
Abstract
Congenital adrenal hyperplasia (CAH) is one of the most common autosomal recessive inherited endocrine disease. Steroid 11β-hydroxylase deficiency (11β-OHD) is the second most common form of CAH. The aim of the study was to study the functional consequences of three novel and one previously described CYP11B1 gene mutations (p.(Arg143Trp), p.(Ala306Val), p.(Glu310Lys) and p.(Arg332Gln)) detected in patients suffering from classical and non-classical 11β-OHD. Functional analyses were performed by using a HEK293 cell in vitro expression system comparing wild type (WT) with mutant 11β-hydroxylase activity. Mutant proteins were examined in silico to study their effect on the three-dimensional structure of the protein. Two mutations (p.(Ala306Val) and p.(Glu310Lys)) detected in patients with classical 11β-OHD showed a nearly complete loss of 11β-hydroxylase activity. The mutations p.(Arg143Trp) and p.(Arg332Gln) detected in patients with non-classical 11β-OHD showed a partial functional impairment with approximately 8% and 6% of WT activity, respectively. Functional mutation analysis allows the classification of novel CYP11B1 mutations as causes of classical and non-classical 11β-OHD. The detection of patients with non-classical phenotypes underscores the importance to screen patients with a phenotype comparable to non-classical 21-hydroxylase deficiency for mutations in the CYP11B1 gene in case of a negative analysis of the CYP21A2 gene. As CYP11B1 mutations are most often individual for a family, the in vitro analysis of novel mutations is essential for clinical and genetic counselling.
Similar content being viewed by others


Introduction
Congenital adrenal hyperplasia (CAH) is a family of inborn errors of steroidogenesis, each characterized by a specific enzyme deficiency that impairs cortisol production. The majority of cases are caused by 21-hydroxylase deficiency (21-OHD) followed by 11β-hydroxylase deficiency (11β-OHD; OMIM _202010) in about 5–8% of cases.1, 2 The 11β-hydroxylase converts 11-deoxycortisol (11-S) to cortisol (F) and 11-deoxycorticosterone (DOC) to corticosterone (B), and its deficit causes an accumulation of steroids with mineralocorticoid activity and an overproduction of androgens. Therefore, the classical form of 11β-OHD, with an estimated frequency of 1:200 000 live births, is characterized by genital ambiguity in affected girls and postnatal hyperandrogenism in both sexes resulting in precocious pseudopuberty with rapid somatic growth, bone age acceleration and hypertension in about two-thirds of patients.3, 4 The milder non-classical forms are very similar to the 21-OHD non-classical forms characterized by mild virilization and precocious pseudopuberty.5, 6 In these mild forms, whose frequency is unknown, hypertension does not occur often.2, 5, 6
The 11β-OHD is caused by mutations of the 11β-hydroxylase gene (CYP11B1), located on chromosome 8q21, approximately 40 kb apart from the highly homologous aldosterone synthase gene (CYP11B2), which has 95% of sequence homology.7 To date, more than 60 missense and nonsense mutations are reported in the Human Gene Mutation Database (www.hgmd.cf.ac.uk) localized all over the gene, with a minor presence in exons 1 and 9.
The aim of the study was to describe and characterize five individuals from four unrelated families: two patients with classical 11β-OHD and three patients with non-classical 11β-OHD. All of them have compound heterozygous mutations, and three novel missense mutations were detected. In vitro studies were conducted for the novel mutations and for one previously reported but not functionally studied mutation. The pathogenicity of the detected mutations was proven and discussed with the help of a three-dimensional molecular model.
Patients
Patient 1
Patient 1 of northern Italian origin was born in 1970 at 36 weeks of gestation (weight 2600 g) after an uneventful pregnancy with normal female external genitalia. She came to attention in Switzerland at the age of 6 years due to facial acne without pubarche or axillary hair growth and advanced bone age (11 years). Hormonal diagnosis of non-classical 11β-OHD was conducted but the results are not available to us. A treatment with hydrocortisone was initiated. She got married and came to our observation at the age of 39 years for genetic counselling because of failure to conceive. After the genetic diagnosis of 11β-OHD was assured, treatment with prednisone was adjusted and the patient spontaneously conceived a healthy female child.
Patient 2
The male patient, now 13.4 years old, is the first son of a family with two affected siblings who was born at term (weight 3150 g, length 52 cm) after an uneventful pregnancy to parents originating from northern Italy. The patient presented with premature pubarche, tall stature and accelerated growth velocity at 5.3 years. The bone age was advanced with 11.5 years at a chronological age of 6.3 years. The test results of the adrenocorticotropic hormone (ACTH) stimulation are shown in Table 1. Non-classical 21-OHD was suspected, and the treatment was started with hydrocortisone and a GnRH analogue to prevent an incipient precocious puberty (testicular volume 3–4 ml). The CYP21A2 gene analysis showed a heterozygous intron 2 splice-site mutation on the paternal allele. Blood pressure values tended to undulate in the high normal range (119/78 mm Hg). The elevated concentration of 11-S and DOC during treatment has later prompted the investigation of the CYP11B1 gene (Table 1). Near-final height is 170.6 cm (SD −0.93) with relevant obesity of 104.4 kg and a BMI of 35.9 (SD +2.93).
Patient 3
The female patient is the sister of patient 2. She was born at term after an uneventful pregnancy and is 10.9 years old now. She presented with premature pubarche at the age of 5.5 years without genital virilization. Results of steroid analysis with ID-LC-MS/MS after ACTH administration are depicted in Table 1. Treatment with hydrocortisone was started at the age of 6 years due to progressive bone age advancement (7 years). Blood pressure was in the normal range for age at that time. To date, she has not menstruated and has a pubertal evaluation of Ph5 and B2-3. She does not carry the intron-2-splicing mutation on the CYP21A2 gene.
Patient 4
The female patient (46, XX), now 22.5 years old, was born at term (weight 2950 g, length 51 cm) to healthy non-consanguineous parents of northern Italian origin. She showed ambiguous, markedly pigmented, external genitalia (Prader genital stage II-III). Ultrasound and X-ray examinations showed normal Mullerian structures and normal adrenals. Hormonal analysis revealed normal 17-OH-progesterone (17-OHP) and F levels for age, with elevated plasma testosterone (T) and ACTH levels (Table 1). There was neither clinical evidence for hypertension nor obvious salt loss. Although CAH different from 21-OHD was suspected, treatment with cortisone acetate (43.0 mg/m2/cortisol Eq) and fludrocortisone (0.025 mg/day) was started as assays for 11-S, DOC or 17-OH-pregnenolone were not available. A subsequent assay performed after 2 days of treatment withdrawal showed high levels of 11-S, which confirmed 11β-OHD. Menarche occurred at 13 years of age; she was regularly menstruating when compliant to therapy and attained an adult height of 165 cm (target height 162 cm).
Patient 5
The female patient (46, XX), now 13.3 years old, was born at term to healthy parents of southern Italian origin with a birth weight of 2980 g and a birth length of 48.5 cm. Neonatal screening for 21-OHD CAH was negative. At the age of 3 months, genital ambiguity was noted (Prader stage III) and she was first admitted to our endocrine clinic. Hormone analysis revealed an elevation in the levels of 17-OHP, Δ4-androstenedione (D4A), T and ACTH (Table 1). Electrolytes and blood pressure were in the normal ranges. Ultrasound investigations showed normal adrenals and ovaries. Genitography depicted a short urogenital sinus with the insertion of the vagina into the sinus at 1.5 cm from the perineal level. Treatment with hydrocortisone was initiated and, although the parents affirmed good compliance, the hormonal control was bad and GnRH analogue treatment had to be started at 6.0 years of age because of clear signs of secondary central precocious puberty. Additional hormone analyses at that time revealed elevated levels of 11-S (Table 1). At present, the girl is regularly menstruating (menarche at 12 years) and reached a final height of 149 cm (SD −2.3; target height 154.5, SD −1.4).
Materials and methods
Molecular genetic analysis of the CYP11B1 gene
The molecular analysis of the CYP11B1 gene was performed on DNA extracted from peripheral blood leukocytes following a standard procedure. The coding sequence of the CYP11B1 gene including exon–intron boundaries was amplified in three partially overlapping fragments using the following specific primers: AF (from nt −80 to nt −59)–AR (from nt 2263 to nt 2281) for exons 1 and 2; BF (from nt 2115 to nt 2136)–BR (from nt 3787 to nt 3811) for exons 3, 4 and 5; CF (from nt 3699 to nt 3723)–CR (from nt 5605 to nt 5624) for exons 6, 7, 8 and 9. The position of nucleotides used for primers is numbered considering +1 as the A of the ATG translation initiation codon of the reference sequences GenBank NG_007954.1. Direct sequencing was carried out using an automated CEQ 8000 Sequencer (Beckman Coulter Inc., Brea, CA, USA) with internal additional primers. Sequence variants were designated according to Human Genome Variation Society recommendations (www.hgvs.org/rec.html) using the reference sequences GenBank NM_000497.3 for cDNA. All detected variants are accessible through the NCBI dbSNP-polymorphism repository ClinVar (www.ncbi.nlm.nih.gov/clinvar).
Site-directed mutagenesis
A pcDNA3.1 expression vector construct with the CYP11B1-cDNA as an insert (pcDNA3.1-CYP11B1 construct) was used as previously described.8 The mutagenesis was performed using the QuikChange XL Site-Directed Mutagenesis Kit according to the manufacturer’s protocol (Stratagene, Amsterdam, The Netherlands). The introduction of the mutations was confirmed by sequencing the complete cDNA. To eliminate any undesired mutation in the vector backbone, the mutated CYP11B1-cDNA fragments were re-cloned into the XbaI/BamHI: the site of a newly restricted pcDNA expression vector.
Transient transfection
Approximately 2.5 × 105 HEK293 cells were plated 24 h before transfection. Cells were transiently transfected with 2.5 μg of each pcDNA3.1-CYP11B1 mutant construct with the addition of 0.5 μg of adrenodoxin (pECE-ADX), 0.5 μg of Adx reductase (pECE-ADR) expression vectors (kindly provided by Professor WL Miller, Department of Pediatrics, University of California, San Francisco, CA, USA) and 0.5 μg of pRK-TK (Promega, Mannheim, Germany) coding for renilla luciferase using the X-fect plasmid transfection reagent according to the manufacturer’s protocol (Clontech Laboratories Inc., Mountain View, CA, USA). HEK293 cells were incubated with the transfection reagents for 4 h at 37 °C with 5% CO2 in DMEM supplemented with 1% antibiotics and 10% fetal calf serum, followed by further 48 h incubation in fresh full DMEM media. The renilla luciferase activity was measured using a standard renilla luciferase assay (Promega).
Enzymatic activity assay
The kinetic constants of CYP11B1 in intact HEK293 cells were determined 48 h after transfection. The cells were incubated for 270 min at 37 °C with 1 ml of full DMEM medium containing 0.1, 0.25, 0.5, 1, 2 and 4 μmol/l DOC and 11-deoxycortisol (11-S) with 10 mM NADPH (Sigma-Aldrich, Hamburg, Germany) as an electron donor for the ADR enzyme.
Hormone measurement
Hormone blood levels in patients were measured with the following: (a) radio immunoassay (RIA, Immunotech, Beckman Coulter Inc.) for 17-OHP with intra- and inter-assay coefficients of variation (c.v.) <10%; (b) immune-chemiluminometric assay (ICMA, Siemens) for D4A with Immunolite 2001, c.v. <8%, and for ACTH with Immunolite 2002, c.v. <10%; and (c) electro-chemiluminescence immunoassay (ECLIA, Roche, Modular) for T and F, with c.v. <7% and <5%, respectively.
Serum concentration of the steroid hormones 11-S and DOC was determined by a validated isotopic dilution–liquid chromatography–tandem mass spectrometry (ID-LC-MS/MS) method. The assay description and validation are reported in detail elsewhere.9 Accuracy ranged between 83.7 and 106.2%; intra- and inter-assay c.v. ranged between 2 and 11% for all analytes. Sensitivity in serum matrix was 0.08 ng/ml for 11-S and DOC. Cortisol, 17OHP, D4A and T in patient 3 were also determined by this method with sensitivity values of 0.24 ng/ml, 0.08 ng/ml, 0.04 ng/ml and 0.02 ng/ml, respectively.
The steroid concentrations of DOC, B, 11-S and F in vitro were simultaneously determined in the cell culture supernatant by using an UPLC-MS/MS method as previously described.10 Accuracy ranged between 94.3 and 98.6%; intra- and inter-assay c.v. ranged between 3.3 and 8.7% for all analytes. The limits of quantification were 0.05 ng/ml for DOC, 0.06 ng/ml for B and 11-S and 0.8 ng/ml for F.
Western blot
Transfected cells were lysed in the lysis buffer (Promega). Total protein content was determined using the Bradford method. Western blot analysis using a rabbit antihuman CYP11B1 antibody (kindly provided by Dr H Takemori, Department of Molecular Physiological Chemistry, Osaka University Medical School, Osaka, Japan) was performed on the basis of a standard protocol to ensure the expression and translation of CYP11B1 wild-type and mutant proteins.8
Statistical analyses
The enzymatic activity assay was performed in at least three independent triplicate experiments, and data are presented as means±SD. Kinetic parameters were established by nonlinear regression, using the Michaelis–Menten equation to determine the Michaelis–Menten constants (Km) and Vmax. Catalytic efficiency (cat. eff.) was defined as the ratio Vmax/Km expressed as the percentage of wild-type activity. The 11β-hydroxylase activity of the mutants was expressed as a percentage of substrate conversion in picomole (pmol) per milligram (mg) of total protein per minute (min), defining CYP11B1 wild-type activity as 100% after correction for total protein and renilla luciferase activity. Enzyme kinetic parameters and enzymatic activity were calculated using the GraphPad Prism software version 5.0 (GraphPad, Inc., San Diego, CA, USA). Height and BMI standard deviation scores (SDS) were calculated according to the Italian Reference Standards.11
Molecular modelling
The detailed generation of the human CYP11B1 three-dimensional structure model using the X-ray structure of the mammalian cytochrome CYP2C5 (Pdb code 1DT6) as a template has been described previously.8, 12 The structural representation was generated with Ribbons software.13
Results
Molecular analysis of the CYP11B1 gene
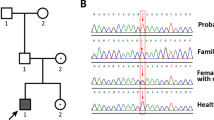
The CYP11B1 gene analysis in patient 1 revealed that she was heterozygous for the mutation c.917C>T (p.(Ala306Val)) and for the novel mutation c.427C>T (p.(Arg143Trp)) (Figure 1). The mutation p.(Ala306Val) has been previously described but was not studied in vitro.14 Parents were not available for genetic analysis, and thus the status of compound heterozygosity is inferred only from the phenotype. Gene analyses in patients 2 and 3 revealed that they were compound heterozygous for the paternal mutation c.896T>C (p.(Leu299Pro))8 and for a novel mutation c.995G>A (p.(Arg332Gln)) inherited from the mother (Figure 1). Gene analysis in patient 4 revealed that she was compound heterozygous for a novel c.928G>A mutation inherited from the father, resulting in the amino-acid change p.(Glu310Lys) in the CYP11B1 protein and for the previously described mutation c.896T>C leading to p.(Leu299Pro)8 inherited from the mother (Figure 1). The analysis in patient 5 revealed that she was compound heterozygous for the paternally inherited mutation c.1066C>T (p.(Gln356Ter)) and for the mutation c.917C>T (p.(Ala306Val)) inherited from the mother (Figure 1). Both mutations have been previously described.14, 15

Molecular genetic analysis of the CYP11B1 gene. (a) Schematic localization of the mutations. Upper: known mutations. Down: novel mutations. *previously reported without functional characterization. (b) Pedigrees of the five patients from four unrelated families with electropherograms of the mutations.
Functional 11 β -hydroxylase in vitro assays
The enzymatic activity of the novel CYP11B1 mutations was functionally analysed using transiently transfected HEK293 cells measuring the conversion of DOC to B and 11-S to F, respectively (Figure 2). The p.(Glu310Lys) mutation showed no residual activity in both pathways. The p.(Ala306Val) mutation showed residual activity of 1.1±0.1% of wild-type activity for the conversion of DOC and 1.2±0.2% of wild-type activity for the conversion of 11-S. Therefore, the p.(Glu310Lys) and p.(Ala306Val) mutations have to be considered as severe mutations causing classical 11β-hydroxylase deficiency. The functional analysis of p.(Arg143Trp) showed 8.1±0.8% of wild-type activity for B production and 8.4±0.8% of wild-type activity for F production. The p.(Arg332Gln) mutation had residual activities of 5.7±0.6% and 5.6±0.4% of wild-type activity for in vitro B and F production, respectively. As both mutants show residual activity above 5% of wild-type activity, they can be considered as mild or non-classical mutations. Therefore, the apparent kinetic constants for the mutations p.(Arg143Trp) and p.(Arg332Gln) have been investigated. For the conversion of DOC to B, both mutations had comparable Km values with significantly lower Vmax values compared with the wild type (Figure 3). The kinetic constants for the conversion of 11S to F had higher Km values and significantly lower Vmax values compared with the wild type (Figure 3). The apparent kinetic constants for the wild-type protein and the mutants are depicted in Table 2. Western blot analysis demonstrated that all mutations had similar translation efficiency compared with the wild-type protein (data not shown).

Residual 11β-hydroxylase activity of the four CYP11B1 mutants compared with wild type measured in intact HEK293 cells expressing the CYP11B1 enzyme. (a) Conversion of DOC to B. (b) Conversion of 11-S to F. Lineweaver–Burk plots of enzymatic activity of non-classical mutations. (c) Conversion of DOC to B. (d) Conversion of 11-S to F.

Multiple CYP11B1 COBALT (NCBI) alignments. The Arg143, Ala306, Glu310 and Arg332 residues of CYP11B1 and corresponding amino acids of the aligned CYPs are shaded. (a) Alignment of human CYP11B1 with human CYP11B2, the mouse, rat and macaca orthologues. (b) Alignment of different human steroidogenic CYP enzymes (type I enzymes, CYP11B1, CYP11B2, CYP11A1; type II enzymes, CYP21A2, CYP17A1, CYP19A1).
Discussion
In the present study, we describe the molecular background of three patients suffering from non-classical 11β-OHD and two individuals showing the classical form of the disease.
Mutants causing non-classical 11β-OHD
Non-classical 11β-OHD is characterized by the reduced ability to synthesize cortisol together with the elevation of adrenal androgens, clinically apparent at childhood or later with, for example, premature pubarche, acne, hirsutism or menstrual irregularity. Genital ambiguity is not noticed in the non-classical form. The frequency of non-classical 11β-OHD is not known but may be underestimated as the phenotype is clinically indistinguishable from non-classical 21-hydroxylase deficiency (21-OHD), which has a frequency of up to 1:800 depending on the population.2 This underscores the importance to screen patients with such a phenotype lacking elevated levels of 17-OHP or mutations within the CYP21A2 gene for 11β-OHD. This can be easily done by baseline and ACTH-stimulated determination of 11-S and DOC or by a 24 h urinary steroid gas chromatography mass spectrometry (GC-MS) profile in order to improve the discrimination between these two conditions.
Today, eight distinct mutations associated with non-classical 11β-OHD have been described.16 Herein, we add two novel mutations causing the non-classical form of the disease. Patient 1 is heterozygous for the mutant p.(Arg143Trp), which showed a residual 11β-hydroxylase activity of approximately 8% of wild-type activity, consistent with non-classical 11β-OHD.12, 17, 18 The kinetic constants reflect steric changes not necessarily affecting substrate binding but reaction velocity. The residue 143 is located in the C helix with a side chain pointing into the solvent surrounding (Figure 4). Helix C is most obviously involved in redox-partner binding, and the disturbance of this contact can cause slower substrate conversion due to insufficient electron flux.19, 20

Ribbon representation of the model of the three-dimensional structure of CYP11B1.8 The central I helix important for a proper haem binding and orientation is coloured in light blue. Side chains of the mutated amino-acid residues p.Arg143, p.Ala306, p.Glu310, p.Arg332 and, in addition, the side chain of p.Glu327, which forms a salt bridge with p.Arg332, are depicted.
The siblings 2 and 3 originate from northern Italy. Both were heterozygous for the mutation p.(Leu299Pro) and the novel p.(Arg332Trp) mutant. They showed a typical non-classical phenotype, which has to be caused by the p.(Arg332Trp) variant as this is the mutation with the higher residual activity of about 6% of wild-type activity. The residue Arg332 is conserved between different species in 11β-hydroxylase and aldosterone synthase as well as in type I P450 enzymes (Figure 3) Again, Arg332 is located in the I helix, and its cationic side chain is part of a salt bridge with the anionic side chain of p.Glu327 (Figure 4). This salt bridge stabilizes helix I and shields the hydrophobic core from the surrounding water. The loss of this salt bridge in case of the p.(Arg332Gln) mutant will destabilize the I helix and hereby reduce haem binding as well as the overall stability of the protein.
Mutants causing classical 11β-OHD
Classical 11β-OHD is characterized by cortisol deficiency and elevated adrenal androgens causing ambiguous external genitalia in the female individuals at birth as detected in patient 4 and 5 in this study. Both showed elevated precursor steroids including 11-S together with low levels of cortisol. No one showed elevated blood pressure at diagnosis at the neonatal age, a symptom typically seen with older age and insufficient or absence of treatment. Patient 4 was compound heterozygous for the novel p.(Glu310Lys) mutation and p.(Leu299Pro). The latter variant has been characterized by Krone et al showing an enzymatic activity of 1.2%.8 As expected from the phenotype of patient 4, the mutant p.(Glu310Lys) has no measureable residual activity. The alignment of different steroidogenic type I and type II cytochrome P450 enzymes reveals that an acidic residue (glutamic acid or aspartic acid) at position 310 is conserved in different species and enzymes showing its relevance for the enzyme function (Figure 3). Glutamic acid 310 is located in the I-helix and directly involved in haem binding (Figure 4). A charge reversal in case of p.(Glu310Lys) results in a loss of haem binding and therefore causes complete loss of activity.
Patient 5 is heterozygous for the mutation p.(Gln356Ter) described by Curnow et al in patients with classical 11β-OHD.15 The second heterozygous mutation was p.(Ala306Val), which has been described in a male Chinese patient with classical phenotype in the heterozygous form.14 As the function of the mutation was not studied, so far we analysed the mutant in vitro and were able to show a severely diminished activity of 1% of wild-type activity. Alanine 306 is conserved in 11β-hydroxylase and aldosterone synthase in different species as well as in the type I enzyme P450 side chain cleavage (CYP11A1) (Figure 3). The residue 306 in type II enzymes is different (21-hydroxylase methionine, 17-hydroxylase threonine and C19 aromatase glutamine), what seems to be an essential difference between mitochondrial and microsomal P450 enzymes. Residue 306 is also located in the I-helix, which is responsible for haem binding (Figure 4). Ala306 is involved in the hydrophobic core of the protein and thereby responsible for the correct orientation of helix I. The increased hydrophobicity of valine in the mutant p.(Ala306Val) will either lead to a destabilization of the whole protein or at least to a reorientation of helix I resulting in the loss of haem binding.
In conclusion, we were able to show the inactivating effect of three novel CYP11B1 mutations as well as one reported mutant. Two mutations are responsible for a non-classical phenotype in which a residual enzyme activity of 6% is enough to prevent genital virilization and prenatal onset of 11β-OHD. As CYP11B1 mutations are most often individual for a family, the in vitro analysis of novel mutations is essential for clinical and genetic counselling.
References
New MI : Inborn errors of adrenal steroidogenesis. Mol Cell Endocrinol 2003; 211: 75–83.
Speiser PW, White PC : Congenital adrenal hyperplasia. N Engl J Med 2003; 349: 776–788.
Peter M, Dubuis JM, Sippell WG : Disorders of the aldosterone synthase and steroid 11beta-hydroxylase deficiencies. Horm Res 1999; 51: 211–222.
White PC, Curnow KM, Pascoe L : Disorders of steroid 11 beta-hydroxylase isozymes. Endocr Rev 1994; 15: 421–438.
Joehrer K, Geley S, Strasser-Wozak EM et al: CYP11B1 mutations causing non-classic adrenal hyperplasia due to 11 beta-hydroxylase deficiency. Hum Mol Genet 1997; 6: 1829–1834.
Peters CJ, Nugent T, Perry LA et al: Cosegregation of a novel homozygous CYP11B1 mutation with the phenotype of non-classical congenital adrenal hyperplasia in a consanguineous family. Horm Res 2007; 67: 189–193.
Mornet E, Dupont J, Vitek A, White PC : Characterization of two genes encoding human steroid 11 beta-hydroxylase (P-450(11) beta). J Biol Chem 1989; 264: 20961–20967.
Krone N, Riepe FG, Gotze D et al: Congenital adrenal hyperplasia due to 11-hydroxylase deficiency: functional characterization of two novel point mutations and a three-base pair deletion in the CYP11B1 gene. J Clin Endocrinol Metab 2005; 90: 3724–3730.
Fanelli F, Belluomo I, Di Lallo VD et al: Serum steroid profiling by isotopic dilution-liquid chromatography-mass spectrometry: comparison with current immunoassays and reference intervals in healthy adults. Steroids 2011; 76: 244–253.
Kulle AE, Welzel M, Holterhus PM, Riepe FG : Implementation of a liquid chromatography tandem mass spectrometry assay for eight adrenal C-21 steroids and pediatric reference data. Horm Res Paediatr 2013; 79: 22–31.
Cacciari E, Milani S, Balsamo A et al: Italian cross-sectional growth charts for height, weight and BMI (2 to 20 yr). J Endocrinol Invest 2006; 29: 581–593.
Krone N, Grischuk Y, Muller M et al: Analyzing the functional and structural consequences of two point mutations (P94L and A368D) in the CYP11B1 gene causing congenital adrenal hyperplasia resulting from 11-hydroxylase deficiency. J Clin Endocrinol Metab 2006; 91: 2682–2688.
Kraulis PJ : MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Cryst 1991; 24: 946–950.
Lee HH, Won GS, Chao HT, Lee YJ, Chung BC : Novel missense mutations, GCC [Ala306]->GTC [Val] and ACG [Thr318]->CCG [Pro], in the CYP11B1 gene cause steroid 11beta-hydroxylase deficiency in the Chinese. Clin Endocrinol (Oxf) 2005; 62: 418–422.
Curnow KM, Slutsker L, Vitek J et al: Mutations in the CYP11B1 gene causing congenital adrenal hyperplasia and hypertension cluster in exons 6, 7 and 8. Proc Natl Acad Sci USA 1993; 90: 4552–4556.
Parajes S, Loidi L, Reisch N et al: Functional consequences of seven novel mutations in the CYP11B1 gene: four mutations associated with nonclassic and three mutations causing classic 11{beta}-hydroxylase deficiency. J Clin Endocrinol Metab 2010; 95: 779–788.
Geley S, Kapelari K, Johrer K et al: CYP11B1 mutations causing congenital adrenal hyperplasia due to 11 beta-hydroxylase deficiency. J Clin Endocrinol Metab 1996; 81: 2896–2901.
Kuribayashi I, Nomoto S, Massa G et al: Steroid 11-beta-hydroxylase deficiency caused by compound heterozygosity for a novel mutation, p.G314R, in one CYP11B1 allele, and a chimeric CYP11B2/CYP11B1 in the other allele. Horm Res 2005; 63 (284-293).
Otyepka M, Skopalik J, Anzenbacherova E, Anzenbacher P : What common structural features and variations of mammalian P450s are known to date? Biochim Biophys Acta 2007; 1770: 376–389.
Bridges A, Gruenke L, Chang YT, Vakser IA, Loew G, Waskell L : Identification of the binding site on cytochrome P450 2B4 for cytochrome b5 and cytochrome P450 reductase. J Biol Chem 1998; 273: 17036–17049.
Acknowledgements
We are grateful to Dr Rita Bernhardt for providing the CYP11B1 cDNA, Dr Walter L Miller for providing the Adx and AR cDNA and Dr Hiroshi Takemori for providing the antihuman-CYP11B rabbit antiserum. We appreciate the expert technical assistance of Tanja Stampe. This work was partially supported by the Italian grant PRIN no. 20083ENLWJ.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Menabò, S., Polat, S., Baldazzi, L. et al. Congenital adrenal hyperplasia due to 11-beta-hydroxylase deficiency: functional consequences of four CYP11B1 mutations. Eur J Hum Genet 22, 610–616 (2014). https://doi.org/10.1038/ejhg.2013.197
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2013.197
Keywords
This article is cited by
-
Pathogenicity of Congenital Adrenal Hyperplasia Induced by the p.P377L Mutation of CYP11B1
Biochemical Genetics (2023)
-
Mass spectrometry: an essential tool to be used in discrimination between causes of congenital adrenal hyperplasia, and its benefits versus radioimmunoassay
Beni-Suef University Journal of Basic and Applied Sciences (2021)
-
Steroid biomarkers for identifying non-classic adrenal hyperplasia due to 21-hydroxylase deficiency in a population of PCOS with suspicious levels of 17OH-progesterone
Journal of Endocrinological Investigation (2020)
-
Non-classical 11β-hydroxylase deficiency caused by compound heterozygous mutations: a case study and literature review
Journal of Ovarian Research (2018)
-
Clinical perspectives in congenital adrenal hyperplasia due to 11β-hydroxylase deficiency
Endocrine (2017)
