
Abstract
The constitutional t(11;22)(q23;q11) is a well-known recurrent non-Robertsonian translocation in humans. Although translocations generally occur in a random fashion, the break points of t(11;22)s are concentrated within several hundred base pairs on 11q23 and 22q11. These regions are characterized by palindromic AT-rich repeats (PATRRs), which appear to be responsible for the genomic instability. Translocation-specific PCR detects de novo t(11;22)s in sperm from healthy males at a frequency of 1/104–105, but never in lymphoblasts, fibroblasts or other human somatic cell lines. This suggests that the generation of t(11;22) rearrangement is linked to gametogenesis, although female germ cells have not been tested. Here, we have studied eight cases of de novo t(11;22) to determine the parental origin of the translocation using the polymorphisms on the relevant PATRRs. All of the eight translocations were found to be of paternal origin. This result implicates a possible novel mechanism of sperm-specific generation of palindrome-mediated chromosomal translocations.
Similar content being viewed by others


Introduction
The constitutional t(11;22)(q23;q11) is a well-known recurrent non-Robertsonian translocation in humans. Carriers of this balanced translocation usually have no clinical symptoms and are often identified after chromosomal malsegregation resulting in the birth of offspring with an unbalanced form of the translocation. The unbalanced individuals have a distinctive phenotype called Emanuel syndrome [MIM 609029], that consists of severe mental retardation, preauricular tag or sinus, ear anomaly, cleft or high-arched palate, micrognathia, heart defects and genital abnormalities in the male.1, 2 Most constitutional translocations generally occur in a random fashion. However, the break points of t(11;22)s are concentrated within several hundred base pairs on 11q23 and 22q11, which are characterized by palindromic AT-rich repeats (PATRRs).3, 4, 5, 6, 7 Recently, molecular cloning of various translocation break points has shown similar palindromic sequences on other chromosomes, such as 17q11, 4q35.1, 1p21.2 and 8q24.1.8, 9, 10, 11 It has been acknowledged that palindrome-mediated genomic instability contributes to a diversity of genomic rearrangements including not only translocations, but also deletions and gene amplification.12, 13 Palindromic sequences have the potential to form stem-loop (hairpin or cruciform) structures by intrastrand base pairing in the single-stranded DNA. Thus, we proposed that such unusual DNA secondary structures give rise to genomic instability that leads to the recurrent translocation.14, 15
As all of the t(11;22) break points are located in a small region within both the PATRR11 (∼450 bp) and the PATRR22 (∼595 bp), we established a PCR detection system for translocation-derivative chromosomes.16 Using this PCR system, de novo t(11;22)s are detected in sperm from healthy males at a frequency of 1/104–105, but never in lymphoblasts, fibroblasts or other human somatic cell lines17, 18, suggesting that the generation of a t(11;22) is linked to meiosis. However, female germ cells have not been tested because the number of human oocytes that can be examined is limited. To investigate whether the translocation is meiosis-specific or male germ cell-specific, we attempted to determine the parental origin of de novo t(11;22) cases.
Materials and methods
Samples
Samples were collected from cases with de novo constitutional t(11;22)s and their parents after obtaining written informed consent. Genomic DNA was extracted from peripheral blood samples, chorionic villus or amniotic fluid samples and saliva samples. The study was approved by the Ethical Review Board for Human Genome Studies at Fujita Health University.
DNA analysis
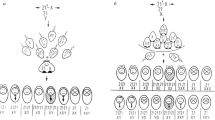
Translocation-specific PCR was performed as previously described.16 The primer sets were designed on both sides of the PATRR on chromosomes 11 and 22 (Figure 1a). We directly sequenced the PCR products on the ABI Prism 3100 Genetic Analyzer.

The PCR system to determine the allele type of the translocated or normal chromosomes. (a) Translocation-specific PCR. Black arrows indicate each proximal and distal unit of the PATRR11, whereas light gray arrows depict the PATRR22. The PCR primers are indicated as arrowheads. The PATRRs on the normal chromosomes were amplified with a PATRR11-specific primer set (black and white arrowheads) or that for PATRR22 (vertical-striped and gray arrowheads). Translocations can be detected using one primer flanking the PATRR11 (black or white arrowheads) and one primer flanking the PATRR22 (vertical-striped or gray arrowheads). Stippled boxes indicate the AT-rich regions flanking the PATRR22. To determine the size of the AT-rich region, we performed nested PCR. The first PCR amplified the PATRR22 or der(22) breakpoint region using specific-primer sets, respectively. Next, internal primers (stippled arrowheads) were used to amplify the AT-rich regions flanking the PATRR22. (b) Determination of the parental origin in family 6. PCR was performed to amplify the AT-rich region adjacent to the PATRR22. The figure shows the ideograms for the normal chromosome 22, and the der(22). M: 1 kb Plus size marker, lane 1: the der(22) of the translocation carrier, lane 2: chromosome 22 of the carrier, lane 3: father, lane 4: mother, lane 5: heterozygote for the 355 bp- and 121 bp-alleles 6: heterozygote for the 179 bp- and 121 bp-alleles, 7: homozygote for the 121 bp-alleles, 8: no DNA. The additional band observed in lane 5 originates from the heteroduplex. Allele type of the der(22) of the carrier corresponds to the paternal type, and that of the PATRR22 of the normal homolog to the maternal type.
Amplification of PATRR11 and PATRR22 was also performed as previously described.7, 16 The PCR primers were as follows; 199F 5′-GAGAGTAAAGAAATAGTTCAGAAAGG-3′ and 190R 5′- CCACAGACTCATTCATGGAACC-3′ for PATRR11, -469F 5′-CCATATGCAGTTATAAATATGTTTCATGATTAT-3′ and +440R 5′-ACAAGTAAACAGGTTTTCAAAGCT-3′ for PATRR22. The PCR condition was to heat at 94°C 2 min, 30 cycles of 10 s of 98°C and 10 min of 60°C. Each PCR product containing the PATRR was cloned into the plasmid vector, pT7Blue (Novagen, Madison, WI, USA), and then sequenced. We used the SURE strain (Stratagene, La Jolla, CA, USA) to maintain the highly unstable PATRR insert.
We determined the genotype of the size polymorphism at the AT-rich region adjacent to the PATRR22 by PCR.7 The PCR primer sets were as follows; 22a 5′-CCCAGTGTGAATTGGGATTCAG-3′ and Rev-22c 5′-CAGTAGTATGGATCCGTTGGAGG-3′. Three different length PCR (355 bp-, 179 bp- and 121 bp-allele) products can be identified. We determined the allele type of the der(22) and PATRR22 separately by means of a nested PCR (Figure 1a).
Results
We studied eight carriers of de novo t(11;22)s and their parents. All t(11;22) carriers were diagnosed by standard karyotyping followed by fluorescence in situ hybridization with appropriate probes. The testing of their parents provided the information that the translocation was de novo in origin, which was also confirmed by translocation-specific PCR.
To examine parental origin of the translocated chromosomes, the PATRRs and flanking regions on chromosome 11, 22, der(11) and der(22) were amplified by PCR and the nucleotide sequences were determined16 (Figure 1a). The highly polymorphic nature of the PATRRs allows one to distinguish between alleles of the translocated and normal chromosomes in the t(11;22) carrier and permitted us to analyze the segregation of the rearranged parental homologs.
In family 1, the allele type of both the proximal part of the PATRR11 on the der(11) and the distal part of the PATRR11 on the der(22) of the translocation carrier corresponded with either of the normal PATRR11s of the father but with neither of the mother (Figure 2 and Supplementary Table 1). On the other hand, the PATRR11 on the normal chromosome 11 of the carrier corresponded with that of the mother but not with that of the father. These results clearly indicate that the translocated chromosomes are of paternal origin. Similarly, the PATRR11 on the der(11) and the der(22) of the translocation carrier in family 2 corresponded with one of the PATRR11s of the father but with neither of the mother. The PATRR11 on the normal chromosome 11 of the carrier was entirely deleted. The mother, but not the father, was found to carry this deleted allele, indicating that the translocated chromosomes are also of paternal origin.

Summary of results for the eight families. Only informative polymorphisms to determine the parental origins were shown. Bold and italic indicate the definitive polymorphisms of paternal and maternal origin, respectively. Numbers at the left indicate the nucleotide positions from the starting point of the PATRR11. In Families 3, 6 and 7, samples from translocation carriers were obtained prenatally from chorionic villi or amniotic fluid. Family 1 was reported previously in Macville et al.28, whereas families 4 and 8 were reported previously in Kurahashi et al.16
In family 3, the PATRR11 on the der(11) and the der(22) of the translocation carrier corresponded with one of the PATRR11s of the father but with neither of the mother. Although the PATRR11 on the normal chromosome 11 of the carrier was not determined, the results indicate that the translocated chromosomes are also of paternal origin. In family 4, although the allele type of the proximal part of the PATRR11 was not informative among the family members, the distal part of the PATRR11 on the der(22) of the carrier corresponded with the one in the father, not with the mother. In family 5, we did not obtain a genomic DNA sample from the father. However, the proximal sequence of the PATRR11 on the der(11) corresponded with neither of the maternal PATRR11s, one of which corresponded with the carrier’s normal PATRR11 on chromosome 11, suggesting that the translocation was also of paternal origin.
Two cases (family 6 and 8), whose allele types of the PATRR11 were not informative, were analyzed for the segregation of the PATRR22, instead. Because of the difficulties in sequencing the PATRR22, we used the size polymorphism of the AT-rich region adjacent to the proximal side of the PATRR227 (Figure 1a). In family 6, the proband carried the 121 bp-allele on the der(22) and the 179 bp-allele on the normal chromosome 22, respectively. The father was found to be a homozygote for the 121 bp-allele, whereas the mother was homozygous for the 179 bp-allele, indicating that the translocated chromosome of the carrier was derived from the father (Figure 1b, 2 and Supplementary Table 2). Similarly, the proband in family 7 was found to carry the 179 bp-allele on the der(22) that was derived from the father. In addition, the information of the PATRR11 also supported the paternal origin in this case (Supplementary Table 1).
In family 8, because even the genotyping of the flanking polymorphism was not informative, we finally performed sequence analysis of the PATRR22. The PATRR22 on the der(11) of the translocation carrier corresponded with one of the PATRR22s of the father but with neither of the mother. The PATRR22 on the normal chromosome 22 of the carrier harbored a small deletion at the center of the PATRR22. The mother, but not father, was also found to carry the deleted PATRR22, indicating that the translocated chromosomes are also of paternal origin (Figure 2 and Supplementary Table 3).
All eight of the translocations were found to be of paternal origin. The difference in paternal origin was significant (Fisher's exact test, P=0.00016). The break points of the eight families were within both the PATRRs but not identical to each other. Thus, we concluded that the paternal origin of de novo t(11;22) in these families can be applicable to other cases of t(11;22).
Discussion
It is well documented that de novo numerical chromosomal abnormalities are preferentially of maternal origin, whereas structural abnormalities arise predominately in paternal germ cells.19, 20 One exception to this generalization is the Robertsonian translocation. Although it is one of the best known non-random constitutional translocations, ∼95% of de novo Robertsonian translocation cases originate in maternal germ cells.21 An oogenesis-specific mechanism has been assumed for Robertsonian translocations. The centromeres of acrocentric chromosomes are brought in close proximity during formation of the nucleolar organizer regions during the prolonged prophase of female meiosis I. Thus, homologous recombination between centromeric repetitive regions may be involved in the generation of Robertsonian translocations. The t(11;22)(q23;q11) is another example of a recurrent constitutional translocation in humans. Despite the similarity between the two PATRRs with regard to AT-richness, no substantial homology is observed between the PATRR11 and the PATRR22.7 Thus, homologous recombination does not appear to be responsible for the t(11;22). In this study, all eight of the de novo t(11;22)s were found to be exclusively of paternal origin. Our result implicates a novel mechanism for sperm-specific generation of the t(11;22).
One way to account for this observation is the difference in the number of cell divisions between spermatogenesis and oogenesis. The number of cell divisions in oogenesis is relatively constant with approximately 22 divisions throughout the female lifetime. In contrast, spermatogenesis reaches roughly 150 divisions by the age of 20 years, with a linear increase of about 23 cell divisions per year. There is a positive relationship between paternal age and de novo gene mutations by replication errors.22 The genomic instability of palindromic DNA appears to be primarily mediated by stalling of the DNA replication fork at a region that forms a hairpin DNA structure.23 The secondary structure-mediated replication errors during the numerous cell divisions in pre-meiotic spermatogenic cells might contribute to male-specific formation of de novo t(11;22)s. Indeed, a positive relationship between paternal age and de novo occurrence of non-recurrent translocation has been reported,24 although such a relationship has not been observed for t(11;22).25
On the other hand, experimental data suggest that a replication-independent DNA cruciform can potentially be a target for a structure-specific nuclease and contribute to palindrome-mediated translocations in humans.26 Cruciform extrusion is energetically prohibited in genomic DNA under standard conditions, because sufficient negative supercoiling is a prerequisite for the formation of a cruciform DNA structure.27 However, successive transitions of chromatin components from histones to protamines might cause dynamic changes in DNA superhelicity. DNA dissociation from histones may involve accumulation of free negative supercoiling that potentially induces cruciform extrusion at the PATRR leading to male-specific formation of de novo t(11;22)s. Thus, conformational changes of the DNA during chromatin remodeling in post-meiotic stages of spermatogenesis might also account for the general fact that structural chromosomal aberrations predominantly originate in paternal gametogenesis. This work implicates a possible novel mechanism of sperm-specific generation of palindrome-mediated chromosomal translocations in humans.
References
Fraccaro M, Lindsten J, Ford CE et al: The 11q;22q translocation: a European collaborative analysis of 43 cases. Hum Genet 1980; 56: 21–51.
Zackai EH, Emanuel BS : Site-specific reciprocal translocation, t(11;22) (q23;q11), in several unrelated families with 3:1 meiotic disjunction. Am J Med Genet 1980; 7: 507–521.
Kurahashi H, Shaikh TH, Hu P et al: Regions of genomic instability on 22q11 and 11q23 as the etiology for the recurrent constitutional t(11;22). Hum Mol Genet 2000; 9: 1665–1670.
Kurahashi H, Emanuel BS : Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet 2001; 10: 2605–2617.
Edelmann L, Spiteri E, Koren K et al: AT-rich palindromes mediate the constitutional t(11;22) translocation. Am J Hum Genet 2001; 68: 1–13.
Tapia-Paez I, Kost-Alimova M, Hu P et al: The position of t(11;22)(q23;q11) constitutional translocation breakpoint is conserved among its carriers. Hum Genet 2001; 109: 167–177.
Kurahashi H, Inagaki H, Hosoba E et al: Molecular cloning of a translocation breakpoint hotspot in 22q11. Genome Res 2007; 17: 461–469.
Nimmakayalu MA, Gotter AL, Shaikh TH et al: A novel sequence-based approach to localize translocation breakpoints identifies the molecular basis of a t(4;22). Hum Mol Genet 2003; 12: 2817–2825.
Kurahashi H, Shaikh T, Takata M et al: The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am J Hum Genet 2003; 72: 733–738.
Gotter AL, Shaikh TH, Budarf ML et al: A palindrome-mediated mechanism distinguishes translocations involving LCR-B of chromosome 22q11.2. Hum Mol Genet 2004; 13: 103–115.
Gotter AL, Nimmakayalu MA, Jalali GR et al: A palindrome-driven complex rearrangement of 22q11.2 and 8q24.1 elucidated using novel technologies. Genome Res 2007; 17: 470–481.
Kuroda-Kawaguchi T, Skaletsky H, Brown L et al: The AZFc region of the Y chromosome features massive palindromes and uniform recurrent deletions in infertile men. Nat Genet 2001; 29: 279–286.
Tanaka H, Bergstrom D, Yao M et al: Widespread and nonrandom distribution of DNA palindromes in cancer cells provides a structural platform for subsequent gene amplification. Nat Genet 2005; 37: 320–327.
Kurahashi H, Inagaki H, Yamada K et al: Cruciform DNA structure underlies the etiology for palindrome-mediated human chromosomal translocations. J Biol Chem 2004; 279: 35377–35383.
Kogo H, Inagaki H, Ohye T et al: Cruciform extrusion propensity of human translocation-mediating palindromic AT-rich repeats. Nucleic Acids Res 2007; 35: 1198–1208.
Kurahashi H, Shaikh TH, Zackai EH et al: Tightly clustered 11q23 and 22q11 breakpoints permit PCR-based detection of the recurrent constitutional t(11;22). Am J Hum Genet 2000; 67: 763–768.
Kurahashi H, Emanuel BS : Unexpectedly high rate of de novo constitutional t(11;22) translocations in sperm from normal males. Nat Genet 2001; 29: 139–140.
Kato T, Inagaki H, Yamada K et al: Genetic variation affects de novo translocation frequency. Science 2006; 311: 971.
Buwe A, Guttenbach M, Schmid M : Effect of paternal age on the frequency of cytogenetic abnormalities in human spermatozoa. Cytogenet Genome Res 2005; 111: 213–228.
Martin RH : Meiotic errors in human oogenesis and spermatogenesis. Reprod Biomed Online 2008; 16: 523–531.
Page SL, Shaffer LG : Nonhomologous Robertsonian translocations form predominantly during female meiosis. Nat Genet 1997; 15: 231–232.
Crow JF : The origins, patterns and implications of human spontaneous mutation. Nat Rev Genet 2000; 1: 40–47.
Leach DR : Long DNA palindromes, cruciform structures, genetic instability and secondary structure repair. Bioessays 1994; 16: 893–900.
Thomas NS, Morris JK, Baptista J et al: De novo apparently balanced translocations in man are predominantly paternal in origin and associated with a significant increase in paternal age. J Med Genet 2009.
Kato T, Yamada K, Inagaki H et al: Age has no effect on de novo constitutional t(11;22) translocation frequency in sperm. Fertil Steril 2007; 88: 1446–1448.
Inagaki H, Ohye T, Kogo H et al: Chromosomal instability mediated by non-B DNA: cruciform conformation and not DNA sequence is responsible for recurrent translocation in humans. Genome Res 2009; 19: 191–198.
Sinden RR : Cruciform structures in DNA: DNA structure and function. San Diego 1994; pp: 134–177.
Macville MV, Loneus WH, Marcus-Soekarman D et al: XX male with sex reversal and a de novo 11;22 translocation. Am J Med Genet A 2006; 140: 1973–1977.
Acknowledgements
This work was supported by grant-in-aids for Scientific Research, from the Ministry of Health, Labour and Welfare and the Ministry of Education, Culture, Sports, Science and Technology of Japan (19590322 and 21590346 to TO, 21390101 to HK), and CA39926 from the National Institutes of Health, USA as well as funds from the Charles EH Upham chair of Pediatrics (BSE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim
Supplementary Information accompanies the paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Ohye, T., Inagaki, H., Kogo, H. et al. Paternal origin of the de novo constitutional t(11;22)(q23;q11). Eur J Hum Genet 18, 783–787 (2010). https://doi.org/10.1038/ejhg.2010.20
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2010.20
Keywords
This article is cited by
-
Constitutional t(8;22)(q24;q11.2) that mimics the variant Burkitt-type translocation in Philadelphia chromosome-positive chronic myeloid leukemia
International Journal of Hematology (2017)
-
Breakpoint analysis of the recurrent constitutional t(8;22)(q24.13;q11.21) translocation
Molecular Cytogenetics (2014)
-
DNA secondary structure is influenced by genetic variation and alters susceptibility to de novo translocation
Molecular Cytogenetics (2011)
-
Advanced age increases chromosome structural abnormalities in human spermatozoa
European Journal of Human Genetics (2011)
