
Abstract
Ubiquitination has emerged as a crucial mechanism that regulates signal transduction in diverse biological processes, including different aspects of immune functions. Ubiquitination regulates pattern-recognition receptor signaling that mediates both innate immune responses and dendritic cell maturation required for initiation of adaptive immune responses. Ubiquitination also regulates the development, activation, and differentiation of T cells, thereby maintaining efficient adaptive immune responses to pathogens and immunological tolerance to self-tissues. Like phosphorylation, ubiquitination is a reversible reaction tightly controlled by the opposing actions of ubiquitin ligases and deubiquitinases. Deregulated ubiquitination events are associated with immunological disorders, including autoimmune and inflammatory diseases.
Similar content being viewed by others

Introduction
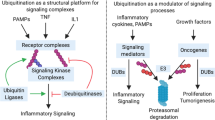
Ubiquitination is a posttranslational mechanism of protein modification involving covalent conjugation of ubiquitin to lysine (K) residues of target proteins, a process that is catalyzed by the sequential action of ubiquitin-activating (E1), ubiquitin-conjugating (E2), and ubiquitin-ligating (E3) enzymes1. Formation of polyubiquitin chains involves isopeptide bond connection between the carboxyl-terminal glycine residue of ubiquitin and an internal K residue or the amino-terminal methionine (M1) of another ubiquitin. The presence of seven Ks, along with the M1, creates a large variety of ubiquitin chains, including K6-, K11-, K27-, K29-, K33-, K48-, K63-, and M1-linked ubiquitin chains, as well as mixed ubiquitin chains. The substrate specificity of ubiquitination is mainly determined by E3s, a family of > 600 mammalian members that recognize protein substrates and assist or directly catalyze the transfer of ubiquitin from E2s to the substrates2,3. Ubiquitination is a reversible and dynamic event, since the conjugated ubiquitin chains can be cleaved by a family of ubiquitin-specific proteases, termed deubiquitinases (DUBs)4. Although the best-known function of ubiquitination is to target substrate proteins for degradation in the 26S proteasome, it is now clear that some ubiquitin chains, such as the K63-linked and M1-linked (also called linear) ubiquitin chains, mediate signal transduction via nondegradative mechanisms5,6. Such ubiquitin chains can function as a platform that facilitates protein-protein interactions in signal transduction. A growing family of proteins with ubiquitin-binding domains (UBDs) recognize target proteins when they are attached with specific types of ubiquitin chains and, thereby, mediate assembly of signaling complexes7. Ubiquitination is indispensable for a large variety of biological processes, including different aspects of immune functions.
The immune system is a complex network of cells, tissues, and organs evolved to protect host from infectious agents. All animals, as well as plants, possess an innate immune system, which senses infections by recognizing essential and conserved components of pathogens known as pathogen-associated molecular patterns (PAMPs)8. Innate immune cells, such as macrophages, dendritic cells (DCs), and neutrophils, express several families of pattern-recognition receptors (PRRs) that recognize the different PAMPs and mediate phagocytosis and signal transduction that triggers production of antimicrobial factors and inflammatory mediators9. Thus, innate immunity provides immediate defense against infections and mediates induction of inflammation that is in turn important for recruiting immune cells to sites of infection. Innate immunity also regulates the induction of adaptive immunity, a more sophisticated immune response available only in mammals and other vertebrates10. The adaptive immunity relies on T and B lymphocytes characterized by the presence of highly specific antigen-recognition receptors, the T cell receptor (TCR) and B cell receptor (BCR). B cells produce antibodies and mediate humoral immunity, whereas T cells, including CD4+ and CD8+ T cells, mediate cellular immunity and provide help for activation of B cells in humoral immune response.
The activation of naive T cells occurs when antigen-presenting cells (APCs), mostly DCs, have been alerted by an infection or other dangerous signals. Following exposure to PAMPs, DCs are stimulated to mature into competent APCs and migrate to the draining lymph nodes, where they activate naive T cells by presenting foreign antigens to the TCR and stimulating costimulatory molecules, such as CD28. PAMP-stimulated DCs and macrophages also release a plethora of cytokines that guide the differentiation of CD4+ T cells to several subsets of T helper (Th) cells involved in different aspects of immune response. Under normal condition, T cells launch robust response to invading pathogens but are tolerant to self-antigens11. Ubiquitination has a crucial role in the regulation of innate and adaptive immune responses as well as immune tolerance. In this review, we discuss recent progress regarding how ubiquitination regulates immune functions, focusing on PRR signaling in innate immunity and inflammation, DC maturation, T cell activation and differentiation, and T cell tolerance.
Ubiquitination in PRR signaling
Innate immune recognition is based on PAMP detection by several families of PRRs, including toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (NLRs), C-type lectin receptors (CLRs), and cytosolic DNA sensors12. TLRs are a family of membrane-bound PRRs that are located on cell surface or intracellular vesicles (Figure 1). Cell-surface TLRs sense pathogens present in the extracellular environment by recognizing various membrane components of the microbes, whereas intracellular TLRs are located on the membrane of endosomes and lysosomes and detect nucleic acids internalized into the lumen of these intracellular vesicles from the extracellular environment13. RLRs, including RIG-I, MDA5, and LGP2, are RNA helicases that sense RNAs from RNA viruses in the cytosol13,14. The NLR family includes a large number of cytoplasmic PRRs characterized by the presence of an N-terminal effector domain, a central nucleotide-binding NOD domain, and a C-terminal leucine-rich repeat domain mediating ligand binding15. An extensively studied function of NLRs is their involvement in the formation and activation of inflammasomes, a signaling pathway mediating activation of caspase-1 and secretion of inflammatory cytokines IL-1 and IL-1816,17,18. In addition, some NLRs also mediate noninflammasome functions, as demonstrated for the NLR family founding members NOD1 and NOD2. NOD1 and NOD2 detect bacterial cell wall peptidoglycan components, γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP) and muramyl dipeptide (MDP), respectively, and mediate antimicrobial innate immunity and inflammatory responses19. CLRs are membrane-associated PRRs that contain a characteristic C-type lectin-like domain and recognize carbohydrates, such as the fungal cell wall component β-1, 3-glucans. A well-recognized function of the CLRs is mediating anti-fungal immunity. The prototypical CLR member, Dectin-1, recognizes the fungal cell wall carbohydrate β-1, 3-glucans and is required for innate immunity against several fungal species20. Cytosolic DNA sensors mediate innate immunity against various bacterial and viral pathogens. More than 10 putative DNA sensors have been identified over the past few years21, and the recently discovered cyclic GMP-AMP synthase (cGAS) appears to have a predominant role in mediating type I interferon (IFN-I) induction by cytoplasmic DNAs22.

Main signaling pathways of PRRs. A common signaling feature of different PRRs is the dependence on adaptor molecules. TLRs, except TLR3, associate with MyD88, which in turn recruits and activates IRAK4, leading to phosphorylation of IRAK1 and IRAK1-mediated activation of the downstream adaptor TRAF6. TLR3 and TLR4 associate with TRIF and activate the downstream adaptor RIP1. Via a ubiquitin-dependent mechanism, TRAF6 and RIP1 both activate TAK1 and its downstream pathways, leading to induction of proinflammatory cytokines. TRIF also activates TBK1 (or IKKε) for the induction of type I IFNs, via a mechanism thought to involve the adaptor TRAF3. RLRs signal via the mitochondrial adaptor MAVS and several TRAF members to activate the TAK1 proinflammatory pathway and the type I IFN pathway, whereas the NLR member NOD2 (as well as NOD1) activates the TAK1 proinflammatory pathway via the signaling adaptor RIP2. STING is a signaling adaptor of cytoplasmic DNA sensors, such as cGAS, which activates STING via synthesizing the second messenger, cyclic dinucleotide cGAMP. STING also senses bacteria-derived cyclic di-nucleotides, c-di-AMP and c-di-GMP.
Signal transduction by PRRs relies on their association with specific adaptors (Figure 1). MyD88 is a common adaptor for all TLRs, except TLR3, whereas TRIF is an adaptor for TLR3 and TLR413. Both MyD88 and TRIF contain a TIR domain, which mediates interaction with the TIR of TLRs. MyD88 also contains a death domain (DD) that mediates interaction with the DD of IL-1R-associated kinases (IRAKs). Upon recruitment to the TLRs, MyD88 assembles a complex with IRAK4, IRAK1, and IRAK2 via DD-DD interactions, causing the activation of IRAK4 and phosphorylation of IRAK1 and IRAK2. Phosphorylated IRAK1/2 then mediates the activation of TRAF6, which in turn functions as both an adaptor and an E3 ligase conjugating K63-linked ubiquitin chains to itself and other proteins. TRAF6 activates the ubiquitin-dependent kinase TAK1, which in turn activates IKK and the MAPKs JNK and p38. Via an IKK-dependent mechanism, TAK1 also activates the kinase TPL2 and its downstream kinase ERK23. These signaling events ultimately lead to the activation of transcription factors AP1 and NF-κB and induction of proinflammatory cytokines (Figure 1). In addition to targeting the TAK/IKK/MAPK signaling axis, TRIF activates the IKK-related kinases TBK1 and IKKε, resulting in the activation of transcription factor IRF3 and induction of type I IFNs. Unlike MyD88, TRIF does not contain a DD, and its signaling in the proinflammatory pathway relies on a receptor-interacting kinase (RIP) homotypic interaction motif (RHIM)24. In response to signals from TLR3 and TLR4, TRIF recruits RIP1 via RHIM-RHIM interaction and stimulates K63-linked polyubiquitination of RIP1, which is important for the recruitment and activation of TAK1 and IKK25. Analogous to TRIF, the NLR members NOD1 and NOD2 recruit RIP2 and stimulate K63-linked ubiquitination of RIP2, which is required for the activation of IKK and MAPKs and induction of proinflammatory cytokines26.
The common signaling adaptor for RLRs is mitochondrial antiviral-signaling protein (MAVS; also named IPS-1, VISA, and CARDIF), which contains a caspase activation and recruitment domain (CARD)27,28,29,30. TRAF3 has been implicated as a signaling adaptor that mediates induction of type I IFNs by RLRs as well as TLRs31,32. However, in some studies, TRAF3 was found to be dispensable for type I IFN induction by RNA viruses, vesicular stomatitis virus (VSV) and Sendai virus (SeV), arguing for the redundant functions of several other TRAF members, including TRAF2, TRAF5, and TRAF633,34. It is possible that different TRAFs may regulate type I IFN induction in different cell types or different PRR pathways. RLRs also stimulate the NF-κB and MAPK pathways and the expression of proinflammatory cytokines.
Stimulator of IFN gene (STING; also known as MITA, MPYS, TREM173, and ERIS) is an endoplasmic reticulum (ER)-localized protein that functions as an adaptor in the cytoplasmic DNA-sensing pathway35,36,37. STING also directly senses cyclic di-nucleotides, including cyclic-di-guanylate monophosphate (c-di-GMP) and cyclic-di-adenylate monophosphate (c-di-AMP), produced by bacteria38,39,40,41,42. Upon stimulation by these bacterial second messengers, STING becomes activated and then stimulates the TBK1/IRF3 signaling pathway to mediate type I IFN induction. The recently identified cytosolic DNA sensor cGAS catalyzes the synthesis of cyclic GMP-AMP (cGAMP), which functions as an endogenous second messenger that stimulates STING activation43,44,45. The cGAS-mediated DNA-sensing pathway responds to cytosolic DNAs derived from both pathogens and self, suggesting its involvement in both innate immunity against infection and the pathogenesis of certain autoimmune diseases22,46,47
Ubiquitination in IKK activation
A common downstream target of the PRRs is IKK, which together with MAPKs mediates induction of proinflammatory cytokines and chemokines (Figure 1). IKK functions through activation of NF-κB transcription factors – dimeric complexes of several structurally related DNA-binding proteins, including RelA (also called p65), RelB, c-Rel, p50, and p52. NF-κB members are normally sequestered in the cytoplasm as inactive complexes via association with inhibitors, IκBs, and the activation of NF-κBs can be mediated by canonical and noncanonical pathways48,49. The canonical NF-κB pathway is a major target of the PRRs, although the noncanonical NF-κB pathway has a modulatory role in specific PRR pathways, particularly the type I IFN pathway50. Activation of canonical NF-κB relies on the typical trimeric IKK complex, composed of catalytic subunits IKKα and IKKβ and a regulatory subunit termed NF-κB essential modulator (NEMO, also called IKKγ). Upon activation, IKK phosphorylates a prototypical IκB member, IκBα, causing K48-linked ubiquitination and proteolysis of IκBα and concomitant nuclear translocation of canonical NF-κB dimers. In addition, IKK also phosphorylates other IκB members, including the IκB-like protein p105, contributing to canonical NF-κB activation23. The noncanonical NF-κB pathway, which specifically activates the p52/RelB dimer, is the subject of several reviews48,49 and will not be discussed here.
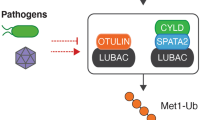
Early studies revealed that IKK activation depends on K63-linked polyubiquitin chain synthesis mediated by TRAF6 and the E2 dimer Ubc13/Uev1A, leading to the identification of TAK1 as a ubiquitin-dependent kinase that activates both IKK and MAPKs51,52. Depending on the stimuli, K63-linked polyubiquitin chain conjugation and IKK activation can also be mediated by other E3 ubiquitin ligases, such as TRAF2, inhibitor of apoptosis 1 (cIAP1), and cIAP253. More recently, linear ubiquitination, catalyzed by the linear ubiquitin chain assembly complex (LUBAC), has been shown to also contribute to the activation of IKK and NF-κB in some receptor signaling pathways6,54. In agreement with these studies, the activation of IKK is negatively regulated by DUBs that cleave K63 and linear ubiquitin chains, such as CYLD, A20, and Otulin4,54,55. Mechanistically, the ubiquitin-dependent activation of TAK1 and IKK relies on the ubiquitin-binding functions of their regulatory proteins: TAB2 (or TAB3) for TAK1 and NEMO for IKK. TAB2 and TAB3 have a UBD that preferentially binds K63-linked polyubiquitin chains56. The ubiquitin-binding function of TAB2 and TAB3 mediates the recruitment of TAK1 to ubiquitinated adaptors, such as TRAF6, and appears to also directly promote the autophosphorylation and activation of TAK153. NEMO also contains a UBD, known as an UBAN (ubiquitin binding in ABIN and NEMO) domain, which binds to both linear ubiquitin chains and K63-linked ubiquitin chains albeit more strongly to the former57,58. The functional significance of the ubiquitin-binding activity of NEMO in IKK activation is underscored by the finding that NEMO mutations in its UBAN domain are associated with impaired NF-κB activation and X-linked ectodermal dysplasia with immunodeficiency59. Notably, NEMO not only binds linear ubiquitin chains but is also conjugated with linear ubiquitin chains60. It has been proposed that linear ubiquitin-conjugated NEMO may be bound by another NEMO molecule, which facilitates the dimerization and activation of IKK61. The role of linear ubiquitination in NF-κB regulation has been extensively studied in the TNF receptor 1 signaling pathway, but accumulating evidence suggests that linear ubiquitination also plays a role in NF-κB activation by PRRs62.
Ubiquitination in MAPK activation
The PRRs activate three major families of MAP kinases (MAPKs), including p38, JNK, and ERK. The activation of p38 and JNK, as seen with the activation of IKK, is largely dependent on TAK163,64. Although ERK is not directly targeted by TAK1, its activation is nevertheless dependent on TAK1 signaling. In this case, the TAK1-activated IKK is required for activation of TPL2, an upstream kinase of the ERK signaling pathway23 (Figure 1). Under normal condition, TPL2 is physically associated with an NF-κB precursor protein, p105; upon activation by TAK1, IKK induces phosphorylation-dependent p105 degradation, causing the release of TPL2 and allowing TPL2 to phosphorylate and activate the ERK kinase, MEK1/265,66,67,68. Thus, TAK1 is a kinase that is required for activation of both IKK and three families of MAPKs23,69. Given the ubiquitin-dependent nature of TAK1 activation, it is not surprising that activation of the MAPK signaling pathways by different PRRs is dependent on ubiquitination. Genetic deficiency in a K63-specific E2 component, Ubc13, impairs activation of TAK1 and MAPKs by TLR ligands and the inflammatory cytokine IL-1β70,71. Ubc13 has an essential role in mediating NEMO ubiquitination in IL-1β-stimulated MEFs, although the role of Ubc13 in TRAF6 ubiquitination was less clear due to the discrepancy between two different studies70,71. While one study suggests that Ubc13 is dispensable for IL-1β-stimulated TRAF6 ubiquitination in MEFs71, another study demonstrates that splenocytes from mice with heterozygous Ubc13 deletion are defective in LPS-stimulated TRAF6 ubiquitination70. Whether the discrepancy is due to cell type differences or experimental variations is unclear. Nevertheless, these studies clearly demonstrate a role for K63-linked ubiquitination in regulating activation of MAPKs by TLRs and IL-1R.
Ubiquitination in TBK1/IKKε activation
TBK1 and its homologue IKKε are IKK-related kinases that mediate phosphorylation and activation of transcription factor IRF3 and induction of type I IFNs72,73,74,75,76. This antiviral signaling pathway is stimulated by various PRRs, including TRIF-dependent TLRs (TLR3 and TLR4), RLRs (RIG-I and MDA5), NLRs (NOD1, NOD2, and Nlrp6), and cytoplasmic DNA sensors77,78. Ubiquitination has a critical role in regulating both signal-induced and homeostatic activation of TBK1 and IKKε. Genetic deficiency in the DUB CYLD causes constitutive activation of TBK1 and IKKε in DCs, rendering the cells hyperresponsive to VSV-induced type I IFN expression79,80. Although this phenotype may involve ubiquitin-dependent activation of RIG-I, CYLD also directly targets TBK1 and IKKε to inhibit their ubiquitination79. A20 has also been shown to inhibit the activation of TBK1 and IKKε, and this function of A20 requires its associated protein Tax1-binding protein 1 (TAX1BP1), which appears to function as an adaptor that facilitates the binding of A20 to TBK1 and IKKε81. Interestingly, A20 inhibits the K63-linked ubiqutination of TBK1 and IKKε in a DUB-independent mechanism, probably through interference of the binding of TBK1 and IKKε with a putative ubiquitin ligase, TRAF381. Several other E3 ligases have been implicated in the conjugation of K63-linked ubiquitin chains to TBK1 and IKKε in the induction of type I IFNs. The ubiquitin ligases mind bomb 1 (MIB1) and MIB2, bind to TBK1 in response to SeV infection or double-stranded RNA stimulation and conjugate K63-linked polyubiquitin chains to TBK182,83. MEFs deficient in MIB1 or in both MIB1 and MIB2 exhibited attenuated induction of IFNβ and IFNβ-target genes in response to SeV infection. Nrdp1 (also called ring finger protein 41 and FLRF) is an E3 with duel roles in regulating TLR-stimulated signaling. On the one hand, it promotes TRIF-dependent activation of TBK1 and IRF3 by mediating K63-linked ubiquitination of TBK1; on the other hand, it catalyzes K48-linked ubiquitination of MyD88 and negatively regulates MyD88-dependent activation of NF-κB and AP184. The ubiquitination of IKKε in cancer cell lines is dependent on cIAP1, cIAP2, and TRAF2, which may form an E3 complex to mediate K63-linked polyubiquitination of IKKε85. However, cIAPs appear to be dispensable for IFN responses in immune cells, since inhibition of cIAPs in macrophages does not affect the TLR4-stimulated expression of type I IFNs86.
K63-linked polyubiquitination of TBK1 and IKKε occurs at two K residues, K30 and K401, which are conserved between these two homologous kinases85,87. Mutation of either K30 or K401 severely inhibits the ubiquitination and kinase activity of IKKε, whereas simultaneous mutations of both residues are required for blocking the ubiquitination and kinase function of TBK1. The ubiqutination of TBK1 requires its dimerization, although how dimerization promotes TBK1 ubiquitination is unknown87. It is also unclear how ubiquitination of TBK1 and IKKε facilitates their catalytic activation. It has been proposed that K63-linked ubiquitination of TBK1 may facilitate the recruitment of a kinase that phosphorylates and activates TBK1 or promote the association of TBK1 with NEMO to form a higher order complex for triggering TBK1 autophosphorylation87,88. Although NEMO mainly functions as the regulatory subunit of IKK, it is also required for the activation of TBK1 and induction of type I IFNs in some PRR pathways33,89.
K48-linked ubiquitination plays a negative role in the regulation of TBK1/IKKε function and type I IFN induction. Several E3 ubiquitin ligases have been shown to target TBK1 or IKKε for degradation. The RING finger E3 ubiquitin ligase Deltex 4 (DTX4) mediates K48-linked ubiquitination and degradation of TBK1 in a mechanism that depends on the NLR member NLRP490. It has been proposed that upon activation by double-stranded RNA and DNA or RNA viruses, phosphorylated TBK1 is bound by NLRP4, which in turn recruits DTX4 and allows this E3 to conjugate K48-linked ubiquitin chains onto K670 of TBK1 to trigger TBK1 degradation90. A more recent study suggests that dual-specificity tyrosine phoshorylation-regulated kinase 2 (DYRK2) phosphorylates TBK1 at serine 527, which is required for the recruitment of NLRP4 and DTX4 to mediate ubiquitin-dependent TBK1 degradation91. Another E3 ligase implicated in the regulation of TBK1 degradation is TRAF-interacting protein (TRIP), which directly binds to and promotes K48-linked ubiquitination of TBK1, thereby negatively regulating type I IFN induction by TRIF- and RIG-I-mediated antiviral pathways92. A tripartite motif-containing (TRIM) family member, TRIM27, functions as an E3 that mediates ubiquitin-dependent degradation of TBK1 in a negative feedback pathway of type I IFN signaling93. During viral infection, type I IFNs induce the expression of sialic acid-binding Ig-like lectin 1 (Siglec1), which interacts with the adaptor molecule DAP12 and recruits the protein tyrosine phosphatase SHP2. SHP2 in turn functions as a scaffolding protein to recruit the E3 ubiquitin ligase TRIM27, leading to ubiquitin-dependent degradation of TBK1 and negative regulation of type I IFN induction93. A recent study suggests that TBK1 is also targeted for ubiquitin-dependent degradation by suppressor of cytokine signaling 3 (SOCS3)94, a member of a protein family that is induced by various cytokine signals and function as important regulators of innate and adaptive immunity95. SOCS proteins exert their immunoregulatory functions via different mechanisms, one of which being forming` an E3 ubiquitin ligase complex with elongins B and C, cullin5, and Rbx-196,97. Within this E3 ligase complex, SOCS proteins function as the substrate-binding subunit and induce degradation of a variety of proteins. SOCS3 binds to TBK1 and mediates K48-linked ubiquitination of TBK1 at K341 and K344, thereby promoting TBK1 degradation and inhibiting IRF3 activation by VSV and influenza A virus94.
Ubiquitination in TLR signaling
Ubiquitination plays a crucial role in the signaling function of the adaptor proteins TRAF6 and RIP1 in the MyD88- and TRIF-dependent TLR pathways (Figure 2). TRAF6 has E3 ubiquitin ligase function and conjugates K63-linked ubiquitin chains in cooperation with the E2 dimer Ubc13/Uev1A52. In addition to ubiquitinating other molecules, TRAF6 undergoes self-conjugation with K63-linked polyubiquitin chains, which serve as a platform to facilitate the recruitment and catalytic activation of TAK1 and IKK5. As discussed in an above section, both TAK1 and IKK have a regulatory subunit, TAB2 or TAB3 for TAK1 and NEMO for IKK, with ubiquitin-binding functions. It is generally believed that TAB2/3 recruits TAK1 to ubiquitinated TRAF6, where TAK1 is activated via autophosphorylation; activated TAK1 then phosphorylates the catalytic subunits of IKK, causing IKK activation (Figure 2). However, this model is questioned by some recent studies using mutant mouse models. Macrophages derived from TAB2/TAB3 double knockout mice have no defects in TLR-stimulated activation of TAK1 or its downstream kinases, IKK and MAPKs98. In B cells, the double deletion of TAB2 and TAB3 inhibits TLR- and CD40-mediated activation of MAPKs, predominantly ERK, without affecting the activation of TAK1 and NF-κB. These findings suggest that the ubiquitin-binding regulatory subunits of TAK1 are either not important or functionally redundant with other ubiquitin-binding factors in TAK1 activation.

Ubiquitin regulation of TLR signaling. In response to MyD88- and TRIF-dependent TLR signals, TRAF6 undergoes self-ubiquitination and RIP1 is ubiquitinated by Peli1, an E3 ligase that is activated via its phosphorylation by IRAK1 and TBK1 or IKKε. It is generally thought that K63-linked ubiquitin chains conjugated to TRAF6 and RIP1 facilitate the recruitment and activation of TAK1 and IKK, which requires the ubiquitin-binding proteins TAB2 (or TAB3) and NEMO. Activated IKK and MAPKs (downstream of TAK1) mediate activation of NF-κB c-Rel and RelA, and members of the AP1 family, which together with TRAF6-stimulated IRF5 transactivate a variety of proinflammatory cytokine genes. As discussed in the text, this signaling model requires further modifications, since some parts are not supported by knockout mouse studies. TLR signaling is subject to control by negative regulators, one of which is TRAF3 that negatively regulates TLR-stimulated proinflammatory signaling by two possible mechanisms: interfering with the signaling function of the MyD88 complex and mediating degradation of IRF5 and c-Rel by forming an E3 complex with TRAF2 and cIAP. At least in the TLR4 signaling pathway, TRAF3 is targeted for ubiquitin-dependent degradation via a mechanism in which Peli1 either cooperates with TRAF6 or functions downstream of TRAF6 to mediate K63-linked ubiquitination and activation of cIAP, allowing the activated cIAP to conjugate K48-linked ubiquitin chains to TRAF3. By mediating TRAF3 degradation, Peli1 promotes proinflammatory TLR signaling.
The role of TRAF6 ubiquitination is also controversial. While one study demonstrates that the auto-ubiquitination site of TRAF6 (K124) is crucial for mediating activation of TAK1 and IKK99, another study suggests that auto-ubiquitination of TRAF6 is dispensable for the recruitment and activation of TAK1100. Nevertheless, both of these studies emphasize the importance of the RING domain of TRAF6 for its signaling function, highlighting a role for the E3 ligase activity of TRAF6. It is likely that TRAF6 also transduces TLR signaling via ubiquitination of other signaling molecules. For example, TRAF6 is known to mediate K63-linked polyubiquitination of IRAK1, which seems to be crucial for IKK activation by TLRs and IL-1R101. A recent study suggests that MyD88 is also conjugated with K63-linked polyubiquitin chains in response to infection by the nontypeable Haemophilus influenzae (NTHi)102. Although it is unclear whether this ubiquitination event is mediated by TRAF6 or another E3, the ubiquitination of MyD88 is negatively regulated by the DUB CYLD and is important for activating downstream proinflammatory signaling. Another intriguing mechanism of TRAF6 function is to activate TAK1 via synthesizing free or unanchored polyubiquitin chains103. Binding of free ubiquitin chains by TAB2 triggers autophosphorylation and catalytic activation of TAK1. It will be important to examine whether the TAB2/TAB3 double deficient cells have a defect in TAK1 activation under conditions that involve unanchored polyubiquitin chains.
Unlike TRAF6, RIP1 does not possess E3 ligase activity. TRAF6 was initially thought to mediate RIP1 ubiquitination; however, TRAF6 is not essential for TRIF-dependent activation of IKK, suggesting an additional E3(s) for RIP1 ubiquitination25,104. Genetic evidence suggests that TLR-stimulated RIP1 ubiquitination requires the E3 ubiquitin ligase Peli1 (also called Pellino1)105, a member of a highly homologous ubiquitin ligase family that also includes Peli2 and Peli3106 (Figure 2). The E3 ligase function of Peli1 can be stimulated via its phosphorylation by the IKK-related kinases, TBK1 and IKKε, or by the kinase IRAK1107. Peli1 deficiency impairs the ubiquitination of RIP1 and attenuates the activation of NF-κB in cells stimulated with TLR3 and TLR4 ligands, poly(I:C) and LPS. Consistently, Peli1 is important for poly(I:C)- and LPS-stimulated expression of proinflammatory cytokines, and Peli1-deficient mice are resistant to LPS-induced septic shock105. In peripheral innate immune cells, Peli1 is mainly required for the TRIF-dependent proinflammatory signaling, which is probably due to functional redundancy between Peli1 and other Peli members in the MyD88-dependent pathway. In support of this idea, the central nervous system (CNS)-resident macrophages, microglia, predominantly express Peli1 and rely on Peli1 for both TRIF- and Myd88-dependent TLR signaling108. In the MyD88 pathway, Peli1 does not seem to activate a major signaling component but rather promotes MyD88 signaling through mediating ubiquitin-dependent degradation of a negative regulator, TRAF3108 (Figure 2).
Both the MyD88- and TRIF-dependent TLR pathways are subject to regulation by negative regulators, among which are the recently reported TRAF members, TRAF2 and TRAF3109,110. Both TRAF2 and TRAF3 negatively regulate TLR-stimulated expression of proinflammatory cytokines in innate immune cells, and genetic deficiency in either TRAF promotes inflammation in mice111,112. The mechanism by which TRAF2 and TRAF3 negatively regulate proinflammatory TLR signaling appears to be complex. TRAF3 may target upstream signaling factors in the MyD88 complex and is thought to inhibit the cytoplasmic translocation of the MyD88 signaling complex, thereby attenuating LPS-stimulated MAPK activation109. Since TRAF3 also negatively regulates proinflammatory cytokine induction by the TRIF-dependent TLR3 ligand poly(I:C), it suggests additional mechanisms of TRAF3 function111. In bone marrow-derived macrophages, TRAF2 and TRAF3 regulate the steady level of c-Rel and IRF5, transcription factors that are important for TLR-stimulated proinflammatory cytokine gene expression111. During M-CSF-induced macrophage differentiation, c-Rel and IRF5 are transcriptionally induced, programing the cells for inflammatory responses to TLR stimulation111,113. TRAF2 and TRAF3 mediate ubiquitin-dependent degradation of c-Rel and IRF5 proteins, thereby preventing aberrant accumulation of these proinflammatory transcription factors111. Deletion of either TRAF2 or TRAF3 greatly elevates the steady state level of c-Rel and IRF5, rendering macrophages hyper-responsive to TLR-stimulated proinflammatory cytokine induction. TRAF2 and TRAF3 are known to associate with cIAP (cIAP1 or cIAP2) and form an E3 ubiquitin ligase complex that promotes ubiquitin-dependent degradation of the kinase NIK in the noncanonical NF-κB signaling pathway48,49. cIAP appears to be also involved in the degradation of c-Rel and IRF5, since a cIAP inhibitor, Smac mimetic, enhances the steady state level of c-Rel and IRF5 in bone marrow-derived macrophages111. Thus, by mediating degradation of two major proinflammatory transcription factors during M-CSF-induced macrophage differentiation, the TRAF/cIAP complex negatively regulates the induction of proinflammatory cytokines in macrophages (Figure 2).
The TLR4 ligand LPS stimulates TRAF3 degradation in both the Raw264.7 macrophage cell line and the CNS-resident macrophages, which likely serves as a mechanism to override the inhibitory function of TRAF386,108. cIAP is thought to function as an E3 ubiquitin ligase that conjugates K48-linked ubiquitin chains to TRAF3 leading to TRAF3 degradation, since the cIAP inhibitor, Smac mimetic, inhibits LPS-stimulated TRAF3 ubiquitination and degradation86. It has been proposed that the K48-linked ubiquitin ligase activity of cIAP is triggered through its K63-linked ubiquitination by TRAF6109. Interestingly, genetic evidence suggests that LPS-stimulated K48 ubiquitination and degradation of TRAF3 in microglia requires the E3 ligase Peli1108 (Figure 2). Peli1 may promote TRAF3 degradation by mediating K63-linked ubiquitination and activation of cIAP, since Peli1 deficiency blocks LPS-stimulated K63-linked ubiquitination of cIAP without affecting the K63-linked ubiquitination of TRAF6108. When expressed in HEK293 cells, both Peli1 and TRAF6 stimulate K63-linked ubiquitination of cIAP, and Peli1 is required for TRAF6-stimulated ubiquitination of cIAP, suggesting that Peli1 may cooperate with TRAF6 or function downstream of TRAF6 in mediating cIAP ubiquitination and activation108 (Figure 2).
The in vivo function of Peli1 in mediating TRAF3 degradation was demonstrated in experimental autoimmune encephalomyelitis (EAE), an animal model of the autoimmune neuroinflammatory disease multiple sclerosis108. Peli1 deficiency causes accumulation of TRAF3 in microglia during EAE induction, which contributes to impaired expression of chemokines and proinflammatory cytokines and ameliorated EAE disease. These findings suggest that microglial TRAF3 is induced and rapidly degraded in a Peli1-dependent manner during EAE induction. These findings also establish Peli1 as a potential drug target in the treatment of multiple sclerosis and other types of inflammatory diseases.
Ubiquitination in RLR signaling
The two major RLR members, RIG-I and MDA5, are both CARD-containing PRRs, and they associate with MAVS via CARD-CARD interactions. The RIG-I/MAVS interaction also requires K63-linked ubiquitination of RIG-I at K172 located in the second CARD114. Viruses and double-stranded RNA induce K63-linked ubiquitination of RIG-I, which facilitates its association with MAVS for MAVS activation114. Two E3 ubiquitin ligases, TRIM25 and RIPLET (also called RNF135), have been shown to mediate RIG-I ubiquitination and type I IFN induction114,115,116. The crucial role of TRIM25 and RIPLET in mediating RIG-I ubiquitination and type I IFN induction has been demonstrated using TRIM25 knockdown and knockout cells and RIPLET knockout mice114,117. Consistently, both TRIM25 and RIPLET are antagonized by a viral protein, non-structural protein 1 (NS1) of influenza A virus, further emphasizing the importance of these E3s in antiviral immunity118,119. Regarding the underlying mechanism, RIPLET conjugates K63-linked ubiquitin chains to K788 at the C-terminal domain of RIG-I, which triggers an open conformation of RIG-I to allow its exposed CARDs to be bound and ubiquitinated by TRIM25, thereby promoting the association of RIG-I with MAVS and TBK1120.
CYLD is a DUB known to negatively regulate RIG-I ubiqutination and RIG-I-mediated IFN gene induction79,80. CYLD deficiency causes constitutive activation of TBK1 and IKKε in DCs, and the CYLD-deficient DCs and MEFs are hyper-responsive to VSV in IFNβ induction80. CYLD binds to RIG-I and inhibits the ubiquitination and signaling function of RIG-I79,80. In addition, CYLD also inhibits the ubiquitination of TBK1 and IKKε, which also contributes to the negative regulation of IFN responses by CYLD79. It is important to note that the role of CYLD in antiviral innate immunity appears to be complex. Despite enhanced RIG-I signaling, the CYLD-deficient cells and mice are more susceptible to VSV infection due to attenuated signaling and antiviral gene expression induced by IFNβ, suggesting a positive role for CYLD in regulating type I IFN receptor function. Several other DUBs have also been implicated in the regulation of RIG-I ubiquitination and virus-induced type I IFN expression; these include A20, USP3, USP15, USP21, and USP25121,122,123,124,125,126. The reason for the involvement of so many DUBs in RIG-I regulation is unclear, but one possibility is that different DUBs may function in distinct cell types. Furthermore, some of these findings are based on cell lines and overexpression systems and yet to be confirmed by in vivo and genetic approaches. There are also some controversies, such as in the USP15 studies. While one study implicates USP15 as a positive regulator of RIG-I signaling, the other suggests an opposite role125,126.
Ubiquitination also has a negative role in regulating RLR signaling. Ubiquitination mediates proteasomal degradation of RIG-I as well as its signaling adaptor MAVS, and this negative-regulatory mechanism involves the ubiquitin ligase RNF125 (also called TRAC-1)127. RNF125 is upregulated by IFNα or its inducer poly(I:C) and then conjugates K48-linked ubiquitin chains to RIG-I, MAVS, and MDA5 to induce their degradation. Thus, RNF125 functions as a feedback regulator in the type I IFN induction signaling circuit. A recent study suggests that the linear ubiquitin ligase complex LUBAC negatively regulates type I IFN induction by the RIG-I and TRIM25 signaling axis128. LUBAC inhibits the RIG-I-mediated signaling and virus-induced type I IFN expression by two different mechanisms. First, LUBAC competes with TRIM25 for RIG-I binding and, thereby, inhibits TRIM25-mediated RIG-I ubiquitination and activation. Second, LUBAC mediates ubiquitin-dependent degradation of TRIM25. Interestingly, LUBAC promotes conjugation of both linear and K48-linked ubiquitin chains to TRIM25. Since LUBAC is known to be a linear ubiquitin chain ligase, it is unclear how it mediates K48-linked ubiquitination of TRIM25. One possibility is that the linear ubiquitin chains conjugated to TRIM25 promote K48-linked ubiquitination by recruiting a K48-specific E3 ligase.
Ubiquitination in NLR signaling
NLRs respond to both PAMPs and various endogenous ligands called danger-associated molecular patterns and mediate activation of inflammasomes as well as noninflammasome functions. Since the topic of inflammasomes has been discussed in several recent reviews16,17,18, this section will focus on the noninflammasome functions of NLRs. The founding members of the NLR family, NOD1 and NOD2, detect the bacterial peptidoglycan components iE-DAP and MDP, respectively, and mediate signaling via the adaptor RIP219. In response to ligand stimulation, NOD1 and NOD2 recruit RIP2 through CARD-CARD homotypic interactions129,130; this in turn triggers K63-linked polyubiquitination of RIP2, thereby facilitating the recruitment and activation of TAK1 and IKK131,132,133,134. Several E3 ubiquitin ligases have been implicated in the regulation of RIP2 ubiquitination and NOD1/2 signaling; these include TRAF2, TRAF5, TRAF6, cIAP1, cIAP2, and XIAP132,133,134,135,136,137. Moreover, a recent study provides genetic evidence for a role of Peli3 in mediating NOD2-induced activation of NF-κB and MAPKs and innate immunity protection from intestinal inflammation138. Analogous to Peli1-mediated RIP1 ubiquitination, Peli3 conjugates K63-linked ubiquitin chains to RIP2 to facilitate proinflammatory signaling. Why the ubiquitination of RIP2 involves so many different E3s is still not fully understood, although it is possible that some of them may be functionally cooperative. Another E3, ITCH, has also been shown to mediate K63-linked ubiquitination of RIP2; however, the function of ITCH in the NOD2 pathway appears to be complex, since it promotes activation of MAPKs that inhibits activation of NF-κB139.
Another important noninflammasome function of NLRs is to negatively regulate signal transduction from other PRRs and inhibit inflammatory responses. NLRP12 (also known as Monarch1 and PYPAF7) is a potent negative regulator of TLR-induced proinflammatory cytokine production and appears to function through inhibiting IRAK1 phosphorylation and NF-κB activation140. NLRP12 also negatively regulates noncanonical NF-κB signaling via a mechanism involving its physical association with NIK and TRAF3141,142. TRAF3 is a NIK-binding protein that mediates ubiquitin-dependent NIK degradation by recruiting NIK to the E3 ubiquitin ligase cIAP48,49. NLRP12 promotes NIK degradation via a yet-to-be defined mechanism, and NLRP12-deficient mice display enhanced susceptibility to colon inflammation and colitis-associated cancer142,143. Of note, the pathogenesis of NLRP12-deficient mice may also involve aberrant activation of T cells. NLRP12 has a T cell-intrinsic role in regulating IL-4 production and the pathogenesis of the T cell-dependent autoimmune disease EAE144.
An atypical NLR member, NLRX1, contains a mitochondrial targeting sequence and associates with the microchondrial adaptor MAVS to inhibit RLR signaling145. NLRX1 interferes with the association of MAVS with RIG-I and attenuates the activation of NF-κB and IRF3 pathways and induction of inflammatory cytokines and type I IFNs in response to RNA viruses145. In response to influenza virus infection, NLRX1-deficient mice exhibit increased expression of IFNβ and IFN-target genes as well as the proinflammatory cytokine IL-6146. In addition to inhibiting the MAVS-RIG-I interaction, NLRX1 binds to TRAF6 and IKK to negatively regulate NF-κB activation by the TLR4 ligand LPS146,147. This function of NLRX1 involves a ubiquitin-dependent dynamic regulation of protein-protein interactions. In unstimulated cells, NLRX1 forms a complex with TRAF6, and upon LPS stimulation, NLRX1 undergoes K63-linked polyubiquitination and is released from TRAF6. Ubiquitinated NLRX1 then associates with the IKK complex, via the ubiquitin-binding activity of NEMO, to inhibit IKK and NF-κB activation147. However, the role of NLRX1 in antiviral signaling is controversial, since two other studies fail to demonstrate a role for NLRX1 in negatively regulating MAVS-dependent antiviral responses148,149.
NLRC3 and NLRC5 are two other NLR members that negatively regulate PRR signaling and inflammatory responses150,151,152. NLRC3 physically associates with TRAF6 and inhibits TLR-stimulated K63-linked polyubiquitination of TRAF6 and activation of the downstream NF-κB signaling pathway152. NLRC3 also binds to stimulator of IFN genes (STING, also called MITA, MPYS, or ERIS), an adaptor of cytoplasmic DNA sensors153. NLRC3-STING binding appears to prevent the proper trafficking of STING to perinuclear and punctated region, a site required for STING activation. NLRC3 also blocks the binding of STING to the downstream kinase TBK1. Thus, NLRC3 deficiency promotes the induction of type I IFNs and proinflammatory cytokines by cytosolic DNA and DNA viruses153. NLRC5 negatively regulates PRR signaling by two different mechanisms151. First, NLRC5 directly suppresses the activation of IKK by interacting with the IKK catalytic subunits, IKKα and IKKα, blocking their association with NEMO and the assembly of the functional IKK complex. Second, NLRC5 binds to the RLRs, RIG-I and MDA5, to interfere with their association with MAVS and, thereby, inhibits the induction of type I IFNs by poly(I:C) and the RNA virus VSV151. However, controversies exist regarding the role of NLRC5 in regulating NF-κB activation and PRR signaling, since some studies failed to detect an effect of NLRC5 deficiency on the induction of type I IFNs or proinflammatory cytokines by viruses or bacteria154,155. The precise reason for the discrepancies is unclear, but it could be due to the cell type-specific functions of NLRC5 or the status of NLRC5 ubiquitination156,157. It has been recently shown that in LPS-stimulated cells, NLRC5 undergoes K63-linked polyubiquitination, which triggers the dissociation of NLRC5 from IKK, relieving the IKK-inhibitory function of NLRC5156. The ubiquitination of NLRC5 is mediated by TRAF2 or TRAF6 and negatively regulated by several potential DUBs, including USP14, USP18, and USP22156.
Ubiquitination in STING signaling
Sensing DNA in the cytoplasm is a crucial innate immune mechanism against various bacterial and viral pathogens and also serves as a mechanism to respond to danger signals elicited due to release of DNA to the cytoplasm21. Upon recognition of DNAs, cytoplasmic DNA sensors activate the adaptor STING, which mediates activation of TBK1/IKKε and IKK pathways, leading to the induction of type I IFNs and proinflammatory cytokines. As indicated already, STING senses bacterial c-di-AMP and c-di-GMP as well as the endogenous cyclic di-nucleotide cGAMP synthesized by cGAS as a response to cytoplasmic DNAs22. Ubiquitination plays an important role in the signaling function of STING. A type I IFN-induced E3 ubiquitin ligase, TRIM56, interacts with STING and targets STING for K63-linked polyubiquitination, which appears to facilitate STING dimerization and its association with the downstream kinase TBK1158. Thus, TRIM56 overexpression enhances IFNβ induction by double-stranded DNAs, whereas TRIM56 knockdown attenuates this response. Another E3 that mediates K63-linked ubiquitination and activation of STING is TRIM32159. TRIM32 interacts with STING at mitochondria and ER and promotes the interaction of STING with TBK1. STING is also ubiquitinated by autocrine motility factor receptor (AMFR), an E3 ligase that is located in the ER membrane and known to catalyze polyubiquitination of a subset of ER-associated protein degradation target proteins160. In response to cytoplasmic DNA stimulation, AMFR interacts with STING in a mechanism that depends on insulin-induced gene 1 (Insig1) and conjugates K27-linked polyubiquitin chains to STING. The K27-linked ubiquitin chains appear to facilitate the recruitment of TBK1 to STING for activation. Genetic deficiencies in AMFR and Insig1 attenuate host defenses against the DNA virus herpes simplex virus 1 (HSV1)161.
Ubiquitination also plays a negative role in regulating STING signaling, and several E3 ubiquitin ligases have been implicated in the control of the STING pathway. RNF5 (also called RMA1) is an E3 that binds to STING in mitochondria upon viral infection and conjugates K48-linked ubiquitin chains to K150 of STING to target STING for proteasomal degradation162. The RNF5-mediated STING degradation is negatively regulated by another E3 ligase, RNF26, which conjugates K11-linked ubiquitin chains to K150 of STING and likely interferes with the conjugation of K48-linked ubiquitin chains by RNF5163. Ubiquitin-dependent degradation of STING is also mediated by TRIM30a, an E3 that is induced by HSV-1 infection in DCs164. In addition to STING, DNA sensors may also be targeted for ubiquitin-dependent degradation. One example is DDX41, a DNA sensor that is ubiquitinated by the E3 ligase TRIM21165. TRIM21 interacts with and mediates K48-linked ubiquitination and degradation of DDX41 to negatively regulate type I IFN induction by double-stranded DNA165.
Ubiquitination in DC maturation and function
DCs are professional APCs that are crucial for both stimulating T cell responses to foreign antigens and maintaining immune tolerance to self-antigens166,167,168. Under steady state condition, DCs exist in immature states and present self-antigens or commensal microbial antigens to T cells to induce T cell tolerance, thereby preventing autoimmune and inflammatory disorders167,168. During an infection, DCs recognize PAMPs via their PRRs and are stimulated to mature and become competent APCs, which present microbial antigens to naive T cells to induce an efficient T cell response required for the clearance of the infection166. Upon PRR stimulation, DCs also secrete various cytokines that regulate the differentiation of CD4+ T cells to different subsets of helper T cells, including Th1, Th2, Th9, Th17, T follicular helper (Tfh) cells, and inducible Treg cells169.
DC maturation involves upregulation of surface MHC class II (MHCII) and costimulatory molecules, such as CD80 and CD86. An important mechanism that controls the surface expression of MHCII and CD86 is ubiquitin-dependent degradation of these molecules in the lysosome170,171,172. Membrane-associated RING-CH-1 (MARCH1) is an E3 ligase that ubiquitinates MHCII and CD86, promoting their endocytosis and lysosomal degradation170,173,174 (Figure 3). MARCH1 is downregulated during DC maturation, and MARCH1 downregulation is opposed by the autocrine action of IL-10 that induces MARCH1 expression170,174,175. Thus, by reducing the surface expression of MHCII and CD86 in DCs, MARCH1 restricts the antigen presentation capability of DCs. However, the biological function of MHCII ubiquitination seems to be complex, since a recent study unexpectedly reveals that DCs lacking MARCH1 or expressing a ubiquitination-defective MHCII mutant have reduced, rather than enhanced, ability to activate naive CD4+ T cells despite the expression of higher levels of MHCII176. Although how MHCII ubiquitination mediates the T cell-activation function of DCs is unknown, the mutant DCs defective in MHCII ubiquitination exhibit reduced expression of integrin β2 and cytokine IL-12, which may partially contribute to their impaired function. Another E3 ubiquitin ligase, Hrd1, promotes MHCII gene transcription by mediating ubiquitin-dependent degradation of the transcriptional repressor B lymphocyte-induced maturation protein 1 (BLIMP1)177. Hrd1 deficiency in DCs impairs DC-mediated priming of CD4+ T cells and renders mice resistant to the induction of EAE, a T cell-dependent autoimmune disease177.

Ubiquitin regulation of DC activation. Dendritic cells (DCs) are primary antigen-presenting cells (APCs) that are activated by signaling from PRRs, such as TLRs, during an infection. Activated DCs undergo maturation, including upregulation of MHCII and the costimulatory molecule CD86 that are required for antigen presentation and T cell costimulation. Activated DCs also produce various cytokines, such as the proinflammatory cytokines IL-12 and IL-23, which in turn regulate the differentiation of antigen-activated T cells. The membrane-bound E3 ubiquitin ligase MARCH1 mediates ubiquitin-dependent internalization and lysosomal degradation of MHCII and CD86, thereby negatively regulating the function of DCs in T cell activation. The rhomboid family protease Rhbdd3 negatively regulates DC activation by binding to NEMO and recruiting the DUB A20 for deconjugation of K63-linked ubiquitin chains from NEMO. Trabid is a member of the A20 subfamily of OTU-containing DUBs that deubiquitinates and stabilizes Jmjd2d, a histone demethylase that removes transcriptionally repressive histone modifications, H3K9me2 and H3K9me3, from the Il12 (Il12a and Il12b) and Il23 (Il23a) promoters to promote the recruitment of c-Rel-containing NF-κB complexes to the κB enhancer element for Il12/Il23 transcription. The E3 ubiquitin ligase catalyzing Jmjd2d ubiquitination is currently unknown.
Ubiquitination also plays an important role in regulating the signaling pathways involved in the induction of DC maturation. K63-linked ubiquitination of NEMO is a crucial mechanism mediating the activation of IKK and NF-κB. A point mutation of NEMO (C417R) in patients with EDI interferes with NEMO ubiquitination and activation of the NF-κB member c-Rel by CD40 ligand. The EDI DCs are defective in CD40-mediated induction of the c-Rel-dependent inflammatory cytokine IL-12 and costimulatory molecules, associated with impaired function in inducing T cell activation178. A rhomboid family protease, Rhbdd3, negatively regulates DC activation by promoting deconjugation of K63-linked polyubiquitin chains from NEMO. Rhbdd3 binds to NEMO via its UBA domain, which recognizes K27 ubiquitin chains attached to K302 of NEMO. Rhbdd3 in turn recruits DUB A20 to NEMO, allowing A20 to cleave the K63-linked ubiquitin chains of NEMO, thereby inhibiting IKK activation (Figure 3). Rhbdd3 deficiency causes aberrant DC activation and production of the inflammatory cytokine IL-6 in response to TLR ligands, and Rhbdd3-deficient mice display perturbed T cell homeostasis and spontaneously develop autoimmunity179.
A recent study has demonstrated a role for ubiquitination in mediating an epigenetic mechanism that regulates DC function180. Trabid (also known as Zranb1), an A20 subfamily of OTU domain-containing DUB, is required for TLR-stimulated expression of the inflammatory cytokines IL-12 and IL-23 in DCs (Figure 3). Previous studies suggest that the NF-κB family member c-Rel plays an important role in TLR-stimulated expression of IL-12 and IL-23181,182. Interestingly, Trabid is dispensable for the induction of c-Rel nuclear translocation but is required for the recruitment of c-Rel to the promoters of Il12a, Il12b, and Il23a genes. Trabid appears to function by deubiquitinating and stabilizing a histone demethylase, Jmjd2d, which in turn regulates histone modifications at the Il12 and Il23 promoters to facilitate c-Rel recruitment (Figure 3). DC-conditional deletion of Trabid impairs IL-12 and IL-23 production in DCs and the generation of Th1 and Th17 subsets of inflammatory T cells, rendering mice refractory to the induction of EAE180. It is currently unclear which E3 ubiquitin ligase mediates the ubiquitination of Jmjd2d.
Ubiquitination in T cell activation
T cells serve as a central component of adaptive immunity. Upon maturation in the thymus, naive CD4+ and CD8+ T cells migrate to the periphery and circulate between the blood stream and lymphoid organs, and become activated when their TCR detects a foreign antigen presented by MHC molecules on APCs. Activated T cells undergo clonal expansion and subsequently differentiate into various subsets of effector T cells that participate in different aspects of immune function183,184. T cell activation requires ligation of both the TCR and costimulatory molecules, particularly CD28. The TCR/CD28 costimulation triggers cascades of signaling events, which regulate both the initial activation and subsequent differentiation of T cells183. TCR stimulation leads to the activation of protein tyrosine kinase Lck, which phosphorylates the TCR signaling chain CD3ζ resulting in the recruitment of tyrosine kinase Zap70 to the TCR complex183. Activated Zap70 phosphorylates a number of signaling adaptors and enzymes, most importantly the scaffold proteins LAT (linker for activation of T cells) and SLP-76 (Src homology 2 domain-containing leukocyte protein of 76 kDa). Upon phosphorylation at different tyrosine residues, LAT and SLP-76 recruit various signaling factors, triggering the activation of phospholipase C, γ 1 (PLCγ1) and its downstream protein kinase C θ (PKCθ), Ras, and calcium pathways (Figure 4). These pathways in turn lead to the activation of transcription factors NF-κB, AP1, and NFAT, which cooperate in the induction of genes involved in T cell proliferation and survival.

Ubiquitin regulation of T cell activation. TCR signaling is initiated by antigen/MHC-stimulated colocalization of the TCR complex with T cell coreceptor, CD4 or CD8, which allows the CD4/CD8-associated protein tyrosine kinase Lck to phosphorylate the signaling chains of TCR. Phosphorylated TCRζ chains recruit and activate the protein tyrosine kinase Zap70, which in turn phosphorylates LAT and SLP-76, triggering the recruitment and activation of key signaling components, such as PLCγ1. PLCγ1 hydrolyzes the membrane lipid PIP2 to generate second messengers, DAG and IP3, which in turn mediate activation of PKCθ, Ras, and calcium pathways, leading to activation of NF-κB, AP1, and NFAT. K63-linked ubiquitination is crucial for activation of NF-κB and AP1 pathways by the intermediate signaling complex, composed of CARMA1, BCL10, and MALT1. This complex associates with an E3 and the E2 dimer Ubc13/Uev1a to conjugate K63-linked ubiquitin chains to BCL10, thereby promoting the recruitment and activation of TAK1 and IKK. Several E3 ubiquitin ligases, including Cbl-b, c-Cbl, GRAIL, and ITCH, negatively regulate TCR-proximal signaling by mediating ubiquitin-dependent degradation of major signaling components. Some E3 ligases, likely including Nrdp1 and c-Cbl, conjugate nondegradative ubiquitin chains, including K33-linked chains, to Zap70, which facilitates the recruitment of protein tyrosine phosphatases Sts1 and Sts2 to dephosphorylate and inhibit Zap70. Peli1 and MDM2 are E3s that mediate ubiquitin-dependent degradation of activated transcription factors, c-Rel and NFATc2, respectively.
TCR-proximal signaling
Ubiquitination plays a crucial role in the regulation of TCR-proximal signaling. Several E3 ubiquitin ligases, including Cbl-b, GRAIL, Itch, and Deltex, negatively regulate the TCR-proximal signaling events by mediating ubiquitination and degradation of key TCR signaling components, including CD3ζ, PKCθ, Zap70, PLCγ1, and PI3 kinase (PI3K)185,186,187,188. Ubiquitination also regulates TCR-stimulated endocytosis and degradation of LAT, although the underlying mechanism is poorly defined189,190. T cells lacking the ubiquitin ligase c-Cbl or expressing a RING mutant form of c-Cbl exhibit a defect in TCR-stimulated LAT internalization, which is associated with elevated cellular levels of LAT189. However, a LAT mutant incapable of undergoing ubiquitination is still efficiently internalized, although it displays moderately increased stability190. It has been proposed that the effect of c-Cbl on LAT may be indirectly mediated through ubiquitination of other proteins that are associated with LAT190. LAT ubiqutination may also regulate its signaling function via a degradation-independent mechanism, since expression of the ubiquitination-resistant LAT mutant in T cells causes enhanced TCR signaling191. A role for non-degradative protein ubiquitination in regulating TCR signaling has been more clearly demonstrated by other studies192,193. Cbl-b and Itch mediate conjugation of K33-linked ubiquitin chains to TCRζ, which inhibits the association of TCRζ with Zap70 via a non-degradative mechanism193. It has also been shown that c-Cbl binds to Zap70 in response to TCR signaling and inhibits Zap70 activation, although it is not clear whether c-Cbl mediates degradation or non-degradative inhibition of Zap70194,195. Recent studies suggest that ubiquitination of Zap70 may negatively regulate Zap70 activity by recruiting the protein tyrosine phosphatase Sts1 (also called TULA-2 or Ubash3b) and its homologue, Sts2 (also called TULA or Ubash3a)196 (Figure 4). In addition to their phosphatase domain, Sts1 and Sts2 contain a UBD and an SH3 domain, which allow these phosphatases to bind substrates (like Zap70) that are dually modified by ubiquitination and tyrosine phosphorylation197. By dephosphorylating Zap70, Sts1 and Sts2 negatively regulate Zap70 activation and TCR-proximal signaling. In CD8+ T cells, the E3 ligase Nrdp1 conjugates K33-linked polyubiquitin chains onto Zap70 and promotes the dephosphorylation of Zap70 by Sts1and Sts2, thereby negatively regulating the early phase of TCR signaling198. Nrdp1 specifically functions in CD8+ T cells, since Nrdp1 deficiency has no effect on the activation of CD4+ T cells, suggesting the involvement of additional E3s in the regulation of non-degradative ubiquitination of Zap70.
Compared to E3 ligases, DUBs involved in the regulation of TCR-proximal signaling are still poorly defined. Nevertheless, a recent study has identified Otud7b as a DUB of Zap70 that facilitates TCR signaling199. Otud7b is a member of the A20 subfamily of OTU domain-containing DUBs implicated in the regulation of the NF-κB pathway200. In B cells, Otud7b suppresses the activation of noncanonical NF-κB signaling by deubiquitinating and stabilizing a negative regulator, TRAF3201. Otud7b deficiency promotes B cell activation and antibody production. Interestingly, in contrast to its negative regulatory role in B cells, Otud7b plays a positive role in promoting TCR/CD28-stimulated T cell activation and differentiation into the Th1 cells199. Otud7b binds to and deubiquitinates Zap70, thereby preventing the interaction of Zap70 with the negative-regulatory phosphatases, Sts1 and Sts2199 (Figure 4). Otud7b-deficient T cells are hypo-responsive to antigen stimulation, and Otud7b knockout mice exhibit attenuated T cell response against bacterial infection and are refractory to EAE, a T cell-dependent autoimmune disease199. These findings establish Otud7b as a DUB that positively regulates TCR-proximal signaling and mediates T cell response in immunity and autoimmunity. Another DUB, USP9X, has also been shown to regulate TCR-proximal signaling events, although the target of USP9X has not been defined202. It appears that the function of USP9X is complex. Despite the hypoproliferative phenotype of USP9X-deficient T cells, USP9X-deficient mice have spontaneous expansion of T cells with an activated phenotype, associated with lupus-like autoimmunity and lymphoproliferative disease202. This latter phenotype may be due to impaired negative selection of thymocytes and enhanced production of self-reacting T cells202.
CBM signaling axis
A well-studied intermediate signaling component in the TCR pathway is the so-called CBM complex, which is composed of the scaffold protein CARMA1 (also called CARD11), the adaptor BCL-10, and the paracaspase MALT1 (mucosa-associated lymphoid tissue lymphoma translocation gene 1). This signaling complex is predominantly involved in the activation of IKK and JNK pathways203, although it is also involved in TCR-stimulated activation of the metabolic kinase mTORC1204,205,206. Upon TCR/CD28 stimulation, CARMA1 becomes phosphorylated by PKCθ, which triggers the formation of the CBM complex and the association with a K63-specific E2 enzyme dimer, Ubc13/Uev1A, and an E3 ligase207,208. It is thought that Ubc13 conjugates K63-linked ubiquitin chains onto adaptor molecules, such as BCL10 and MALT1, which facilitate the recruitment and activation of TAK1 and its downstream kinases IKK and MAPKs. Indeed, Ubc13 deletion in T cells inhibits the activation of TAK1 and MAPKs JNK and p38, and partially impairs the activation of IKK209. Ubc13 also mediates ubiquitination of the IKK-regulatory subunit NEMO, which may also contribute to IKK activation207,208,209. The functional significance of K63-linked ubiquitination in T cells is underscored by the drastic reduction of peripheral T cells in T cell-conditional Ubc13 knockout mice despite normal development of thymocytes209. When stimulated with TCR/CD28 agonistic antibodies or T cell mitogens, the Ubc13-deficient thymocytes exhibit defective proliferation. The E3 ligase mediating K63-linked ubiquitination in the CBM complex is yet to be definitively defined by genetic approach. Early in vitro studies suggest that TRAF6 is an E3 that physically interacts with MALT1 and ubiquitinates NEMO, thereby mediating TCR-stimulated IKK activation207. However, TRAF6 deletion in T cells does not inhibit TCR-stimulated activation of NF-κB210, indicating functional redundancy between TRAF6 and other TRAF members or the involvement of other E3s. Regarding the latter possibility, a more recent study identified MIB2 as an E3 ligase that binds to BCL10 and is recruited to the CBM complex in the EL-4 T cell line211. MIB2 ubiquitinates NEMO and promotes activation of TAK1 and IKK, and MIB2 knockdown in the Jurkat T cell line inhibits the induction of NF-κB reporter gene by TCR/CD28211. However, it remains to be examined whether MIB2 plays a role in CBM-mediated NF-κB activation and T cell activation under in vivo conditions. Deletion of the MIB2 homolog, MIB1, in mice impairs the development of T cells and marginal zone B cells212. However, this function of MIB1 is not cell-intrinsic but rather mediated through regulation of Notch ligands in thymic and splenic stromal cells. Furthermore, in contrast to MIB1, MIB2 is dispensable for the development of T cells and marginal zone B cells212. It remains to be examined whether MIB2 regulates peripheral T cell activation and NF-κB activation in these cells.
In line with the positive role of K63 ubiquitination in mediating TCR signaling, T cell activation is controlled by specific DUBs known to cleave K63-linked ubiquitin chains4. These DUBs act on either the CBM components or downstream signaling factors, including TAK1 and IKK. CYLD is a crucial DUB that regulates the development and homeostasis of T cells213,214,215,216. CYLD controls the threshold of T cell activation by negatively regulating TCR/CD28-stimulated activation of TAK1 and its downstream kinases JNK and IKK214. Loss of CYLD causes accumulation of ubiquitinated TAK1 and aberrant TAK1 activation. CYLD deletion renders T cells hyper-responsive to TCR and CD28 costimulation, and CYLD-deficient mice exhibit perturbed T cell homeostasis characterized by increased frequencies of T cells with effector or memory phenotypes. These mutant animals spontaneously develop, or are pre-disposed to, colitis likely due to aberrant immune responses to commensal microflora in the gut214,217. T cell adoptive transfer studies suggest an important role for T cells in mediating the intestinal inflammation in these mutant animals214. The DUB USP18 also plays a role in regulating TCR signaling, which involves deubiquitination of TAK1 and its regulatory protein TAB1218. However, in contrast to CYLD, which controls homeostatic activation of TAK1, USP18 regulates TAK1 signaling during the differentiation of Th17 subset of CD4+ effector T cells218.
Another DUB that regulates the strength and duration of IKK/NF-κB activation by the TCR and CD28 signals is A20219,220. A20 cleaves the K63-linked ubiquitin chains from MALT1 to inhibit the recruitment and activation of IKK220. In the Jurkat T cell line, A20 knockdown allows strong activation of IKK/NF-κB by the TCR signal without CD28 costimulation220. Consistently, mice with conditional deletion of A20 in peripheral T cells display autoimmune phenotypes, including lymphadenopathy and T cell infiltrations in peripheral organs221. A20-deficient CD8+ T cells show enhanced production of cytokines, including IL-12 and IFN-γ, in response to antigen stimulation and display stronger antitumor activity in a transplantable B16 melanoma model221. A20 deficiency causes a higher level of nuclear c-Rel, coupled with a reduced cytoplasmic IκBα level, in CD8+ T cells stimulated with anti-CD3 and anti-CD28 antibodies, emphasizing a role for A20 in negatively regulating NF-κB signaling in T cells221.
DUBs may also play positive roles in the regulation of CBM signaling. One example is USP9X. In addition to facilitating TCR-proximal signaling, USP9X interacts with the CBM complex and facilitates CBM-mediated activation of NF-κB, although the precise underlying mechanism has not been defined. It has been proposed that USP9X may maintain the integrity of the BCL10-MALT1 complex by deubiquitinating BCL10222.
Transcription factor regulation
Ubiquitination also controls the activation and fate of downstream transcription factors in the TCR pathway. One example is the ubiquitin-dependent regulatory mechanism for c-Rel223, an NF-κB member with a crucial role in regulating T cell activation and differentiation181,224,225,226,227. Activation of c-Rel in T cells requires the costimulation of TCR and CD28228,229. Compared to RelA, c-Rel is also less sensitive to IκBα-mediated feedback regulation. Ubiquitin-dependent degradation appears to be an important mechanism of c-Rel regulation during T cell activation, and the c-Rel ubiquitination is mediated by the E3 ligase Peli1223 (Figure 4). Although Peli1 is known for its role in mediating K63-linked polyubiquitination in TLR signaling105,108, Peli1 also has the ability to conjugate K48-linked ubiquitin chains223,230. Peli1, but not Peli2 or Peli3, is abundantly expressed in T cells and further upregulated along with T cell activation. Peli1 deficiency inhibits c-Rel ubiquitination and causes accumulation of nuclear c-Rel in activated T cells223. Consistently, Peli1 deficiency renders T cells hyper-responsive to TCR/CD28 stimulation and less sensitive to Treg-mediated inhibition, and Peli1-deficient mice develop autoimmune symptoms.
A more recent study has demonstrated a mechanism that regulates ubiquitin-dependent degradation of an NF-AT family member, NF-ATc2, which involves the E3 ubiquitin ligase MDM2231. MDM2 is known as an oncogenic E3 ligase that mediates degradation and functional inhibition of the tumor suppressor p53232. However, in T cells, MDM2 negatively regulates TCR signaling by targeting NF-ATc2 for ubiquitination and degradation. During T cell activation, MDM2 itself also undergoes ubiquitin-dependent degradation, a molecular event that facilitates nuclear accumulation of NF-ATc2 and induction of cytokines, particularly IFN-γ231. Under normal condition, a DUB, Usp15, deubiquitinates MDM2 to inhibit MDM2 degradation, thereby negatively regulating the generation of IFN-γ-producing Th1 effector T cells. USP15 deficiency promotes Th1 responses against bacterial infections and tumor challenge in a transplantable melanoma model231. These studies suggest that pharmacological inhibition of USP15 may be an approach to promote antitumor immunity. However, it is important to note that chronic production of IFN-γ in Usp15 knockout mice may promote the induction of PD1 ligand (PD-L1) and other immunosuppressive factors in tumor microenvironment, which in turn facilitate tumor growth, as demonstrated with a chemical-induced fibrosarcoma model233.
Ubiquitination in T cell tolerance and autoimmunity
Under normal condition, T cells respond to foreign antigens derived from invading pathogens but are tolerant to self-antigens, thus preventing the development of autoimmunity234. The induction of T cell tolerance can occur during T cell development in the thymus, in which self-reactive T cells are deleted by the mechanism of negative selection. In addition to this so-called central tolerance, peripheral tolerance is required for suppressing the autoimmune action of self-reacting T cells that have escaped from the central tolerance. Both the central tolerance and peripheral tolerance of T cells are subject to regulation by ubiquitination.
Central tolerance
Thymic epithelial cells (TECs) provide an essential microenvironment for the development and selection of thymocytes. While cortical TECs (cTECs) are important for positive selection of thymocytes capable of recognizing self-MHC with appropriate affinity, medullary TECs (mTECs) are crucial for negative selection to eliminate self-reactive T cells11. A hallmark of mTECs is the expression of autoimmune regulator (AIRE), which was initially identified by positional cloning from patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED)235. In mTECs, AIRE functions as a transcription factor mediating promiscuous expression of a large variety of tissue-restricted self-antigens that are required for thymocyte negative selection236. AIRE contains two plant homeodomains (PHDs), one of which (PHD1) has E3 ubiquitin ligase activity237,238. The E3 ubiquitin ligase activity of AIRE is thought to play a role in mediating the function of AIRE in central tolerance238; however, whether AIRE is an E3 remains controversial, since another study could not demonstrate the E3 activity of the PHD1 of AIRE239.
The E3 ligase TRAF6 is another master regulator of central tolerance, which functions by mediating the development of thymic stroma240. TRAF6 deficiency in mice impairs the maturation of mTECs and reduces the expression of AIRE, thereby interfering with deletion of self-reacting T cells and causing autoimmunity240. The TEC-specific function of TRAF6 has been demonstrated by both fetal thymic stroma tissue transplantation and generation of TEC-conditional TRAF6 knockout mice240,241. The mechanism of TRAF6 function in mTECs involves the activation of canonical NF-κB and, thereby, induction of RelB expression240. Cell lines derived from TRAF6-deficient mTECs express much lower levels of RelB compared to wild-type mTEC lines. Defects in mTEC development and central tolerance induction have also been seen in RelB knockout mice or mice defective in noncanonical NF-κB signaling required for RelB activation242. Precisely how RelB and nonnonical NF-κB signaling regulate mTEC development is still poorly understood.
Cell-intrinsic mechanism of peripheral tolerance
Although central tolerance plays a crucial role in eliminating autoimmune T cells, this mechanism alone is insufficient for preventing autoimmunity, since not all self-antigens are expressed in the thymus11. Some autoimmune T cells escape from negative selection in the thymus and exist in peripheral lymphoid tissues, which are controlled by peripheral tolerance. The maintenance of peripheral T cell tolerance requires both intrinsic and extrinsic mechanisms243. A major extrinsic mechanism of peripheral tolerance is mediated by the Treg cells, which will be discussed in the following section. Intrinsic mechanism of T cell tolerance relies on various factors that negatively regulate TCR signaling and control the threshold of T cell activation. Under normal condition, naive T cell activation requires stimulation of both the TCR and costimulatory receptors, particularly CD28. T cells are efficiently activated by foreign antigens, which are typically presented by activated APCs expressing costimulatory molecules. However, when stimulated by self-antigens without costimulation, T cells become functionally inactivated and enter a state called anergy244. Genetic deficiencies in key negative regulators may lower the threshold of T cell activation and render T cells responsive to self-antigens, thus breaking the tolerance and leading to the development of autoimmunity.
As discussed in a previous section, several E3 ligases are involved in the negative regulation of TCR-proximal signaling. In fact, many of these E3 molecules, including c-Cbl, Cbl-b, GRAIL, and Itch, are involved in the regulation of T cell anergy245. Under tolerance conditions, such as TCR ligation in the absence of costimulation, these E3 molecules are induced via the calcium pathway and prevent T cell activation by suppressing TCR signaling185,188,192,246. Genetic deficiencies in such E3 ligases impair the induction of T cell anergy and also reduce the sensitivity of T cells to Treg-mediated suppression, causing spontaneous development of autoimmunity in mice. TRAF6 is an adaptor E3 that regulates both central tolerance and peripheral tolerance. TRAF6-deficient T cells exhibit hyper-sensitivity to TCR stimulation and diminished requirement for CD28 costimulation, associated with enhanced activation of the PI3K-AKT signaling pathway210. These mutant T cells also have a defect in anergy induction and become refractory to Treg-mediated suppression210,247. Peripheral T cell tolerance is also regulated by E3 ligases targeting downstream signaling factors of the TCR pathway. As discussed already, Peli1 is an E3 that mediates ubiquitin-dependent degradation of the NF-κB member c-Rel and controls the induction of IL-2 and IFN-γ. Peli1-deficient T cells are less sensitive to in vitro inhibition by Treg cells and TGF-β and display aberrant homeostasis and activation, and Peli1-deficient mice spontaneously develop autoimmunity characterized by lymphadenopathy, tissue infiltration of lymphocytes, and accumulation of autoimmune antibodies in the serum223.
Roquin family of proteins, Roquin-1 and Roquin-2, are RNA-binding RING-type E3 ubiquitin ligases that mediate the decay of different mRNAs, including those encoding ICOS and OX-40 in T cells248. An initial study reveals that mice carrying a point mutation of Roquin-1, termed sanroque mice, develop a lupus-like autoimmune disease characterized by enlargement of spleen and lymph nodes, accumulation of autoimmune antibodies, and generation of excessive numbers of Tfh cells and germinal centers249. Deregulated Tfh cell production in the sanroque mice contributes to the development of autoimmunity, and this abnormality is attributed to the aberrant expression of ICOS250,251. Subsequent gene targeting studies suggest that Roquin-1 and Roquin-2 have functional redundancies in the regulation of T cell tolerance and Tfh cell differentiation252,253,254. Regarding the molecular mechanism, Roquin proteins recognize the constitutive decay element (CDE) in the 3′-untranslated region of target mRNAs and promote target mRNA degradation by recruiting the Ccr4-Caf1-Not deadenylase complex255. Since about 50 mRNAs contain CDE and 95 mRNAs can be significantly enriched by Roquin immunoprecipitation, it has been postulated that the complex immune phenotype of the Roquin knockout mice and saroque mice may involve deregulated degradation of multiple mRNAs255. It is unclear whether the ubiquitin ligase activity of Roquin proteins is required for their regulation of mRNA degradation and T cell functions. Nevertheless, Roquin-2 has been shown to mediate ubiquitination and degradation of apoptosis signal-regulating kinase 1 (ASK1), which is important for promoting cell death induced by reactive oxygen species256. Similar functions have been demonstrated for the Roquin-2 ortholog, RLE-1 (regulation of longevity by E3), in Caenorhabditis elegans256. A recent structural study suggests that RNA binding by Roquin-2 may influence its ubiquitin ligase activity257.
Treg development and function
Treg cells form a subpopulation of T cells that is required for maintaining peripheral tolerance and preventing autoimmunity258. Treg cells develop in the thymus and can also arise from naive CD4+ T cells in the periphery. DC-mediated antigen presentation is crucial for the development of Treg cells in the thymus259. MARCH1, a membrane-associated E3 ubiquitin ligase regulating MHCII ubiquitination in DCs, has a positive role in regulating thymic Treg development260. As discussed above, MARCH1 mediates ubiquitination of MHCII and the costimulatory molecule CD86, promoting their endocytosis and lysosomal degradation170,173,174. MARCH1 deficiency causes increased surface expression of MHCII, which is associated with reduced production of thymus-derived Treg cells260. Similarly, knock-in mice expressing a ubiquitination-defective form of MHCII also have reduced number of thymic Treg cells, further emphasizing the role of MHCII expression in regulation of Treg development. The mechanism by which MHCII ubiquitination regulates Treg development is unknown, but it does not seem to involve the control of MHCII/peptide avidity260. It has been proposed that ubiquitinated MHCII may have altered surface dynamics or serve as signaling molecules, which conditions DCs for Treg cell generation.
A TRAF family member, TRAF3, has been shown to negatively regulate thymic Treg development, although it remains to be determined whether TRAF3 acts as an E3 ligase or an adaptor in this function261,262. TRAF3 regulates the conversion of CD25+Foxp3− Treg precursors to the CD25+Foxp3+ mature Treg cells262, a step of Treg development that requires the action of the cytokine IL-2263,264. It appears that TRAF3 inhibits IL-2 receptor signaling by promoting the association of IL-2 receptor with a negative regulatory tyrosine phosphatase, TCPTP262. Interestingly, although T cell-conditional TRAF3 knockout mice have elevated numbers of Treg cells in both the thymus and periphery, these mutant animals display perturbed T cell homeostasis characterized by an increase in the frequency of CD4+ T cells with effector- or memory-like surface markers265. This is apparently because of a positive role for TRAF3 in regulating the immunosuppressive function of established Treg cells266. Ablation of TRAF3 in Treg cells using the Foxp3-Cre deletion system causes perturbation of T cell homeostasis with a predominant increase in the frequency of Th1 type of effector/memory-like T cells266. The Treg-specific TRAF3 deficiency does not reduce, but rather moderately increases, the overall frequency of Treg cells; however, the TRAF3 deficiency attenuates antigen-stimulated production of follicular Treg (Tfr) cells, resulting in upregulated germinal center formation and antibody production266. TRAF3 plays a role in mediating MAPK activation and induction of ICOS, which in turn is important for antigen-stimulated Tfr generation266. Thus, TRAF3 has different roles in regulating the development and peripheral function of Treg cells.
The inducible production of Treg cells from naive CD4+ T cells in the periphery is also regulated by ubiquitination. Cbl-b, which is a negative regulator of naive T cell activation, is a positive regulator of induced regulatory T (iTreg) cell induction by TGF-β, since Cbl-b deficiency in mice impairs iTreg cell generation both in vitro and in vivo267,268. Cbl-b appears to negatively regulate PI3K activation, thereby preventing the phosphorylation and inactivation of the downstream transcription factors Foxo3a and Foxo1. These Foxo transcription factors bind to the promoter of Foxp3 to directly promote Foxp3 expression and iTreg generation267. TGF-β-induced Foxp3 expression also requires the E3 ubiquitin ligase Itch, which mediates non-degradative ubiquitination of the transcription factor TIEG1, which in turn appears to enhance the transactivation function of TIEG1 at the Foxp3 promoter269.
K63-linked ubiquitination plays a vital role in regulating the immunosuppressive function of Treg cells. Treg-specific deletion of the K63-specific E2 enzyme Ubc13 impairs the suppressive function of Treg cells, causing activation of effector T cells and development of autoimmune symptoms in mice270. Ubc13 deletion in established Treg cells has no effect on Foxp3 expression or the frequency of Treg cells. However, the Ubc13-deficient Treg cells lose the immunosuppressive functions in vivo, causing T cell activation and autoimmune symptoms. When adoptively transferred, along with naive T cells, into Rag1 knockout mice, the Ubc13-deficient Treg cells failed to suppress the activation of naive T cells. These mutant Treg cells display the effector functions of Th1 and Th17 inflammatory T cells, characterized by the production of IFN-γ and IL-17. This function of Ubc13 involves the activation of IKK in the Treg cells, since expression of a constitutively active form of IKKβ could rescue the function of Ubc13-deficient Treg cells270. The Ubc13/IKK pathway is required for maintaining abundant expression of SOCS1, which in turn is required for preventing Treg cells from acquiring inflammatory effector functions of Th1 and Th17 cells. The promoter region of SOCS1 gene contains a STAT-NF-κB composite site and is cooperatively stimulated by the TCR-CD28 stimuli and inflammatory cytokines (IL-6 and IFN-γ)270. These results not only demonstrate an important role for K63-linked ubiquitination and IKK signaling in the maintenance of Treg function, but also highlight a paradoxical role for K63-linked ubiquitination in T cell responses: while required for T cell activation, it is also important for regulating the immunosuppressive function of Treg cells.
K48-linked ubiquitination also regulates the immuosuppressive function of Treg cells. In particular, the Treg master transcription factor Foxp3 is subject to tight regulation by ubiquitin-dependent degradation271. The ubiquitin ligase Stub1 has been shown to physically associate with Foxp3 and conjugate K48-linked ubiquitin chains to Foxp3 when Treg cells are stimulated with proinflammatory cytokines and the TLR4 ligand LPS272. Conversely, the DUB Usp7 deubiquitinates and stabilizes Foxp3 to promote Treg functions. USP7 knockdown or inhibition by a DUB inhibitor reduces the level of Foxp3 protein and compromises the function of Treg cells273. In thymic Treg precursor cells, the E3 ubiquitin ligase Cbl-b cooperates with Stub1 to mediate TCR/CD28-stimulated Foxp3 degradation274. Cbl-b deficiency partially rescues the defect of CD28-deficient mice in natural Treg development. The E3 ligase GRAIL is highly expressed in Treg cells and contributes to the immunosuppressive function of Treg cells275,276,277. GRAIL-deficient Treg cells have normal expression Foxp3 and various surface markers, but these mutant Treg cells are partially impaired in their ability to suppress the activation of naive T cells277. GRAIL deficiency also promotes the expression of Th17 effector cytokines (IL-17 and IL-21) and transcription factors (RORα and RORγ) in Treg cells, which appears to involve upregulated expression of the transcription factor NFATc1277.
The E3 ligase von Hippel-Lindau (VHL) also has an essential role in maintaining the immunoresppressive function of Treg cells. Treg-conditional ablation of VHL using the Foxp3-Cre system leads to tissue inflammation due to loss of Treg functions. The VHL-deficient Treg cells display a Th1-like inflammatory phenotype producing high amounts of IFN-γ. This phenotype involves aberrant accumulation of the hypoxia-inducible factor 1α (HIF-1α), which appears to directly bind IFN-γ promoter and mediate IFN-γ induction in the VHL-deficient Treg cells278.
Concluding remarks
This review highlights some of the major functions of ubiquitination in the regulation of innate and adaptive immune responses. To date, a large number of E3 ubiquitin ligases and DUBs have been characterized as important immune regulators, and they participate in different aspects of immune functions, ranging from PRR signaling in innate immunity and inflammation to DC maturation and T cell activation and differentiation. Ubiquitination also plays a crucial role in regulating central and peripheral immune tolerance to prevent the development of autoimmunity. Studies using mutant mouse models have been instrumental in elucidating the physiological functions of E3 ubiquitin ligases and DUBs, and this area of research will continue adding important information to our understanding of the role of ubiquitination in the immune system.
A major challenge in future studies will be to characterize the molecular mechanisms underlying the immunoregulatory functions of the E3s and DUBs. Identification of the substrates of these enzymes in physiologically relevant immune cells represents an important, and challenging, task. Equally important is to understand how non-degradative ubiquitination regulates the function of protein kinases and other signaling components. An increasing number of UBD-containing proteins have been identified, and biochemical and physiological characterization of these ubiquitin-binding proteins will provide additional insight into the signaling mechanism of ubiquitination.
References
Hershko A, Ciechanover A . The ubiquitin system. Annu Rev Biochem 1998; 67:425–479.
Deshaies RJ, Joazeiro CA . RING domain E3 ubiquitin ligases. Annu Rev Biochem 2009; 78:399–434.
Berndsen CE, Wolberger C . New insights into ubiquitin E3 ligase mechanism. Nat Struct Mol Biol 2014; 21:301–307.
Sun SC . Deubiquitylation and regulation of the immune response. Nat Rev Immunol 2008; 8:501–511.
Liu S, Chen ZJ . Expanding role of ubiquitination in NF-κB signaling. Cell Res 2011; 21:6–21.
Iwai K . Diverse ubiquitin signaling in NF-κB activation. Trends Cell Biol 2012; 22:355–364.
Husnjak K, Dikic I . Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem 2012; 81:291–322.
Janeway CA Jr . Approaching the asymptote? Evolution and revolution in immunology. Cold Spring Harb Symp Quant Biol 1989; 54 Pt 1:1–13.
Brubaker SW, Bonham KS, Zanoni I, Kagan JC . Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 2015; 33:257–290.
Iwasaki A, Medzhitov R . Control of adaptive immunity by the innate immune system. Nat Immunol 2015; 16:343–353.
Xing Y, Hogquist KA . T-cell tolerance: central and peripheral. Cold Spring Harb Perspect Biol 2012; 4: pii: a006957.
Wu J, Chen ZJ . Innate immune sensing and signaling of cytosolic nucleic acids. Annu Rev Immunol 2014; 32:461–488.
Kawai T, Akira S . The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 2010; 11:373–384.
Yoneyama M, Fujita T . RNA recognition and signal transduction by RIG-I-like receptors. Immunol Rev 2009; 227:54–65.
Martinon F, Mayor A, Tschopp J . The inflammasomes: guardians of the body. Annu Rev Immunol 2009; 27:229–265.
Davis BK, Wen H, Ting JP . The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol 2011; 29:707–735.
Latz E, Xiao TS, Stutz A . Activation and regulation of the inflammasomes. Nat Rev Immunol 2013; 13:397–411.
Man SM, Kanneganti TD . Regulation of inflammasome activation. Immunol Rev 2015; 265:6–21.
Caruso R, Warner N, Inohara N, Nunez G . NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 2014; 41:898–908.
Dambuza IM, Brown GD . C-type lectins in immunity: recent developments. Curr Opin Immunol 2015; 32:21–27.
Dempsey A, Bowie AG . Innate immune recognition of DNA: a recent history. Virology 2015; 479-480:146–152.
Cai X, Chiu YH, Chen ZJ . The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 2014; 54:289–296.
Sun SC, Ley SC . New insights into NF-κB regulation and function. Trends Immunol 2008; 29:469–478.
Meylan E, Burns K, Hofmann K, et al. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κ B activation. Nat Immunol 2004; 5:503–507.
Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA . Rip1 mediates the Trif-dependent toll-like receptor 3- and 4-induced NF-κB activation but does not contribute to interferon regulatory factor 3 activation. J Biol Chem 2005; 280:36560–36566.
Moynagh PN . The roles of Pellino E3 ubiquitin ligases in immunity. Nat Rev Immunol 2014; 14:122–131.
Kawai T, Takahashi K, Sato S, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol 2005; 6:981–988.
Meylan E, Curran J, Hofmann K, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005; 437:1167–1172.
Seth RB, Sun L, Ea CK, Chen ZJ . Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell 2005; 122:669–682.
Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB . VISA is an adapter protein required for virus-triggered IFN-β signaling. Mol Cell 2005; 19:727–740.
Häcker H, Redecke V, Blagoev B, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006; 439:204–207.
Oganesyan G, Saha SK, Guo B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 2006; 439:208–211.
Zeng W, Xu M, Liu S, Sun L, Chen ZJ . Key role of Ubc5 and lysine-63 polyubiquitination in viral activation of IRF3. Mol Cell 2009; 36:315–325.
Liu S, Chen J, Cai X, et al. MAVS recruits multiple ubiquitin E3 ligases to activate antiviral signaling cascades. eLife 2013; 2:e00785.
Ishikawa H, Barber GN . STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008; 455:674–678.
Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC . MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol 2008; 28:5014–5026.
Zhong B, Yang Y, Li S, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008; 29:538–550.
Burdette DL, Monroe KM, Sotelo-Troha K, et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011; 478:515–518.
Jin L, Hill KK, Filak H, et al. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol 2011; 187:2595–2601.
Huang YH, Liu XY, Du XX, Jiang ZF, Su XD . The structural basis for the sensing and binding of cyclic di-GMP by STING. Nat Struct Mol Biol 2012; 19:728–730.
Shang G, Zhu D, Li N, et al. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat Struct Mol Biol 2012; 19:725–727.
Shu C, Yi G, Watts T, Kao CC, Li P . Structure of STING bound to cyclic di-GMP reveals the mechanism of cyclic dinucleotide recognition by the immune system. Nat Struct Mol Biol 2012; 19:722–724.
Sun L, Wu J, Du F, Chen X, Chen ZJ . Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339:786–791.
Wu J, Sun L, Chen X, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013; 339:826–830.
Zhang X, Shi H, Wu J, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 2013; 51:226–235.
Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V . TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 2014; 192:5993–5997.
Gray EE, Treuting PM, Woodward JJ, Stetson DB . Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi-Goutieres syndrome. J Immunol 2015; 195:1939–1943.
Sun SC . Non-canonical NF-κB signaling pathway. Cell Res 2011; 21:71–85.
Sun SC . The noncanonical NF-κB pathway. Immunol Rev 2012; 246:125–140.
Jin J, Hu H, Li HS, et al. Noncanonical NF-κB pathway controls the production of type I interferons in antiviral innate immunity. Immunity 2014; 40:342–354.
Deng L, Wang C, Spencer E, et al. Activation of the IκB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell 2000; 103:351–361.
Wang C, Deng L, Hong M, Akkaraju GR, Inoue JI, Chen ZJ . TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 2001; 412:346–351.
Chen ZJ . Ubiquitination in signaling to and activation of IKK. Immunol Rev 2012; 246:95–106.
Tokunaga F . Linear ubiquitination-mediated NF-κB regulation and its related disorders. J Biochem 2013; 154:313–323.
Harhaj EW, Dixit VM . Deubiquitinases in the regulation of NF-κB signaling. Cell Res 2011; 21:22–39.
Kanayama A, Seth RB, Sun L, et al. TAB2 and TAB3 activate the NF-κB pathway through binding to polyubiquitin chains. Mol Cell 2004; 15:535–548.
Kensche T, Tokunaga F, Ikeda F, Goto E, Iwai K, Dikic I . Analysis of nuclear factor-κB (NF-κB) essential modulator (NEMO) binding to linear and lysine-linked ubiquitin chains and its role in the activation of NF-κB. J Biol Chem 2012; 287:23626–23634.
Rahighi S, Ikeda F, Kawasaki M, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-κB activation. Cell 2009; 136:1098–1109.
Hubeau M, Ngadjeua F, Puel A, et al. New mechanism of X-linked anhidrotic ectodermal dysplasia with immunodeficiency: impairment of ubiquitin binding despite normal folding of NEMO protein. Blood 2011; 118:926–935.
Tokunaga F, Sakata S, Saeki Y, et al. Involvement of linear polyubiquitylation of NEMO in NF-κB activation. Nat Cell Biol 2009; 11:123–132.
Iwai K, Fujita H, Sasaki Y . Linear ubiquitin chains: NF-κB signalling, cell death and beyond. Nat Rev Mol Cell Biol 2014; 15:503–508.
Shimizu Y, Taraborrelli L, Walczak H . Linear ubiquitination in immunity. Immunol Rev 2015; 266:190–207.
Shim JH, Xiao C, Paschal AE, et al. TAK1, but not TAB1 or TAB2, plays an essential role in multiple signaling pathways in vivo. Genes Dev 2005; 19:2668–2681.
Thiefes A, Wolter S, Mushinski JF, et al. Simultaneous blockade of NFκB, JNK, and p38 MAPK by a kinase-inactive mutant of the protein kinase TAK1 sensitizes cells to apoptosis and affects a distinct spectrum of tumor necrosis factor [corrected] target genes. J Biol Chem 2005; 280:27728–27741.
Waterfield M, Wei J, Reiley W, Zhang MY, Sun SC . IKKb Is an essential component of the Tpl2 signaling pathway. Mol Cell Biol 2004; 24:6040–6048.
Waterfield M, Zhang M, Norman LP, Sun SC . NF-κB1/p105 regulates lipopolysaccharide-stimulated MAP kinase signaling by governing the stability and function of the Tpl2 kinase. Mol Cell 2003; 11:685–694.
Beinke S, Deka J, Lang V, et al. NF-κB1 p105 negatively regulates TPL-2 MEK kinase activity. Mol Cell Biol 2003; 23:4739–4752.
Beinke S, Robinson MJ, Hugunin M, Ley SC . Lipopolysaccharide activation of the TPL-2/MEK/extracellular signal-regulated kinase mitogen-activated protein kinase cascade is regulated by IκB kinase-induced proteolysis of NF-κB1 p105. Mol Cell Biol 2004; 24:9658–9667.
Arthur JS, Ley SC . Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol 2013; 13:679–692.
Fukushima T, Matsuzawa S, Kress CL, et al. Ubiquitin-conjugating enzyme Ubc13 is a critical component of TNF receptor-associated factor (TRAF)-mediated inflammatory responses. Proc Natl Acad Sci USA 2007; 104:6371–6376.
Yamamoto M, Okamoto T, Takeda K, et al. Key function for the Ubc13 E2 ubiquitin-conjugating enzyme in immune receptor signaling. Nat Immunol 2006; 7:962–970.
Fitzgerald KA, McWhirter SM, Faia KL, et al. IKKε and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol 2003; 4:491–496.
Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J . Triggering the interferon antiviral response through an IKK-related pathway. Science 2003; 300:1148–1151.
McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T . IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proc Natl Acad Sci USA 2004; 101:233–238.
Hemmi H, Takeuchi O, Sato S, et al. The roles of two IκB kinase-related kinases in lipopolysaccharide and double stranded RNA signaling and viral infection. J Exp Med 2004; 199:1641–1650.
Perry AK, Chow EK, Goodnough JB, Yeh WC, Cheng G . Differential requirement for TANK-binding kinase-1 in type I interferon responses to toll-like receptor activation and viral infection. J Exp Med 2004; 199:1651–1658.
McNab F, Mayer-Barber K, Sher A, Wack A, O'Garra A . Type I interferons in infectious disease. Nat Rev Immunol 2015; 15:87–103.
Wang P, Zhu S, Yang L, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science 2015; 350:826–830.
Friedman CS, O'Donnell MA, Legarda-Addison D, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep 2008; 9:930–936.
Zhang M, Wu X, Lee AJ, et al. Regulation of IκB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem 2008; 283:18621–18626.
Parvatiyar K, Barber GN, Harhaj EW . TAX1BP1 and A20 inhibit antiviral signaling by targeting TBK1-IKKi kinases. J Biol Chem 2010; 285:14999–15009.
Li S, Wang L, Berman M, Kong YY, Dorf ME . Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 2011; 35:426–440.
Ye JS, Kim N, Lee KJ, Nam YR, Lee U, Joo CH . Lysine 63-linked TANK-binding kinase 1 ubiquitination by mindbomb E3 ubiquitin protein ligase 2 is mediated by the mitochondrial antiviral signaling protein. J Virol 2014; 88:12765–12776.
Wang C, Chen T, Zhang J, et al. The E3 ubiquitin ligase Nrdp1 'preferentially' promotes TLR-mediated production of type I interferon. Nat Immunol 2009; 10:744–752.
Zhou AY, Shen RR, Kim E, et al. IKKε-mediated tumorigenesis requires K63-linked polyubiquitination by a cIAP1/cIAP2/TRAF2 E3 ubiquitin ligase complex. Cell Rep 2013; 3:724–733.
Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M . Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol 2010; 11:70–75.
Tu D, Zhu Z, Zhou AY, et al. Structure and ubiquitination-dependent activation of TANK-binding kinase 1. Cell Rep 2013; 3:747–758.
Wang L, Li S, Dorf ME . NEMO binds ubiquitinated TANK-binding kinase 1 (TBK1) to regulate innate immune responses to RNA viruses. PLoS One 2012; 7:e43756.
Zhao T, Yang L, Sun Q, et al. The NEMO adaptor bridges the nuclear factor-κB and interferon regulatory factor signaling pathways. Nat Immunol 2007; 8:592–600.
Cui J, Li Y, Zhu L, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol 2012; 13:387–395.
An T, Li S, Pan W, et al. DYRK2 Negatively Regulates Type I Interferon Induction by Promoting TBK1 Degradation via Ser527 Phosphorylation. PLoS Pathog 2015; 11:e1005179.
Zhang M, Wang L, Zhao X, et al. TRAF-interacting protein (TRIP) negatively regulates IFN-β production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J Exp Med 2012; 209:1703–1711.
Zheng Q, Hou J, Zhou Y, Yang Y, Xie B, Cao X . Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res 2015; 25:1121–1136.
Liu D, Sheng C, Gao S, et al. SOCS3 Drives Proteasomal Degradation of TBK1 and Negatively Regulates Antiviral Innate Immunity. Mol Cell Biol 2015; 35:2400–2413.
Yoshimura A, Naka T, Kubo M . SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007; 7:454–465.
Piessevaux J, Lavens D, Peelman F, Tavernier J . The many faces of the SOCS box. Cytokine Growth Factor Rev 2008; 19:371–381.
Zhang JG, Farley A, Nicholson SE, et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA 1999; 96:2071–2076.
Ori D, Kato H, Sanjo H, et al. Essential roles of K63-linked polyubiquitin-binding proteins TAB2 and TAB3 in B cell activation via MAPKs. J Immunol 2013; 190:4037–4045.
Lamothe B, Besse A, Campos AD, Webster WK, Wu H, Darnay BG . Site-specific Lys-63-linked tumor necrosis factor receptor-associated factor 6 auto-ubiquitination is a critical determinant of IκB kinase activation. J Biol Chem 2007; 282:4102–4112.
Walsh MC, Kim GK, Maurizio PL, Molnar EE, Choi Y . TRAF6 autoubiquitination-independent activation of the NFκB and MAPK pathways in response to IL-1 and RANKL. PLoS One 2008; 3:e4064.
Conze DB, Wu CJ, Thomas JA, Landstrom A, Ashwell JD . Lys63-linked polyubiquitination of IRAK-1 is required for interleukin-1 receptor- and toll-like receptor-mediated NF-κB activation. Mol Cell Biol 2008; 28:3538–3547.
Lee BC, Miyata M, Lim JH, Li JD . Deubiquitinase CYLD acts as a negative regulator for bacterium NTHi-induced inflammation by suppressing K63-linked ubiquitination of MyD88. Proc Natl Acad Sci USA 2016; 113:E165–171.
Xia ZP, Sun L, Chen X, et al. Direct activation of protein kinases by unanchored polyubiquitin chains. Nature 2009; 461:114–119.
Gohda J, Matsumura T, Inoue J . Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathway in TLR signaling. J Immunol 2004; 173:2913–2917.
Chang M, Jin W, Sun SC . Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat Immunol 2009; 10:1089–1095.
Jin W, Chang M, Sun SC . Peli: a family of signal-responsive E3 ubiquitin ligases mediating TLR signaling and T-cell tolerance. Cell Mol Immunol 2012; 9:113–122.
Goh ET, Arthur JS, Cheung PC, Akira S, Toth R, Cohen P . Identification of the protein kinases that activate the E3 ubiquitin ligase Pellino 1 in the innate immune system. Biochem J 2012; 441:339–346.
Xiao Y, Jin J, Chang M, et al. Peli1 promotes microglia-mediated CNS inflammation by regulating Traf3 degradation. Nat Med 2013; 19:595–602.
Hacker H, Tseng PH, Karin M . Expanding TRAF function: TRAF3 as a tri-faced immune regulator. Nat Rev Immunol 2011; 11:457–468.
Yang XD, Sun SC . Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol Rev 2015; 266:56–71.
Jin J, Xiao Y, Hu H, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nat Commun 2015; 6:5930.
Lalani AI, Moore CR, Luo C, et al. Myeloid cell TRAF3 regulates immune responses and inhibits inflammation and tumor development in mice. J Immunol 2015; 194:334–348.
Lacey DC, Achuthan A, Fleetwood AJ, et al. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J Immunol 2012; 188:5752–5765.
Gack MU, Shin YC, Joo CH, et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007; 446:916–920.
Gao D, Yang YK, Wang RP, et al. REUL is a novel E3 ubiquitin ligase and stimulator of retinoic-acid-inducible gene-I. PLoS One 2009; 4:e5760.
Oshiumi H, Matsumoto M, Hatakeyama S, Seya T . Riplet/RNF135, a RING finger protein, ubiquitinates RIG-I to promote interferon-β induction during the early phase of viral infection. J Biol Chem 2009; 284:807–817.
Oshiumi H, Miyashita M, Inoue N, Okabe M, Matsumoto M, Seya T . The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010; 8:496–509.
Gack MU, Albrecht RA, Urano T, et al. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009; 5:439–449.
Rajsbaum R, Albrecht RA, Wang MK, et al. Species-specific inhibition of RIG-I ubiquitination and IFN induction by the influenza A virus NS1 protein. PLoS Pathog 2012; 8:e1003059.
Oshiumi H, Miyashita M, Matsumoto M, Seya T . A distinct role of Riplet-mediated K63-Linked polyubiquitination of the RIG-I repressor domain in human antiviral innate immune responses. PLoS Pathog 2013; 9:e1003533.
Lin R, Yang L, Nakhaei P, et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem 2006; 281:2095–2103.
Zhong H, Wang D, Fang L, et al. Ubiquitin-specific proteases 25 negatively regulates virus-induced type I interferon signaling. PLoS One 2013; 8:e80976.
Fan Y, Mao R, Yu Y, et al. USP21 negatively regulates antiviral response by acting as a RIG-I deubiquitinase. J Exp Med 2014; 211:313–328.
Cui J, Song Y, Li Y, et al. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res 2014; 24:400–416.
Pauli EK, Chan YK, Davis ME, et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci Signal 2014; 7:ra3.
Zhang H, Wang D, Zhong H, et al. Ubiquitin-specific protease 15 negatively regulates virus-induced type I interferon signaling via catalytically-dependent and -independent mechanisms. Sci Rep 2015; 5:11220.
Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K . Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci USA 2007; 104:7500–7505.
Inn KS, Gack MU, Tokunaga F, et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell 2011; 41:354–365.
Inohara N, Koseki T, del Peso L, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-κB. J Biol Chem 1999; 274:14560–14567.
Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G . Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-κB. J Biol Chem 2001; 276:4812–4818.
Abbott DW, Wilkins A, Asara JM, Cantley LC . The Crohn's disease protein, NOD2, requires RIP2 in order to induce ubiquitinylation of a novel site on NEMO. Curr Biol 2004; 14:2217–2227.
Abbott DW, Yang Y, Hutti JE, Madhavarapu S, Kelliher MA, Cantley LC . Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol 2007; 27:6012–6025.
Yang Y, Yin C, Pandey A, Abbott D, Sassetti C, Kelliher MA . NOD2 pathway activation by MDP or Mycobacterium tuberculosis infection involves the stable polyubiquitination of Rip2. J Biol Chem 2007; 282:36223–36229.
Hasegawa M, Fujimoto Y, Lucas PC, et al. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-κB activation. EMBO J 2008; 27:373–383.
Bertrand MJ, Doiron K, Labbe K, Korneluk RG, Barker PA, Saleh M . Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 2009; 30:789–801.
Damgaard RB, Nachbur U, Yabal M, et al. The ubiquitin ligase XIAP recruits LUBAC for NOD2 signaling in inflammation and innate immunity. Mol Cell 2012; 46:746–758.
Krieg A, Correa RG, Garrison JB, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci USA 2009; 106:14524–14529.
Yang S, Wang B, Humphries F, et al. Pellino3 ubiquitinates RIP2 and mediates Nod2-induced signaling and protective effects in colitis. Nat Immunol 2013; 14:927–936.
Tao M, Scacheri PC, Marinis JM, Harhaj EW, Matesic LE, Abbott DW . ITCH K63-ubiquitinates the NOD2 binding protein, RIP2, to influence inflammatory signaling pathways. Curr Biol 2009; 19:1255–1263.
Williams KL, Lich JD, Duncan JA, et al. The CATERPILLER protein monarch-1 is an antagonist of toll-like receptor-, tumor necrosis factor α-, and Mycobacterium tuberculosis-induced pro-inflammatory signals. J Biol Chem 2005; 280:39914–39924.
Lich JD, Williams KL, Moore CB, et al. Monarch-1 suppresses non-canonical NF-κB activation and p52-dependent chemokine expression in monocytes. J Immunol 2007; 178:1256–1260.
Allen IC, Wilson JE, Schneider M, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 2012; 36:742–754.
Zaki MH, Vogel P, Malireddi RK, et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011; 20:649–660.
Lukens JR, Gurung P, Shaw PJ, et al. The NLRP12 sensor negatively regulates autoinflammatory disease by modulating interleukin-4 production in T cells. Immunity 2015; 42:654–664.
Moore CB, Bergstralh DT, Duncan JA, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature 2008; 451:573–577.
Allen IC, Moore CB, Schneider M, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity 2011; 34:854–865.
Xia X, Cui J, Wang HY, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity 2011; 34:843–853.
Rebsamen M, Vazquez J, Tardivel A, Guarda G, Curran J, Tschopp J . NLRX1/NOD5 deficiency does not affect MAVS signalling. Cell Death Differ 2011; 18:1387.
Soares F, Tattoli I, Wortzman ME, Arnoult D, Philpott DJ, Girardin SE . NLRX1 does not inhibit MAVS-dependent antiviral signalling. Innate immunity 2013; 19:438–448.
Benko S, Magalhaes JG, Philpott DJ, Girardin SE . NLRC5 limits the activation of inflammatory pathways. J Immunol 2010; 185:1681–1691.
Cui J, Zhu L, Xia X, et al. NLRC5 negatively regulates the NF-κB and type I interferon signaling pathways. Cell 2010; 141:483–496.
Schneider M, Zimmermann AG, Roberts RA, et al. The innate immune sensor NLRC3 attenuates Toll-like receptor signaling via modification of the signaling adaptor TRAF6 and transcription factor NF-κB. Nat Immunol 2012; 13:823–831.
Zhang L, Mo J, Swanson KV, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 2014; 40:329–341.
Kumar H, Pandey S, Zou J, et al. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J Immunol 2011; 186:994–1000.
Yao Y, Wang Y, Chen F, et al. NLRC5 regulates MHC class I antigen presentation in host defense against intracellular pathogens. Cell Res 2012; 22:836–847.
Meng Q, Cai C, Sun T, et al. Reversible ubiquitination shapes NLRC5 function and modulates NF-κB activation switch. J Cell Biol 2015; 211:1025–1040.
Tong Y, Cui J, Li Q, Zou J, Wang HY, Wang RF . Enhanced TLR-induced NF-κB signaling and type I interferon responses in NLRC5 deficient mice. Cell Res 2012; 22:822–835.
Tsuchida T, Zou J, Saitoh T, et al. The ubiquitin ligase TRIM56 regulates innate immune responses to intracellular double-stranded DNA. Immunity 2010; 33:765–776.
Zhang J, Hu MM, Wang YY, Shu HB . TRIM32 protein modulates type I interferon induction and cellular antiviral response by targeting MITA/STING protein for K63-linked ubiquitination. J Biol Chem 2012; 287:28646–28655.
Fang S, Ferrone M, Yang C, Jensen JP, Tiwari S, Weissman AM . The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc Natl Acad Sci USA 2001; 98:14422–14427.
Wang Q, Liu X, Cui Y, et al. The E3 ubiquitin ligase AMFR and INSIG1 bridge the activation of TBK1 kinase by modifying the adaptor STING. Immunity 2014; 41:919–933.
Zhong B, Zhang L, Lei C, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity 2009; 30:397–407.
Qin Y, Zhou MT, Hu MM, et al. RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog 2014; 10:e1004358.
Wang Y, Lian Q, Yang B, et al. TRIM30α is a negative-feedback regulator of the intracellular DNA and DNA virus-triggered response by targeting STING. PLoS Pathog 2015; 11:e1005012.
Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu YJ . The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat Immunol 2013; 14:172–178.
Steinman RM, Hawiger D, Nussenzweig MC . Tolerogenic dendritic cells. Annu Rev Immunol 2003; 21:685–711.
Mayer CT, Berod L, Sparwasser T . Layers of dendritic cell-mediated T cell tolerance, their regulation and the prevention of autoimmunity. Front Immunol 2012; 3:183.
Hammer GE, Ma A . Molecular control of steady-state dendritic cell maturation and immune homeostasis. Annu Rev Immunol 2013; 31:743–791.
Walsh KP, Mills KH . Dendritic cells and other innate determinants of T helper cell polarisation. Trends Immunol 2013; 34:521–530.
Baravalle G, Park H, McSweeney M, et al. Ubiquitination of CD86 is a key mechanism in regulating antigen presentation by dendritic cells. J Immunol 2011; 187:2966–2973.
Shin JS, Ebersold M, Pypaert M, Delamarre L, Hartley A, Mellman I . Surface expression of MHC class II in dendritic cells is controlled by regulated ubiquitination. Nature 2006; 444:115–118.
van Niel G, Wubbolts R, Ten Broeke T, et al. Dendritic cells regulate exposure of MHC class II at their plasma membrane by oligoubiquitination. Immunity 2006; 25:885–894.
Corcoran K, Jabbour M, Bhagwandin C, Deymier MJ, Theisen DL, Lybarger L . Ubiquitin-mediated regulation of CD86 protein expression by the ubiquitin ligase membrane-associated RING-CH-1 (MARCH1). J Biol Chem 2011; 286:37168–37180.
De Gassart A, Camosseto V, Thibodeau J, et al. MHC class II stabilization at the surface of human dendritic cells is the result of maturation-dependent MARCH I down-regulation. Proc Natl Acad Sci USA 2008; 105:3491–3496.
Tze LE, Horikawa K, Domaschenz H, et al. CD83 increases MHC II and CD86 on dendritic cells by opposing IL-10-driven MARCH1-mediated ubiquitination and degradation. J Exp Med 2011; 208:149–165.
Ishikawa R, Kajikawa M, Ishido S . Loss of MHC II ubiquitination inhibits the activation and differentiation of CD4 T cells. Int Immunol 2014; 26:283–289.
Yang H, Qiu Q, Gao B, Kong S, Lin Z, Fang D . Hrd1-mediated BLIMP-1 ubiquitination promotes dendritic cell MHCII expression for CD4 T cell priming during inflammation. J Exp Med 2014; 211:2467–2479.
Temmerman ST, Ma CA, Borges L, et al. Impaired dendritic-cell function in ectodermal dysplasia with immune deficiency is linked to defective NEMO ubiquitination. Blood 2006; 108:2324–2331.
Liu J, Han C, Xie B, et al. Rhbdd3 controls autoimmunity by suppressing the production of IL-6 by dendritic cells via K27-linked ubiquitination of the regulator NEMO. Nat Immunol 2014; 15:612–622.
Jin J, Xie X, Xiao Y, et al. Epigenetic regulation of the expression of Il12 and Il23 and autoimmune inflammation by the deubiquitinase Trabid. Nat Immunol 2016; 17:259–268.
Hilliard BA, Mason N, Xu L, et al. Critical roles of c-Rel in autoimmune inflammation and helper T cell differentiation. J Clin Invest 2002; 110:843–850.
Sanjabi S, Hoffmann A, Liou HC, Baltimore D, Smale ST . Selective requirement for c-Rel during IL-12 P40 gene induction in macrophages. Proc Natl Acad Sci USA 2000; 97:12705–12710.
Smith-Garvin JE, Koretzky GA, Jordan MS . T cell activation. Annu Rev Immunol 2009; 27:591–619.
Zhu J, Yamane H, Paul WE . Differentiation of effector CD4 T cell populations. Annu Rev Immunol 2010; 28:445–489.
Heissmeyer V, Macian F, Im SH, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol 2004; 5:255–265.
Huang F, Gu H . Negative regulation of lymphocyte development and function by the Cbl family of proteins. Immunol Rev 2008; 224:229–238.
Hsu TS, Hsiao HW, Wu PJ, Liu WH, Lai MZ . Deltex1 promotes protein kinase Cθ degradation and sustains Casitas B-lineage lymphoma expression. J Immunol 2014; 193:1672–1680.
Park Y, Jin HS, Aki D, Lee J, Liu YC . The ubiquitin system in immune regulation. Adv Immunol 2014; 124:17–66.
Balagopalan L, Barr VA, Sommers CL, et al. c-Cbl-mediated regulation of LAT-nucleated signaling complexes. Mol Cell Biol 2007; 27:8622–8636.
Balagopalan L, Ashwell BA, Bernot KM, et al. Enhanced T-cell signaling in cells bearing linker for activation of T-cell (LAT) molecules resistant to ubiquitylation. Proc Natl Acad Sci USA 2011; 108:2885–2890.
Rodriguez-Pena AB, Gomez-Rodriguez J, Kortum RL, et al. Enhanced T-cell activation and differentiation in lymphocytes from transgenic mice expressing ubiquitination-resistant 2KR LAT molecules. Gene Ther 2015; 22:781–792.
Jeon MS, Atfield A, Venuprasad K, et al. Essential role of the E3 ubiquitin ligase Cbl-b in T cell anergy induction. Immunity 2004; 21:167–177.
Huang H, Jeon MS, Liao L, et al. K33-linked polyubiquitination of T cell receptor-ζ regulates proteolysis-independent T cell signaling. Immunity 2010; 33:60–70.
Lupher ML Jr, Reedquist KA, Miyake S, Langdon WY, Band H . A novel phosphotyrosine-binding domain in the N-terminal transforming region of Cbl interacts directly and selectively with ZAP-70 in T cells. J Biol Chem 1996; 271:24063–24068.
Rao N, Lupher ML Jr, Ota S, Reedquist KA, Druker BJ, Band H . The linker phosphorylation site Tyr292 mediates the negative regulatory effect of Cbl on ZAP-70 in T cells. J Immunol 2000; 164:4616–4626.
Carpino N, Chen Y, Nassar N, Oh HW . The Sts proteins target tyrosine phosphorylated, ubiquitinated proteins within TCR signaling pathways. Mol Immunol 2009; 46:3224–3231.
Carpino N, Turner S, Mekala D, et al. Regulation of ZAP-70 activation and TCR signaling by two related proteins, Sts-1 and Sts-2. Immunity 2004; 20:37–46.
Yang M, Chen T, Li X, et al. K33-linked polyubiquitination of Zap70 by Nrdp1 controls CD8+ T cell activation. Nat Immunol 2015; 16:1253–1262.
Hu H, Wang H, Xiao Y, et al. Otud7b facilitates T cell activation and inflammatory responses by regulating Zap70 ubiquitination. J Exp Med 2016; 213:399–414.
Evans PC, Taylor ER, Coadwell J, Heyninck K, Beyaert R, Kilshaw PJ . Isolation and characterization of two novel A20-like proteins. Biochem J 2001; 357:617–623.
Hu H, Brittain GC, Chang JH, et al. OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature 2013; 494:371–374.
Naik E, Webster JD, DeVoss J, Liu J, Suriben R, Dixit VM . Regulation of proximal T cell receptor signaling and tolerance induction by deubiquitinase Usp9X. J Exp Med 2014; 211:1947–1955.
Blonska M, Lin X . CARMA1-mediated NF-κB and JNK activation in lymphocytes. Immunol Rev 2009; 228:199–211.
Nakaya M, Xiao Y, Zhou X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014; 40:692–705.
Hamilton KS, Phong B, Corey C, et al. T cell receptor-dependent activation of mTOR signaling in T cells is mediated by Carma1 and MALT1, but not Bcl10. Sci Signal 2014; 7:ra55.
Shi JH, Sun SC . TCR signaling to NF-κB and mTORC1: Expanding roles of the CARMA1 complex. Mol Immunol 2015; 68:546–557.
Sun L, Deng L, Ea CK, Xia ZP, Chen ZJ . The TRAF6 ubiquitin ligase and TAK1 kinase mediate IKK activation by BCL10 and MALT1 in T lymphocytes. Mol Cell 2004; 14:289–301.
Zhou H, Wertz I, O'Rourke K, et al. Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 2004; 427:167–171.
Yamamoto M, Sato S, Saitoh T, et al. Cutting edge: pivotal function of Ubc13 in thymocyte TCR signaling. J Immunol 2006; 177:7520–7524.
King CG, Kobayashi T, Cejas PJ, et al. TRAF6 is a T cell-intrinsic negative regulator required for the maintenance of immune homeostasis. Nat Immunol 2006; 12:1088–1092.
Stempin CC, Chi L, Giraldo-Vela JP, High AA, Hacker H, Redecke V . The E3 ubiquitin ligase mind bomb-2 (MIB2) protein controls B-cell CLL/lymphoma 10 (BCL10)-dependent NF-κB activation. J Biol Chem 2011; 286:37147–37157.
Song R, Kim YW, Koo BK, et al. Mind bomb 1 in the lymphopoietic niches is essential for T and marginal zone B cell development. J Exp Med 2008; 205:2525–2536.
Reiley WW, Zhang M, Jin W, et al. Regulation of T cell development by the deubiquitinating enzyme CYLD. Nat Immunol 2006; 7:411–417.
Reiley WW, Jin W, Lee AJ, et al. Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J Exp Med 2007; 204:1475–1485.
Lee AJ, Zhou X, Chang M, et al. Regulation of natural killer T-cell development by deubiquitinase CYLD. EMBO J 2010; 29:1600–1612.
Tsagaratou A, Trompouki E, Grammenoudi S, Kontoyiannis DL, Mosialos G . Thymocyte-specific truncation of the deubiquitinating domain of CYLD impairs positive selection in a NF-κB essential modulator-dependent manner. J Immunol 2010; 185:2032–2043.
Zhang J, Stirling B, Temmerman ST, et al. Impaired regulation of NF-κB and increased susceptibility to colitis-associated tumorigenesis in CYLD-deficient mice. J Clin Invest 2006; 116:3042–3049.
Liu X, Li H, Zhong B, et al. USP18 inhibits NF-κB and NFAT activation during Th17 differentiation by deubiquitinating the TAK1-TAB1 complex. J Exp Med 2013; 210:1575–1590.
Coornaert B, Baens M, Heyninck K, et al. T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-κB inhibitor A20. Nat Immunol 2008; 9:263–271.
Duwel M, Welteke V, Oeckinghaus A, et al. A20 negatively regulates T cell receptor signaling to NF-κB by cleaving Malt1 ubiquitin chains. J Immunol 2009; 182:7718–7728.
Giordano M, Roncagalli R, Bourdely P, et al. The tumor necrosis factor α-induced protein 3 (TNFAIP3, A20) imposes a brake on antitumor activity of CD8 T cells. Proc Natl Acad Sci USA 2014; 111:11115–11120.
Park Y, Jin HS, Liu YC . Regulation of T cell function by the ubiquitin-specific protease USP9X via modulating the Carma1-Bcl10-Malt1 complex. Proc Natl Acad Sci USA 2013; 110:9433–9438.
Chang M, Jin W, Chang JH, et al. The ubiquitin ligase Peli1 negatively regulates T cell activation and prevents autoimmunity. Nat Immunol 2011; 12:1002–1009.
Köntgen F, Grumont RJ, Strasser A, et al. Mice lacking the c-rel proto-oncogene exhibit defects in lymphocyte proliferation, humoural immunity, and interleukin-2 expression. Genes Dev 1995; 9:1965–1977.
Liou HC, Jin Z, Tumang J, Andjelic S, Smith KA, Liou ML . c-Rel is crucial for lymphocyte proliferation but dispensable for T cell effector function. Int Immunol 1999; 11:361–371.
Chen G, Hardy K, Bunting K, Daley S, Ma L, Shannon MF . Regulation of the IL-21 gene by the NF-κB transcription factor c-Rel. J Immunol 2010; 185:2350–2359.
Liou HC, Smith KA . The roles of c-rel and interleukin-2 in tolerance: a molecular explanation of self-nonself discrimination. Immunol Cell Biol 2011; 89:27–32.
Maggirwar SB, Harhaj EW, Sun SC . Regulation of the interleukin-2 CD28 responsive element by NF-ATp and various NF-kB/Rel transcription factors. Mol Cell Biol 1997; 17:2605–2614.
Zhou XY, Yashiro-Ohtani Y, Nakahira M, et al. Molecular mechanisms underlying differential contribution of CD28 versus non-CD28 costimulatory molecules to IL-2 promoter activation. J Immunol 2002; 168:3847–3854.
Ordureau A, Smith H, Windheim M, et al. The IRAK-catalysed activation of the E3 ligase function of Pellino isoforms induces the Lys63-linked polyubiquitination of IRAK1. Biochem J 2008; 409:43–52.
Zou Q, Jin J, Hu H, et al. USP15 stabilizes MDM2 to mediate cancer-cell survival and inhibit antitumor T cell responses. Nat Immunol 2014; 15:562–570.
Li Q, Lozano G . Molecular pathways: targeting Mdm2 and Mdm4 in cancer therapy. Clin Cancer Res 2013; 19:34–41.
Zou Q, Jin J, Xiao Y, et al. T cell intrinsic USP15 deficiency promotes excessive IFN-γ production and an immunosuppressive tumor microenvironment in MCA-induced fibrosarcoma. Cell Rep 2015; 13:2470–2479.
Goodnow CC, Sprent J, Fazekas de St Groth B, Vinuesa CG . Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 2005; 435:590–597.
Anderson MS, Venanzi ES, Klein L, et al. Projection of an immunological self-shadow within the thymus by the aire protein. Sciece 2002; 298:1395–1401.
Abramson J, Goldfarb Y . AIRE: From promiscuous molecular partnerships to promiscuous gene expression. Eur J Immunol 2016; 46:22–33.
Kumar PG, Laloraya M, Wang CY, et al. The autoimmune regulator (AIRE) is a DNA-binding protein. J Biol Chem 2001; 276:41357–41364.
Uchida D, Hatakeyama S, Matsushima A, et al. AIRE functions as an E3 ubiquitin ligase. J Exp Med 2004; 199:167–172.
Bottomley MJ, Stier G, Pennacchini D, et al. NMR structure of the first PHD finger of autoimmune regulator protein (AIRE1). Insights into autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) disease. J Biol Chem 2005; 280:11505–11512.
Akiyama T, Maeda S, Yamane S, et al. Dependence of self-tolerance on TRAF6-directed development of thymic stroma. Science 2005; 308:248–251.
Bonito AJ, Aloman C, Fiel MI, et al. Medullary thymic epithelial cell depletion leads to autoimmune hepatitis. J Clin Invest 2013; 123:3510–3524.
Zhu M, Fu Y . The complicated role of NF-κB in T-cell selection. Cell Mol Immunol 2010; 7:89–93.
Tzachanis D, Lafuente EM, Li L, Boussiotis VA . Intrinsic and extrinsic regulation of T lymphocyte quiescence. Leuk Lymphoma 2004; 45:1959–1967.
Schwartz RH . T cell anergy. Annu Rev Immunol 2003; 21:305–334.
Mueller DL . E3 ubiquitin ligases as T cell anergy factors. Nat Immunol 2004; 5:883–890.
Valdor R, Macian F . Induction and stability of the anergic phenotype in T cells. Semin Immunol 2013; 25:313–320.
King CG, Buckler JL, Kobayashi T, et al. Cutting edge: requirement for TRAF6 in the induction of T cell anergy. J Immunol 2008; 180:34–38.
Heissmeyer V, Vogel KU . Molecular control of Tfh-cell differentiation by Roquin family proteins. Immunol Rev 2013; 253:273–289.
Vinuesa CG, Tangye SG, Moser B, Mackay CR . Follicular B helper T cells in antibody responses and autoimmunity. Nat Rev Immunol 2005; 5:853–865.
Yu D, Tan AH, Hu X, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature 2007; 450:299–303.
Linterman MA, Rigby RJ, Wong RK, et al. Follicular helper T cells are required for systemic autoimmunity. J Exp Med 2009; 206:561–576.
Bertossi A, Aichinger M, Sansonetti P, et al. Loss of Roquin induces early death and immune deregulation but not autoimmunity. J Exp Med 2011; 208:1749–1756.
Pratama A, Ramiscal RR, Silva DG, et al. Roquin-2 shares functions with its paralog Roquin-1 in the repression of mRNAs controlling T follicular helper cells and systemic inflammation. Immunity 2013; 38:669–680.
Vogel KU, Edelmann SL, Jeltsch KM, et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity 2013; 38:655–668.
Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G . Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 2013; 153:869–881.
Maruyama T, Araki T, Kawarazaki Y, et al. Roquin-2 promotes ubiquitin-mediated degradation of ASK1 to regulate stress responses. Sci Signal 2014; 7:ra8.
Zhang Q, Fan L, Hou F, Dong A, Wang YX, Tong Y . New insights into the RNA-binding and E3 ubiquitin ligase activities of Roquins. Sci Rep 2015; 5:15660.
Sakaguchi S, Yamaguchi T, Nomura T, Ono M . Regulatory T cells and immune tolerance. Cell 2008; 133:775–787.
Proietto AI, van Dommelen S, Zhou P, et al. Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc Natl Acad Sci USA 2008; 105:19869–19874.
Oh J, Wu N, Baravalle G, et al. MARCH1-mediated MHCII ubiquitination promotes dendritic cell selection of natural regulatory T cells. J Exp Med 2013; 210:1069–1077.
Xie P, Kraus ZJ, Stunz LL, Liu Y, Bishop GA . TNF receptor-associated factor 3 is required for T cell-mediated immunity and TCR/CD28 signaling. J Immunol 2011; 186:143–155.
Yi Z, Lin WW, Stunz LL, Bishop GA . The adaptor TRAF3 restrains the lineage determination of thymic regulatory T cells by modulating signaling via the receptor for IL-2. Nat Immunol 2014; 15:866–874.
Burchill MA, Yang J, Vang KB, et al. Linked T cell receptor and cytokine signaling govern the development of the regulatory T cell repertoire. Immunity 2008; 28:112–121.
Lio CW, Hsieh CS . A two-step process for thymic regulatory T cell development. Immunity 2008; 28:100–111.
Yi Z, Stunz LL, Lin WW, Bishop GA . TRAF3 regulates homeostasis of CD8+ central memory T cells. PLoS One 2014; 9:e102120.
Chang JH, Hu H, Jin J, et al. TRAF3 regulates the effector function of regulatory T cells and humoral immune responses. J Exp Med 2014; 211:137–151.
Harada Y, Harada Y, Elly C, et al. Transcription factors Foxo3a and Foxo1 couple the E3 ligase Cbl-b to the induction of Foxp3 expression in induced regulatory T cells. J Exp Med 2010; 207:1381–1391.
Wohlfert EA, Gorelik L, Mittler R, Flavell RA, Clark RB . Cutting edge: deficiency in the E3 ubiquitin ligase Cbl-b results in a multifunctional defect in T cell TGF-β sensitivity in vitro and in vivo. J Immunol 2006; 176:1316–1320.
Venuprasad K, Huang H, Harada Y, et al. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol 2008; 9:245–253.
Chang JH, Xiao Y, Hu H, et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat Immunol 2012; 13:481–490.
van Loosdregt J, Coffer PJ . Post-translational modification networks regulating FOXP3 function. Trends Immunol 2014; 35:368–378.
Chen Z, Barbi J, Bu S, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity 2013; 39:272–285.
van Loosdregt J, Fleskens V, Fu J, et al. Stabilization of the transcription factor Foxp3 by the deubiquitinase USP7 increases Treg-cell-suppressive capacity. Immunity 2013; 39:259–271.
Zhao Y, Guo H, Qiao G, Zucker M, Langdon WY, Zhang J . E3 ubiquitin ligase Cbl-b regulates thymic-derived CD4+CD25+ regulatory T cell development by targeting Foxp3 for ubiquitination. J Immunol 2015; 194:1639–1645.
MacKenzie DA, Schartner J, Lin J, et al. GRAIL is up-regulated in CD4+ CD25+ T regulatory cells and is sufficient for conversion of T cells to a regulatory phenotype. J Biol Chem 2007; 282:9696–9702.
Schartner JM, Singh AM, Dahlberg PE, Nettenstrom L, Seroogy CM . Recurrent superantigen exposure in vivo leads to highly suppressive CD4+CD25+ and CD4+CD25− T cells with anergic and suppressive genetic signatures. Clin Exp Immunol 2009; 155:348–356.
Nurieva RI, Zheng S, Jin W, et al. The E3 ubiquitin ligase GRAIL regulates T cell tolerance and regulatory T cell function by mediating T cell receptor-CD3 degradation. Immunity 2010; 32:670–680.
Lee JH, Elly C, Park Y, Liu YC . E3 Ubiquitin ligase VHL regulates hypoxia-inducible factor-1α to maintain regulatory T cell stability and suppressive capacity. Immunity 2015; 42:1062–1074.
Acknowledgements
Work performed in our laboratory is supported by grants from the US National Institutes of Health (AI057555, AI064639, GM84459, and AI104519), the Cancer Prevention and Research Institute of Texas (RP150235 and RP140244), and the Thousand Young Talents Plan of China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Hu, H., Sun, SC. Ubiquitin signaling in immune responses. Cell Res 26, 457–483 (2016). https://doi.org/10.1038/cr.2016.40
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2016.40
