinical trials will be needed to fully explore biomarkers of response.
In summary, we have demonstrated that vertical inhibition of IGF-1 signalling with combined IGF-1R and MEK 1/2 inhibition is feasible and well-tolerated in patients, and impacts pERK and pS6 downstream activity, producing clinical benefit in a subset of patients with heavily pre-treated advanced solid tumours. Additional studies aim to further explore biomarkers to identify those patients most likely to benefit from this treatment strategy.
Change history
06 January 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Adjei A, Cohen R, Franklin W, Morris C, Wilson D, Molina J, Hanson L, Gore L, Chow L, Leong S, Maloney L, Gordon G, Simmons H, Marlow A, Litwiler K, Brown S, Poch G, Kane K, Haney J, Eckhardt S (2008) Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol 26: 2139–2146.
Banerji U, Camidge D, Verheul H, Agarwal R, Sarker D, Kaye S, Desar I, Timmer-Bonte J, Eckhardt S, Lewis K, Brown K, Cantarini M, Morris C, George S, Smith P, Herpen CV (2010) The first-in-human study of the hydrogen sulfate (Hyd-sulfate) capsule of the MEK1/2 inhibitor AZD6244 (ARRY-142886): a phase I open-label multicenter trial in patients with advanced cancer. Clin Cancer Res 16: 1613–1623.
Baserga R (1999) The IGF-1 receptor in cancer research. Exp Cell Res 253: 1–6.
Bertrand F, Steelman L, Chappell W, Abrams S, Shelton J, White E, Ludwig D, Mccubrey J (2006) Synergy between an IGF-1R antibody and Raf/MEK/ERK and PI3K/Akt/mTOR pathway inhibitors in suppressing IGF-1R-mediated growth in hematopoietic cells. Leukemia 20: 1254–1260.
Britten C (2013) PI3K and MEK inhibitor combinations: examining the evidence in selected tumor types. Cancer Chemother Pharmacol 71: 1395–1409.
Buck E, Eyzaguirre A, Rosenfeld-Franklin M, Thomson S, Mulvihill M, Barr S, Brown E, O'connor M, Yao Y, Pachter J, Miglarese M, Epstein D, Iwata K, Haley J, Gibson N, Ji Q (2008) Feedback mechanisms promote cooperativity for small molecule inhibitors of epidermal and insulin-like growth factor receptors. Cancer Res 68: 8322–8332.
Chang L, Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37–40.
Chen R, Sweet-Cordero E (2013) Two is better than one: combining igf1r and mek blockade as a promising novel treatment strategy against KRAS-mutant lung cancer. Cancer Discov 3: 491–493.
Flanigan S, Pitts T, Newton T, Kulikowski G, Tan A, Mcmanus M, Spreafico A, Kachaeva M, Selby H, Tentler J, Eckhardt S, Leong S (2013) Overcoming IGF1R/IR resistance through inhibition of MEK signaling in colorectal cancer models. Clin Cancer Res 19: 6219–6229.
Gabrielsson J, Weiner D (2012) Non-compartmental analysis. Methods Mol Biol 929: 377–389.
Higano C, Yu Y, Whiting S (2007) A phase I, first in man study of weekly IMC-A12, a fully human insulin like growth factor-I receptor IgG1 monocolonal antibody, in patients with advanced solid tumors. J Clin Oncol 25 (suppl): abstract 3505.
Imclone Systems Inc (2006) Anti-insulin-like growth factor-I receptor (IGF-1R) monoclonal antibody investigator’s brochure.
Ji Q-S (2007) Preclinical Characterization of OSI-906: A Novel IGF-1R Inhibitor in Clinical Trials (Abstract #C192). AACR-NCI-EORTC International Conference on Molecular Targets and Cancer Therapeutics. San Francisco, CA, USA.
Jin M, Buck E, Mulvihill M (2013) Modulation of insulin-like growth factor-1 receptor and its signaling network for the treatment of cancer: current status and future perspectives. Oncology Reviews 7: e3.
Khokhlatchev A, Canagarajah B, Wilsbacher J, Robinson M, Atkinson M, Goldsmith E, Cobb M (1998) Phosphorylation of the MAP kinase ERK2 promotes its homodimerization and nuclear translocation. Cell 93: 605–615.
Leijen S, Soetekouw P, Evans TJ, Nicolson M, Schellens J, Learoyd M, Grinsted L, Zazulina V, Pwint T, Middleton M (2011) A phase I, open-label, randomized crossover study to assess the effect of dosing of the MEK 1/2 inhibitor Selumetinib (AZD6244; ARRY-142866) in the presence and absence of food in patients with advanced solid tumors. Cancer Chemother Pharmacol 68: 1619–1628.
Liu D, Xing M (2008) Potent inhibition of thyroid cancer cells by the MEK inhibitor PD0325901 and its potentiation by suppression of the PI3K and NF-kappaB pathways. Thyroid 18: 853–864.
Mckian K, Haluska P (2009) Cixutumumab. Expert Opin Investig Drugs 18: 1025–1033.
Molina-Arcas M, Hancock D, Sheridan C, Kumar M, Downward J (2013) Coordinate Direct Input of Both KRAS and IGF1 Receptor to Activation of PI3 kinase in KRAS-Mutant Lung Cancer. Cancer Discov 3: 548–563.
O'neil B, Goff L, Kauh J, Strosberg J, Bekaii-Saab T, Lee R, Kazi A, Moore D, Learoyd M, Lush R, Sebti S, Sullivan D (2011) Phase II study of the mitogen-activated protein kinase 1/2 inhibitor selumetinib in patients with advanced hepatocellular carcinoma. J Clin Oncol 29: 2350–2356.
O'reilly K, Rojo F, She Q, Solit D, Mills G, Smith D, Lane H, Hofmann F, Ludwig D, Baselga J, Rosen N (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66: 1500–1508.
Poulikakos P, Solit D (2011) Resistance to MEK inhibitors: should we co-target upstream? Sci Signal 4: pe16.
Renshaw J, Taylor K, Bishop R, Valenti M, Brandon ADH, Gowan S, Eccles S, Ruddle R, Johnson L, Raynaud F, Selfe J, Thway K, Pietsch T, Pearson A, Shipley J (2013) Dual blockade of the PI3K/AKT/mTOR (AZD8055) and RAS/MEK/ERK (AZD6244) pathways synergistically inhibits rhabdomyosarcoma cell growth in vitro and in vivo. Clin Cancer Res 19: 5940–5951.
Roberts P, Usary J, Darr D, Dillon P, Pfefferle A, Whittle M, Duncan J, Johnson S, Combest A, Jin J, Zamboni W, Johnson G, Perou C, Sharpless N (2012) Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res 18: 5290–5303.
Rothenberg M, Poplin E, Sandler A (2007) Phase I dose-escalation study of the anti-IGF-IR recombinant human IgG1 monoclonal antibody (Mab) IMC-A12, administered every other week to patients with advanced solid tumors. American Association for Cancer Research–National Cancer Institute–European Organisation for Research and Treatment of Cancer International Conference on Molecular Targets and Cancer Therapeutics San Francisco, CA, USA.
Rowinsky E, Youssoufian H, Tonra J, Solomon P, Burtrum D, Ludwig D (2007) IMC-A12, a human IgG1 monoclonal antibody to the insulin-like growth factor I receptor. Clin Cancer Res 13: 5549s.
Rusconi P, Caiola E, Broggini M (2012) RAS/RAF/MEK inhibitors in oncology. Curr Med Chem 19: 1164–1176.
Schöffski P, Adkins D, Blay J, Gil T, Elias A, Rutkowski P, Pennock G, Youssoufian H, Gelderblom H, Willey R, Grebennik D (2013) An open-label, phase 2 study evaluating the efficacy and safety of the anti-IGF-1R antibody cixutumumab in patients with previously treated advanced or metastatic soft-tissue sarcoma or Ewing family of tumours. Eur J Cancer 49: 3219–3228.
Shelton J, Steelman L, White E, Mccubrey J (2004) Synergy between PI3K/Akt and Raf/MEK/ERK pathways in IGF-1R mediated cell cycle progression and prevention of apoptosis in hematopoietic cells. Cell Cycle 3: 372–379.
Slomiany M, Black L, Kibbey M, Tingler M, Day T, Rosenzweig S (2007) Insulin-like growth factor-1 receptor and ligand targeting in head and neck squamous cell carcinoma. Cancer Lett 248: 269–279.
Storer B (1989) Design and analysis of phase I clinical trials. Biometrics 45: 925–937.
Turke A, Song Y, Costa C, Cook R, Arteaga C, Asara J, Engelman J (2012) MEK inhibition leads to PI3K/AKT activation by relieving a negative feedback on ERBB receptors. Cancer Res 72: 3228–3237.
Villanueva J, Vultur A, Lee J, Somasundaram R, Fukunaga-Kalabis M, Cipolla A, Wubbenhorst B, Xu X, Gimotty P, Kee D, Santiago-Walker A, Letrero R, D'andrea K, Pushparajan A, Hayden J, Brown K, Laquerre S, Mcarthur G, Sosman J, Nathanson K et al (2010) Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18: 683–695.
Yanochko G, Eckhart W (2006) Type I insulin-like growth factor receptor over-expression induces proliferation and anti-apoptotic signaling in a three-dimensional culture model of breast epithelial cells. Breast Cancer Res 8: R18.
Yeh T, Marsh V, Bernat B, Ballard J, Colwell H, Evans R, Parry J, Smith D, Brandhuber B, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee P, Winkler J, Koch K, Wallace E (2007) Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res 13: 1576–1583.
Zinn R, Gardner E, Marchionni L, Murphy S, Dobromilskaya I, Hann C, Rudin C (2013) ERK phosphorylation is predictive of resistance to IGF-1R inhibition in small cell lung cancer. Mol Cancer Ther 12: 1131–1139.
Acknowledgements
This work is supported by NIH grants U01 CA070095, with added support from NCI ACTNOW, P30 CA006973, and AstraZeneca.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
MR’s spouse is employed by Amplimmune, a subsidiary of AstraZeneca, and previously held stock in a company that AstraZeneca acquired. This arrangement has been reviewed and approved by the Johns Hopkins University in accordance with its conflict of interest policies. GB and CW are employees of AstraZeneca. The remaining authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Wilky, B., Rudek, M., Ahmed, S. et al. A phase I trial of vertical inhibition of IGF signalling using cixutumumab, an anti-IGF-1R antibody, and selumetinib, an MEK 1/2 inhibitor, in advanced solid tumours. Br J Cancer 112, 24–31 (2015). https://doi.org/10.1038/bjc.2014.515
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2014.515
Keywords
This article is cited by
-
Selumetinib: a selective MEK1 inhibitor for solid tumor treatment
Clinical and Experimental Medicine (2022)
-
Randomized Phase II Trial of Capecitabine and Lapatinib with or without IMC-A12 (Cituxumumab) in Patients with HER2-Positive Advanced Breast Cancer Previously Treated with Trastuzumab and Chemotherapy: NCCTG N0733 (Alliance)
Breast Cancer Research and Treatment (2021)
-
Investigating the potential clinical benefit of Selumetinib in resensitising advanced iodine refractory differentiated thyroid cancer to radioiodine therapy (SEL-I-METRY): protocol for a multicentre UK single arm phase II trial
BMC Cancer (2019)
-
Picropodophyllin (PPP) is a potent rhabdomyosarcoma growth inhibitor both in vitro and in vivo
BMC Cancer (2017)
-
A phase I study evaluating cixutumumab, a type 1 insulin-like growth factor receptor inhibitor, given every 2 or 3 weeks in Japanese patients with advanced solid tumors
Cancer Chemotherapy and Pharmacology (2016)
