
Abstract
Tumor necrosis factor (TNF) family ligands play essential roles in regulating a variety of cellular processes including proliferation, differentiation and survival. Expression of Drosophila TNF ortholog Eiger (Egr) induces JNK-dependent cell death, while the roles of caspases in this process remain elusive. To further delineate the Egr-triggered cell death pathway, we performed a genetic screen to identify dominant modifiers of the Egr-induced cell death phenotype. Here we report that Egr elicits a caspase-mediated cell death pathway independent of JNK signaling. Furthermore, we show NOPO, the Drosophila ortholog of TRIP (TRAF interacting protein) encoding an E3 ubiquitin ligase, modulates Egr-induced Caspase-mediated cell death through transcriptional activation of pro-apoptotic genes reaper and hid. Finally, we found Bendless and dUEV1a, an ubiquitin-conjugating E2 enzyme complex, regulates NOPO-triggered cell death. Our results indicate that the Ben-dUEV1a complex constitutes a molecular switch that bifurcates the Egr-induced cell death signaling into two pathways mediated by JNK and caspases respectively.
Similar content being viewed by others

Introduction
Tumor necrosis factor (TNF) family ligands are type II transmembrane proteins that play key roles in regulating a variety of physiological processes including cell proliferation, differentiation, survival and immunity 1, 2, 3. Mammalian studies revealed that TNF stimulates activation of nuclear factor-κB (NF-κB), caspase, c-Jun N-terminal kinase (JNK) and p38 signaling. Upon phosphorylation by the mitogen-activated protein (MAP) kinase cascade, JNK translocates into the nucleus, phosphorylates and activates transcription factors such as Jun and Fos, and finally leads to caspase-mediated apoptosis 4, 5. This signaling pathway is highly conserved in Drosophila, in which the TNF orthlog Eiger (Egr) triggers cell death through its receptor Wengen (Wgn), the TNF receptor-associated factor 2 (dTRAF2), the JNKK kinase dTAK1, the JNK kinase Hemipterous (Hep) and Basket (Bsk) that encodes the Drosophila JNK 6, 7, 8. Although considerable progress has been made to characterize the molecular mechanism underlying cell death induced by TNF-JNK signaling 9, whether caspases are involved in this process has remained controversial 6, 7.
TRIP (TRAF interacting protein) encodes a RING domain-containing E3 ubiquitin ligase 10 that negatively regulates NF-κB activation through physical interaction with the tumor suppressor CYLD or Syk in vitro 11, 12. Targeted disruption of TRIP in mice, or loss-of-function mutation of its Drosophila ortholog no poles (NOPO), resulted in early embryonic lethality 13, 14, suggesting TRIP/NOPO plays important roles in embryonic development. Recently, it was shown that NOPO physically interacts with the ubiquitin E2 complex consisting of Bendless (Ben)-dUEV1A heterodimer and regulates genomic integrity in Drosophila 14.
In this study, we performed a genetic screen for dominant modifiers of the Egr-triggered cell death phenotype, and found that Egr induced a caspase-dependent but JNK-independent cell death pathway. Furthermore, we showed that NOPO modulated Egr-induced caspase activation and cell death through up-regulation of the proapoptotic genes reaper (rpr) and head involution defective (hid). Finally, we demonstrated that the Ben-dUEV1A E2 complex modulated NOPO activity in the cell death signaling pathway. These data, together with previous results, suggest that the Ben-dUEV1A E2 complex selects dTRAF2 or NOPO as its ubiquitin E3 ligase partner and switches Egr-induced cell death signaling into two independent downstream pathways, one mediated by dTRAF2-JNK and another by NOPO-caspases.
Results and Discussion
Egr triggers two independent cell death pathways
Ectopic expression of Egr, the Drosophila TNF ortholog, in the developing eye (GMR>Egr) triggered cell death and produced a small eye phenotype (Figure 1A and 1B) 6, 7. Previous studies showed that this phenotype was suppressed when JNK signaling was blocked, suggesting that Egr-induced cell death depends on JNK activation 6, 7. Consistent with this hypothesis, expression of a constitutive active form of Hep, the Drosophila JNK kinase, in the developing eye (GMR>HepCA) induced JNK-mediated cell death and produced a similar small eye phenotype (Figure 1H) 7. However, we found that the small eye phenotype induced by GMR>Egr (Figure 1B), but not that by GMR>HepCA (Figure 1H), was partially suppressed by the deficiency Df(3L)H99 that deletes three proapoptotic genes, rpr, hid and grim (Figure 1C and 1I), or co-expression of the inhibitor of apoptosis protein DIAP1 (Figure 1D and 1J) or a dominant negative form of the caspase-9 homolog Drosophila Nedd-2-like caspase (DRONC) (Figure 1E and 1K). In addition, expression of Egr, but not HepCA, activated hid (Figure 1F and 1L) and rpr (Figure 1G and 1M) transcription in third instar larval eye discs. The rpr gene was also activated in hemizygous dTAK1 males or when Puc was co-expressed (Supplementary information, Figure S2F-S2I), suggesting that the JNK pathway is dispensable for Egr-triggered rpr activation. Finally, Egr-induced cell death was not completely suppressed in hemizygous dTRAF2 or dTAK1 mutant males (Supplementary information, Figure S2B and S2C), or by the co-expression of BskDN or Puc (Supplementary information, Figure S2D and S2E), suggesting that the dTRAF2-dTAK1-Hep-Bsk pathway is not the sole downstream mediator of Egr-induced cell death. Taken together, these results indicate that Egr induces cell death through two independent pathways, one mediated by JNK signaling and another by caspase activation.

Egr elicits a JNK-independent apoptotic pathway in Drosophila. (A-E) Compared to wild type (A, GMR-Gal4/+), GMR>Egr triggered a cell death phenotype (B, UAS-Egr/+; GMR-Gal4/+) that could be partially suppressed by removing one copy of the genes hid, reaper and grim using Df(3L)H99 (C, UAS-Egr/+; GMR-Gal4/Df(3L)H99), expression of DIAP1 (D, UAS-Egr/+; GMR-Gal4/UAS-DIAP1) or a dominant negative form of DRONC (E, UAS-Egr/UAS-DRONCDN; GMR-Gal4/UAS-DRONCDN). (F-G) Expression of Egr in developing eye induces hid and reaper expression. hid (F, GMR-Gal4 UAS-Egr/W05014) and reaper (G, rpr-lacZ/+; GMR-Gal4 UAS-Egr/+) expression were detected posterior to the morphological furrow in GMR>Egr eye discs. (H-K) GMR>HepCA-triggered apoptosis in the eye (H, GMR-Gal4 UAS-HepCA/+) cannot be rescued by Df(3L)H99(I, GMR-Gal4 UAS-HepCA/Df(3L)H99), expression of DIAP1 (J, GMR-Gal4 UAS-HepCA/UAS-DIAP1) or a dominant negative form of DRONC (K, GMR-Gal4 UAS-HepCA/UAS-DRONCDN). (L-M) Expression of hid (L, GMR-Gal4 UAS-HepCA/W05014) and reaper (M, rpr-lacZ/+; GMR-Gal4 UAS-HepCA/+) are not detected posterior to the morphological furrow in GMR>HepCA eye discs. For all the genetic interaction experiments, over 50 adult eyes and wings, 30 eye and wing discs were examined for each genotype, and the results have been highly consistent. A representative picture of each genotype is shown in this and following figures.
nopo is required for Egr-induced cell death
To investigate the genetic mechanism underlying Egr-induced caspase activation, we performed a whole genome deficiency screen using the Bloomington deficiency kit to search for dominant modifiers of the GMR>Egr small eye phenotype (Figure 2C). One of the suppressors was mapped cytologically between 55B12 and 55C1, a region uncovered by two overlapping deficiencies, Df(2R)Exel7153 and Df(2R)BSC337 (Figure 2A). Incorporating such deficiency into GMR>Egr flies mildly suppressed the small eye phenotype (Figure 2E and 2F), while Df(2R)ED3485, an adjacent deficiency (Figure 2A), failed to do so (Figure 2D). This region contains six genes including nopo (Figure 2A), the Drosophila ortholog of TRIP encoding an E3 ubiquitin ligase 14, as predicted by the Drosophila genome project 15. Consistently, we found mutation of endogenous nopo (Figure 2G and Supplementary information, Figure S3) or expression of a nopo RNAi (Figure 2H) partially suppressed the small eye phenotype of GMR>Egr, but not that of GMR>HepCA (Supplementary information, Figure S4), suggesting nopo modulates Egr-induced cell death in parallel or upstream of Hep. The knockdown effect of nopo RNAi was verified by quantitative reverse transcription polymerase chain reaction (qRT-PCR) (Supplementary information, Figure S5A) and its ability to suppress the GMR>NOPO eye phenotype (Supplementary information, Figure S5B-S5D).

NOPO regulates Egr-induced cell death. (A) A schematic depiction of the genomic region surrounding the nopo locus. The P element 5-SZ-3004 and three deficiencies Df(2R)ED3485, Df(2R)Exel7153 and Df(2R)BSC33 are indicated. (B-H) Light micrographs of Drosophila adult eyes are shown. Compared to wild type (B, GMR-Gal4/+), the GMR>Egr small eye phenotype (C, UAS-Egr/+; GMR-Gal4/+) remains unaffected by Df(2R)ED3485 (D, UAS-Egr/+; GMR-Gal4/Df(2R)ED3485), but is partially suppressed by deficiency Df(2R)Exel7153 (E, UAS-Egr/+; GMR-Gal4/Df(2R)Exel7153) or Df(2R)BSC337 (F, UAS-Egr/+; GMR-Gal4/Df(2R)BSC337) that deletes genes including nopo. This phenotype is also suppressed in heterozygous nopo mutant (G, UAS-Egr/nopo5-SZ-3004; GMR-Gal4/+) or by expression of a nopo RNAi (H, UAS-Egr/+; GMR-Gal4/UAS-nopo-IR). (I-K) Light micrographs of Drosophila adult wings are shown. Compared to wild-type wing (I, ptc-Gal4/+), ptc>Egr-induced notch phenotype along the wing margin (J, ptc-Gal4 UAS-Egr/+) is suppressed by expression of a nopo RNAi (K, ptc-Gal4 UAS-Egr/UAS-nopo-IR).
We further characterized the genetic interaction between nopo and Egr in the developing wing. Ectopic expression of Egr along the anterior posterior compartment boundary of wing disc produces a notch phenotype in adult wing margin (Figure 2I and 2J). This phenotype was fully suppressed by the RNAi of nopo (Figure 2K). Taken together, these data suggest that nopo is required for Egr-induced cell death in Drosophila.
NOPO induces caspase-mediated cell death independently of JNK
To investigate the role of NOPO in Egr-induced cell death signaling, we expressed NOPO in the developing eye or wing under the control of GMR (GMR>NOPO) or scalloped (sd) (sd>NOPO) promoter. GMR>NOPO produced a small and rough eye phenotype (Figure 3A and 3B), resulting from extensive cell death posterior to the morphogenetic furrow in third instar eye discs (Figure 3C and 3D), as shown by acridine orange (AO) staining, a dye that detects dying cells 16. Consistently, sd>NOPO triggered strong apoptosis along the dorsal ventral compartment boundary in wing discs (Figure 3G and 3H), and generated a small wing phenotype missing most of the wing margin tissue (Figure 3E and 3F). We also checked additional Gal4 drivers expressed in the wing discs, and found that MS1096>NOPO produced a similar but milder phenotype (Supplementary information, Figure S6B and S6E), whereas NOPO expression under the engrailed (en) promoter, which drives NOPO expression exclusively in the posterior compartment (Supplementary information, Figure S6F'), induced apoptosis in the posterior area of wing discs (Supplementary information, Figure S6F) and resulted in a significant reduction of the posterior/anterior compartment ratio in adult wings (Supplementary information, Figure S6C and S6G).

Ectopic NOPO expression induces caspase-dependent but JNK-independent cell death. (A-H) Ectopic expression of NOPO induces cell death. Compared to wild-type eyes (A, GMR-Gal4/+) or wings (E, sd-Gal4/+), NOPO expression driven by GMR-Gal4 or sd-Gal4 produces small and rough eyes (B, UAS-NOPO/+; GMR-Gal4/+) or small wings missing most of the margin tissue (F, sd-Gla4/UAS-NOPO), which are resulted from increased cell death indicated by acridine orange staining, in third instar larval eye discs (C, GMR-Gal4/+; and D, UAS-NOPO/+; GMR-Gal4/+) or wing discs (G, sd-Gal4/+; and H, sd-Gla4/UAS-NOPO).(I-P) The GMR>NOPO phenotype is caspase-dependent but JNK-independent. The GMR>NOPO eye phenotype could not be suppressed by the expression of a dominant negative form of dTAK1 (I, UAS-NOPO/+; GMR-Gal4/UAS-dTAK1DN), or a hep RNAi (J, UAS-NOPO/+; GMR-Gal4/UAS-hep-IR), or a dominant negative form of Bsk (K, UAS-NOPO/+; GMR-Gal4/UAS-BskDN), or Puc (L, UAS-NOPO/+; GMR-Gal4/UAS-puc), but suppressed by the expression of DIAP1 (M, UAS-NOPO/+; GMR-Gal4/UAS-DIAP1), or a dominant negative form of DRONC (N, UAS-NOPO/+; GMR-Gal4/UAS-DRONCDN), or p35 (O, UAS-NOPO/+; GMR-Gal4/UAS-p35), or removing one copy of reaper, hid and grim (P, UAS-NOPO/+; GMR-Gal4/ Df(3L)H99). (Q-T) Ectopic NOPO expression induces reaper and hid transcription. X-Gal staining of a reaper-LacZ (Q, R) and a hid-LacZ (S, T) reporter in wild-type (Q, S) or NOPO-expressing (R, T) imaginal discs are shown. Genotypes: Q, rpr-lacZ/+; GMR-Gal4/+; R, UAS-NOPO/+; rpr-lacZ/+; GMR-Gal4/+; S, sd-Gal4/W05014; T, UAS-NOPO/+; sd-Gal4/W05014.
We further used the GMR>NOPO eye phenotype (Figure 3B) to characterize the genetic interaction between NOPO and the two signaling pathways, namely JNK and caspases, that mediate Egr-induced cell death. We found the GMR>NOPO phenotype was not suppressed by the expression of a dominant negative form of dTAK1 (Figure 3I), a hep RNAi (Figure 3J), a dominant negative form of Bsk that encodes the Drosophila JNK (Figure 3K) or the Bsk inhibitor Puckered (Puc) (Figure 3L). On the other hand, we observed significant rescue of the GMR>NOPO defective eye phenotype upon overexpression of DIAP1, an inhibitor for initiator caspase activity (Figure 3M) 17, a dominant negative form of DRONC (Figure 3N), or p35 (Figure 3O), a viral protein that inhibits downstream effector caspases 18. Taken together, these results demonstrate that NOPO regulates the caspases-mediated, but JNK-independent cell death signaling pathway.
Since p53 also induces caspases-mediated apoptosis in Drosophila 19, we tested whether p53 is required for NOPO-induced cell death. To this end, the GMR>NOPO eye phenotype was not suppressed by the expression of p53259H and p53CT (Supplementary information, Figure S7), two dominant negative forms of p53 19, 20, indicating that NOPO modulates cell death independent of p53 activity.
NOPO activates rpr and hid expression
Developmental cell death in Drosophila is mostly mediated by three closely linked pro-apoptotic genes rpr, hid and grim, which encode small proteins that inactivate DIAP1 21, 22. We found the GMR>NOPO eye phenotype (Figure 3) was significantly suppressed in flies heterozygous for the deficiency Df(3L)H99 (Figure 3P), which simultaneously removes the three pro-apoptotic genes, suggesting their roles in NOPO-induced cell death. Consistent with this result, ectopic NOPO expression induced rpr and hid transcription in larval eye and wing discs (Figure 3Q-3T), and rpr expression in adult eyes, as revealed by qRT-PCR (Supplementary information, Figure S8). Thus, we conclude that nopo modulates Egr-induced caspase activation through transcriptional up-regulation of rpr and hid.
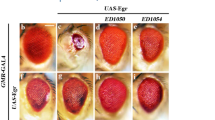
Bendless and dUEV1a regulate NOPO-induced apoptosis
NOPO has been found to interact with Ben, the Drosophila ortholog of Ubc13, in the yeast two-hybrid experiments 14, 23, and it is postulated that Ben could associate with dUEV1a to form a heterodimer that functions as an E2 ubiquitin-conjugating enzyme 24. Recent reports showed that Ben-dUEV1a and NOPO form an E2-E3 ubiquitination complex that regulates genomic integrity during early embryogenesis 14. In support with the physical interaction data between Ben-dUEV1a and NOPO, we found that loss of ben or duev1a significantly suppressed the GMR>NOPO eye phenotype (Figure 4C, 4D and Supplementary information, Figure S9), suggesting the ectopic NOPO activity crucially depends on the E2 function of the endogenous Ben-dUEV1a complex. In addition, while expression of Ben or dUEV1a alone failed to enhance the GMR>NOPO eye phenotype (Figure 4E and 4F), co-expression of Ben and dUEV1a, which by itself did not lead to any obvious defect (Figure 4H), significantly enhanced the GMR>NOPO eye phenotype (Figure 4G), indicating that Ben and dUEV1a might function as a complex to regulate NOPO-induced cell death in vivo. Our unpublished results revealed that Ben and dUEV1a also regulate Egr-induced JNK activation through physical interaction with dTRAF2, another ubiquitin E3 ligase. Taken these data together, our genetic experiments demonstrate an essential role of NOPO in regulating TNF-induced cell death, which is conserved from Drosophila to mammals. We propose a model that the Ben-dUEV1a E2 complex constitutes a molecular switch which, by choosing dTRAF2 or NOPO as its E3 partner, transduces Egr-induced cell death signaling through two independent pathways, one mediated by dTRAF2-JNK and another by NOPO-caspase (Figure 4I).

Bendless and dUEV1a regulate NOPO-induced apoptosis. (A-D) Light micrographs of Drosophila adult eyes are shown. Compared to the wild-type eye (A, GMR-Gal4/+), NOPO-induced cell death (B, UAS-NOPO/+; GMR-Gal4/+) could be suppressed by RNAi inactivation of ben (C, UAS-NOPO/+; GMR-Gal4/UAS-ben-IR) or duev1a (D, UAS-NOPO/+; GMR-Gal4/UAS-duev1a-IR). (E-H) Expression of either Ben (E, UAS-NOPO/+; UAS-BenT8/+; GMR-Gal4/+) or dUEV1a (F, UAS-NOPO/+; GMR-Gal4/UAS-dUEV1a) has no effect on the GMR>NOPO phenotype, co-expression of both, though appears normal by itself (H, UAS-Ben/+; GMR-Gal4/UAS-dUEV1a), significantly enhances the NOPO-induced cell death phenotype (G, UAS-NOPO/+; UAS-Ben/+; GMR-Gal4/UAS-dUEV1a). (I) A model for the role of NOPO in Egr-induced cell death signaling.
Materials and Methods
Drosophila strains
GMR-Gal4, UAS-DIAP1, UAS-p35, GUS-p53259H, GUS-p53CT, Df(3L)H99, sd-Gal4, hid-lacZ (w05014) and the deficiency kit were obtained from Bloomington stock center, UAS-nopo-IR, UAS-ben-IR, UAS-duev1a-IR were obtained from Vienna stock center, nopo5-SZ-3004 was obtained from Szeged Drosophila Stock Center. UAS-Egr, UAS-DRONCDN 6, reaper-lacZ 25, UAS-HepCA 26, ben1 27, UAS-NOPO, nopoExc142 14, ptc-Gal4, UAS-dTAK1DN, UAS-hep-IR, UAS-BskDN, UAS-Puc, pucE69, dTRAF2EX1.1 8, UAS-BenT8 28, dTAK11 29 were previously described.
X-Gal staining
Eye and wing discs were dissected from third instar larvae in PBST and stained for β-galactosidase activity as described 30.
AO staining
Eye and wing discs were dissected from third instar larvae in PBST and incubated in 1×10−5 M AO for 5 min at room temperature prior to imaging.
qRT-PCR
Thirty adult heads were collected from freshly eclosed flies of indicated genotypes. Total RNA was isolated using TRIzol (Invitrogen), and RT-PCR was performed as previously described 31. Primers for reaper are sense 5′-GAGCAGAAGGAGCAGACGAT-3′ and antisense 5′-CGATATTTGCCGGACTTTCT-3′, primers for nopo are sense 5′-CATCGTAAAATCGCCATCAGTCA-3′ and antisense 5′-GCTGCTCCTGCAGTGAACCAAC-3′, primers for actin5c are sense 5′-AAGTTGCTGCTCTGGTTGTC-3′ and antisense 5′-GGGTACTTCAGGGTGAGGATA-3′
References
Locksley RM, Killeen N, Lenardo MJ . The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 2001; 104:487–501.
Wajant H . The FasL-Fas Signaling Network. Sci STKE 2003; 2003:cm5.
Varfolomeev EE, Ashkenazi A . Tumor necrosis factor: an apoptosis JuNKie? Cell 2004; 116:491–497.
Davis RJ . Signal transduction by the JNK group of MAP kinases. Cell 2000; 103:239–252.
Weston CR, Davis RJ . The JNK signal transduction pathway. Curr Opin Genet Dev 2002; 12:14–21.
Igaki T, Kanda H, Yamamoto-Goto Y, et al. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 2002; 21:3009–3018.
Moreno E, Yan M, Basler K . Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 2002; 12:1263–1268.
Xue L, Igaki T, Kuranaga E, et al. Tumor suppressor CYLD regulates JNK-induced cell death in Drosophila. Dev Cell 2007; 13:446–454.
Igaki T, Pastor-Pareja JC, Aonuma H, Miura M, Xu T . Intrinsic tumor suppression and epithelial maintenance by endocytic activation of Eiger/TNF signaling in Drosophila. Dev Cell 2009; 16:458–465.
Besse A, Campos AD, Webster WK, Darnay BG . TRAF-interacting protein (TRIP) is a RING-dependent ubiquitin ligase. Biochem Biophys Res Commun 2007; 359:660–664.
Regamey A, Hohl D, Liu JW, et al. The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor kappaB activation by tumor necrosis factor. J Exp Med 2003; 198:1959–1964.
Zhou Q, Geahlen RL . The protein-tyrosine kinase Syk interacts with TRAF-interacting protein TRIP in breast epithelial cells. Oncogene 2009; 28:1348–1356.
Park ES, Choi S, Kim JM, et al. Early embryonic lethality caused by targeted disruption of the TRAF-interacting protein (TRIP) gene. Biochem Biophys Res Commun 2007; 363:971–977.
Merkle JA, Rickmyre JL, Garg A, et al. no poles encodes a predicted E3 ubiquitin ligase required for early embryonic development of Drosophila. Development 2009; 136:449–459.
Adams MD, Celniker SE, Holt RA, et al. The genome sequence of Drosophila melanogaster. Science 2000; 287:2185–2195.
Abrams JM, White K, Fessler LI, Steller H . Programmed cell death during Drosophila embryogenesis. Development 1993; 117:29–43.
Hawkins CJ, Wang SL, Hay BA . A cloning method to identify caspases and their regulators in yeast: identification of Drosophila IAP1 as an inhibitor of the Drosophila caspase DCP-1. Proc Natl Acad Sci USA 1999; 96:2885–2890.
Hay BA, Wolff T, Rubin GM . Expression of baculovirus P35 prevents cell death in Drosophila. Development 1994; 120:2121–2129.
Brodsky MH, Nordstrom W, Tsang G, et al. Drosophila p53 binds a damage response element at the reaper locus. Cell 2000; 101:103–113.
Ollmann M, Young LM, Di Como CJ, et al. Drosophila p53 is a structural and functional homolog of the tumor suppressor p53. Cell 2000; 101:91–101.
Holley CL, Olson MR, Colon-Ramos DA, Kornbluth S . Reaper eliminates IAP proteins through stimulated IAP degradation and generalized translational inhibition. Nat Cell Biol 2002; 4:439–444.
Martins LM, Iaccarino I, Tenev T, et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem 2002; 277:439–444.
Giot L, Bader JS, Brouwer C, et al. A protein interaction map of Drosophila melanogaster. Science 2003; 302:1727–1736.
Zhou R, Silverman N, Hong M, et al. The role of ubiquitination in Drosophila innate immunity. J Biol Chem 2005; 280:34048–34055.
Nordstrom W, Chen P, Steller H, Abrams JM . Activation of the reaper gene during ectopic cell killing in Drosophila. Dev Biol 1996; 180:213–226.
Kanda H, Igaki T, Kanuka H, Yagi T, Miura M . Wengen, a member of the Drosophila tumor necrosis factor receptor superfamily, is required for Eiger signaling. J Biol Chem 2002; 277:28372–28375.
Thomas JB, Wyman RJ . Mutations altering synaptic connectivity between identified neurons in Drosophila. J Neurosci 1984; 4:530–538.
Uthaman SB, Godenschwege TA, Murphey RK . A mechanism distinct from highwire for the Drosophila ubiquitin conjugase bendless in synaptic growth and maturation. J Neurosci 2008; 28:8615–8623.
Geuking P, Narasimamurthy R, Basler K . A genetic screen targeting the tumor necrosis factor/Eiger signaling pathway: identification of Drosophila TAB2 as a functionally conserved component. Genetics 2005; 171:1683–1694.
Xue L, Noll M . Dual role of the Pax gene paired in accessory gland development of Drosophila. Development 2002; 129:339–346.
Wang MC, Bohmann D, Jasper H . JNK signaling confers tolerance to oxidative stress and extends lifespan in Drosophila. Dev Cell 2003; 5:811–816.
Acknowledgements
We thank Z Wang (Chinese Academy of Sciences), L Lee (Vanderbilt University, USA), R Wyman (Yale University, USA), K Basler (University of Zurich, Switzerland), M Miura (University of Tokyo, Japan), T Igaki (Kobe University, Japan), D Bohmann and H Jasper (University of Rochester, USA), RK Murphey (Florida Atlantic University, USA), Bloomington, Harvard, VDRC, Szeged, and NIG stock centers for fly stocks, M Ho (Tongji University, China) and members of Xue lab for discussion and comments. This work is supported by grants from the National Natural Science Foundation of China (30971681 and 31071294), the National Basic Research Program of China (973 Program; 2010CB944901 and 2011CB943903), Shanghai Pujiang Program (09PJ1409400), the Innovation Program of Shanghai Municipal Education Commission (10ZZ27), Program for New Century Excellent Talents in University (NCET-10-0608) and Shanghai Committee of Science and Technology (09DZ2260100) to L X.
Author information
Authors and Affiliations
Corresponding author
Additional information
( Supplementary information is linked to the online version of the paper on the Cell Research website.)
Supplementary information
Supplementary information, Figure S1
Constitutive activation of Hep induces JNK- dependent cell death. (PDF 71 kb)
Supplementary information, Figure S2
Egr induces JNK independent cell death and rpr activation. (PDF 87 kb)
Supplementary information, Figure S3
Egr induced cell death was partially suppressed in nopo null mutants. (PDF 46 kb)
Supplementary information, Figure S4
Loss of NOPO could not suppress GMR>HepCA triggered cell death. (PDF 40 kb)
Supplementary information, Figure S5
Expression of nopo RNAi down-regulates nopo transcription. (PDF 131 kb)
Supplementary information, Figure S6
Ectopic NOPO expression induces cell death in developing wings. (PDF 61 kb)
Supplementary information, Figure S7
NOPO induced cell death is independent of p53. (PDF 56 kb)
Supplementary information, Figure S8
Ectopic NOPO expression up-regulates reaper transcription. (PDF 119 kb)
Supplementary information, Figure S9
NOPO-induced cell death could be suppressed by loss of endogenous ben. (PDF 63 kb)
Rights and permissions
About this article
Cite this article
Ma, X., Huang, J., Yang, L. et al. NOPO modulates Egr-induced JNK-independent cell death in Drosophila. Cell Res 22, 425–431 (2012). https://doi.org/10.1038/cr.2011.135
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2011.135
Keywords
This article is cited by
-
Slik maintains tissue homeostasis by preventing JNK-mediated apoptosis
Cell Division (2023)
-
The TNFR Wengen regulates the FGF pathway by an unconventional mechanism
Nature Communications (2023)
-
ZnT7 RNAi favors RafGOFscrib−/−-induced tumor growth and invasion in Drosophila through JNK signaling pathway
Oncogene (2021)
-
Toll signaling promotes JNK-dependent apoptosis in Drosophila
Cell Division (2020)
-
Using genetic variation in Aedes aegypti to identify candidate anti-dengue virus genes
BMC Infectious Diseases (2019)
