
Abstract
Idiopathic basal ganglia calcification (IBGC) is characterized by brain calcification and a wide variety of neurologic and psychiatric symptoms. In families with autosomal dominant inheritance, three causative genes have been identified: SLC20A2, PDGFRB, and, very recently, PDGFB. Whereas in clinical practice sporadic presentation of IBGC is frequent, well-documented reports of true sporadic occurrence are rare. We report the case of a 20-year-old woman who presented laryngeal dystonia revealing IBGC. Her healthy parents’ CT scans were both normal. We identified in the proband a new nonsense mutation in exon 4 of PDGFB, c.439C>T (p.Gln147*), which was absent from the parents’ DNA. This mutation may result in a loss-of-function of PDGF-B, which has been shown to cause IBGC in humans and to disrupt the blood-brain barrier in mice, resulting in brain calcification. The c.439C>T mutation is located between two previously reported nonsense mutations, c.433C>T (p.Gln145*) and c.445C>T (p.Arg149*), on a region that could be a hot spot for de novo mutations. We present the first full demonstration of the de novo occurrence of an IBGC-causative mutation in a sporadic case.
Similar content being viewed by others
Introduction
Idiopathic basal ganglia calcification (IBGC) is defined by the presence of brain calcification affecting at least the basal ganglia, after the exclusion of known causes. There is wide intra- and inter-familial diversity of symptoms and ages of onset. The most common signs are cognitive impairment, psychiatric signs, movement disorders, gait disorder, dysarthria, cerebellar syndrome, pyramidal signs, and seizures.1 Most familial cases are dominantly inherited. Three causative genes have recently been discovered. First, loss-of-function mutations in SLC20A2, causing IBGC in approximately 40% families,2, 3 might have an impact on the inorganic phosphate metabolism in the brain. Then, we recently identified PDGFRB, which encodes the transmembrane receptor platelet derived growth factor receptor β (PDGFRβ), as a second IBGC causative gene.4 Third, loss of function of PDGF-B (encoded by the PDGFB gene), the main ligand of PDGFRβ, has recently been shown to cause IBGC in humans and brain calcification through a disruption of the blood–brain barrier integrity in mice.5 Sporadic presentation of IBGC is not uncommon. Nevertheless, true sporadic cases, defined by the absence of basal ganglia calcification in the parents’ CT scans after the age of 50,6 were rarely reported.7 We report the case of a 20-year-old woman presenting laryngeal dystonia revealing IBGC, due to a de novo nonsense mutation within PDGFB.
Materials and Methods
The proband and her parents gave informed, written consent. The study was approved by our local ethics committee. Genomic DNA was extracted from whole peripheral blood using the Flexigen DNA kit (Qiagen, Hilden, Germany). In the proband’s DNA, the entire coding sequence and intron/exon boundaries of SLC20A2 (NM_006749.3), PDGFRB (NM_002609.3), and PDGFB (NM_002608.2) were PCR-amplified. PCR products were sequenced using the BigDye V3.1 Terminator Kit (Applied Biosystems, Courtaboeuf, France) on an automated sequencer (ABI 3100; Applied Biosystems). After the identification of the nonsense mutation in exon 4 of PDGFB (the exons were numbered according to the reference NM_002608.2 in this report; exon 4 corresponds to exon 5 using NG_012111.1 as reference) in the proband, we performed PCR amplification and sequencing of this exon in the parents.
To screen the presence of the mutation in controls, we used the 6503 exomes provided by the Exome Variant Server (EVS, NHLBI GO exome sequencing project, accessed in August, 2013), and a series of 173 in-house exomes from individuals with no neuropsychiatric disorder. To ascertain the parenthood, we used four informative microsatellites (D1S439, D9S1784, D14S986, and D19S913) and genotyped the proband and her parents using a multiplex fluorescent PCR.
To search for nonsense-mediated RNA decay (NMD), we reverse transcribed total mRNA collected in PAXgene Blood RNA tubes (Qiagen) from the patient and her parents (Fisher Scientific, Illkirch, France) and performed PCR sequencing of the complementary DNA of Exon 4 of PDGFB. We then designed a reverse transcription-quantitative multiplex PCR of short fluorescent fragments (RT-QMPSF) of two amplicons of PDGFB, with TOP1 and SF3A1 genes as controls.
All primer sequences are reported in Supplementary Material. The mutation was submitted to the Leiden Open Variation Database (LOVD).
Results
Case report
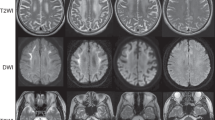
A 20-year-old woman was hospitalized for dysphagia and stridor. She had no familial medical history, and her personal medical history was marked by migraine without aura, leading to the identification of faint calcification of both lenticular nuclei in a CT scan performed at 10 years old during a migraine episode. Direct laryngoscopy showed bilateral vocal cord dysfunction: when the patient breathed in, the vocal cords moved in adduction. Three days later, she presented an acute exacerbation of dyspnea, with vocal cord paralysis in adduction, leading to oro-tracheal intubation. As it was not possible to deprive the patient from the ventilator, she underwent a tracheotomy. Neurological examination, laryngeal CT scan, and electromyography were all normal. No loco-regional cause was found. The cerebral CT scan showed calcification of both lenticular and caudate nuclei, and FLAIR-weighted magnetic resonance imaging only showed several punctiform hyperintensities in the supratentorial white matter and discrete anterior periventricular hyperintensities (Figure 1). An extensive etiological assessment of brain calcification (according to Nicolas et al4) was negative. We retained the diagnosis of IBGC. Her healthy parents had normal CT-scans, performed at the age of 52 years (mother) and 66 years (father). Muscle biopsy (performed as a search for mitochondrial disease) did not show any calcification.

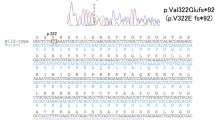
Electropherograms and brain imaging. Electropherograms show a heterozygous substitution of a C by a T in the proband, and a wild-type allele (C) in both parents (a). Brain CT scans on axial (b-1) and coronal (b-2) views show moderate calcifications in the lenticular nuclei (white arrows) and faint calcifications in the caudate nuclei (black arrow). On T2*-weighted MRI, calcifications appear hypointense (b-3) while no signal modification is seen in the lenticular nuclei in FLAIR-weighted images, showing hyperintensities on anterior periventricular regions (b-4) and subcortical white matter (b-5, b-6).
Genetics
No potentially pathogenic variant was identified in SLC20A2 and PDGFRB in the proband. We found a heterozygous single-nucleotide substitution in exon 4 of PDGFB, c.439C>T, predicted to introduce a premature stop codon at position 147 in the protein (p.Gln147*). This mutation was not retrieved in the parents and was absent in controls. Microsatellite analysis was compatible with paternity. We retrieved the mutation at the RNA level, r.439c>u (NM_002608.2). We did not find any decrease in the expression of the mutated allele in the proband by RT–PCR sequencing and RT–QMPSF (Supplementary Data). However, as no SNP was present in the PDGFB-coding sequence, we could not use a more sensitive approach, such as an SNaPshot technique, to detect NMD.
In addition, with the aim to find more mutations of PDGFB, we also sequenced the entire coding region and exon–intron boundaries of PDGFB in the 12 index cases from IBGC families and the 17 cases with sporadic presentation of IBGC with no mutation within PDGFRB and SLC20A2 from our French case series.1 We only found two mutations: the first one was already reported within the original article,5 and the second one is the currently presented mutation.
Discussion
Our patient presented laryngeal dystonia, which is defined by involuntary contraction of the vocal cords, responsible for dysphonia.8 Severe laryngeal dystonia may also be responsible for life-threatening dyspnea. Laryngeal dystonia is a rare manifestation of IBGC9 and such a severe and acute revelation of IBGC has never been reported, to our knowledge.
IBGC is usually inherited according to an autosomal dominant pattern. As clinical expression of brain calcification is not constant, clinically sporadic presentation of IBGC may be due to mutations inherited from an asymptomatic parent in some cases. We demonstrate here that a true sporadic occurrence of IBGC, due to a causative de novo mutation, can occur.
The mutation occurred at position c.439 (p.147), just between two recently reported nonsense mutations in PDGFB (resulting in p.Gln145* and p.Arg149*). Interestingly, the c.445C>T (p.Arg149*) mutation also most likely occurred de novo, although, to our knowledge, paternity was not verified.5 This suggests a clustering of de novo mutations in exon 4 of the PDGFB gene. Such a phenomenon has been studied using pangenomic data from concordant monozygotic twins with autism, and a mutability index (MI) was calculated for each exon as an estimate of relative mutation rate at single-nucleotide resolution.10 The mean MI is 2.41 for exon 4 of PDGFB (chr22:37,957,573_37,957,779, hg18), the genome-wide arithmetic mean of MI being 1, and the mean MI of all other exons of PDGFB being less than 1.54, except for exon 5 (2.15). In accordance with these estimates, our data suggest that exon 4 of PDGFB could be a hotspot for single-nucleotide substitutions. It is also well-recognized that an older paternal age is an extrinsic factor favoring de novo point mutations.10 Although the parental origin of the mutated allele could not be documented in our case, we note that the father was aged 44 and the mother 29 at their daughter’s date of birth.
Surprisingly, we did not find further mutations of PDGFB with the exception of the previously reported one,5 among the familial cases and those with sporadic presentation of our case series. De novo occurrence of a PDGFB mutation is therefore probably a quite rare event among the IBGC cases. Nevertheless, among the seven mutations reported within PDGFB so far, two occurred de novo, which is a relevant result.
This c.439C>T (p.Gln147*) mutation might not be subject to efficient NMD because it is located close to the exon4–exon5 junction,11 which could explain that we did not find mRNA decay. However, the mutation is clearly detrimental, as it is predicted to result in a shortened protein with loss of functionally critical domains including interface-contributing residues of the PDGF-B:PDGFRβ complex.12
To conclude, we demonstrate true sporadic occurrence of IBGC due to a de novo nonsense mutation. Our results further support the involvement of the loss of function of PDGF-B in IBGC and suggest a possible hotspot for single-nucleotide substitutions in exon 4 of PDGFB.
References
Nicolas G, Pottier C, Charbonnier C et al: Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 2013; 136: 3395–3407.
Wang C, Li Y, Shi L et al: Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet 2012; 44: 254–256.
Hsu SC, Sears RL, Lemos RR et al: Mutations in SLC20A2 are a major cause of familial idiopathic basal ganglia calcification. Neurogenetics 2013; 14: 11–22.
Nicolas G, Pottier C, Maltete D et al: Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 2013; 80: 181–187.
Keller A, Westenberger A, Sobrido MJ et al: Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 2013; 45: 1077–1082.
de Oliveira MF, Steinberg SS, de Oliveira JR : The challenging interpretation of genetic and neuroimaging features in basal ganglia calcification. Gen Hosp Psychiatry 2013; 35: 210–211.
Smits MG, Gabreels FJ, Froeling PG, de Abreu RA, Thijssen HO, Renier WO : Calcium-phosphate metabolism in autosomal recessive idiopathic strio-pallido-dentate calcinosis and Cockayne’s syndrome. Clin Neurol Neurosurg 1983; 85: 145–153.
Colosimo C, Suppa A, Fabbrini G, Bologna M, Berardelli A : Craniocervical dystonia: clinical and pathophysiological features. Eur J Neurol 2010; 17Suppl 1: 15–21.
Wszolek ZK, Baba Y, Mackenzie IR et al: Autosomal dominant dystonia-plus with cerebral calcifications. Neurology 2006; 67: 620–625.
Michaelson JJ, Shi Y, Gujral M et al: Whole-genome sequencing in autism identifies hot spots for de novo germline mutation. Cell 2012; 151: 1431–1442.
Schweingruber C, Rufener SC, Zund D, Yamashita A, Muhlemann O : Nonsense-mediated mRNA decay—mechanisms of substrate mRNA recognition and degradation in mammalian cells. Biochim Biophys Acta 2013; 1829: 612–623.
Shim AH, Liu H, Focia PJ, Chen X, Lin PC, He X : Structures of a platelet-derived growth factor/propeptide complex and a platelet-derived growth factor/receptor complex. Proc Natl Acad Sci USA 2010; 107: 11307–11312.
Acknowledgements
We thank Tracey Avequin for her help in editing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Nicolas, G., Jacquin, A., Thauvin-Robinet, C. et al. A de novo nonsense PDGFB mutation causing idiopathic basal ganglia calcification with laryngeal dystonia. Eur J Hum Genet 22, 1236–1238 (2014). https://doi.org/10.1038/ejhg.2014.9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.9
Keywords
This article is cited by
-
Genetic analyses identify pleiotropy and causality for blood proteins and highlight Wnt/β-catenin signalling in migraine
Nature Communications (2022)
-
PDGF receptor mutations in human diseases
Cellular and Molecular Life Sciences (2021)
-
Functional evaluation of PDGFB-variants in idiopathic basal ganglia calcification, using patient-derived iPS cells
Scientific Reports (2019)
-
The role of de novo mutations in adult-onset neurodegenerative disorders
Acta Neuropathologica (2019)
-
Clinical and radiological diversity in genetically confirmed primary familial brain calcification
Scientific Reports (2017)
