
Abstract
β-Thalassemia is the most common autosomal recessive single-gene disorder in Sardinia, where approximately 10.3% of the population is a carrier. Prenatal diagnosis is carried out at 12 weeks of gestation via villocentesis and is commonly aimed at ascertaining the presence or absence of the HBB variant c.118C>T, which is the most common in Sardinia. In this study, we describe for the first time the application of semiconductor sequencing to the non-invasive prenatal diagnosis of β-thalassemia in 37 couples at risk for this variant. In particular, by using a haplotyping-based approach with a hidden Markov model (HMM) and a dedicated pipeline, the two parental haplotypes most likely inherited by the foetus could be established in 30 out of 37 cffDNA samples. Specifically, the paternally inherited haplotype was correctly determined in all 30 of the samples, while the maternal haplotype was incorrectly predicted in six of the 30 genotyped samples. The lack of informative SNPs hampered the inference of both parental haplotypes in the remaining seven samples. As shown in previous studies, we have confirmed that the haplotyping-based approach represents a valuable resource, as it improves the detection of both parental haplotypes inherited by the foetus. In general, our results are encouraging, as we have proven that NIPD is also feasible in couples who are at risk for a monogenic disorder and share the same variant.
Similar content being viewed by others


Introduction
Approximately twenty years have passed since Dennis Lo first described the presence of cell-free foetal DNA (cffDNA) in maternal circulation.1 In this period, great improvements in the technologies used in molecular diagnostics have allowed researchers to finely characterize the structure and biology of cffDNA.
It is now well established that cffDNA originates from apoptotic syncytiotrophoblasts, a feature that correlates with its highly fragmented behaviour.2 cffDNA normally represents a minor fraction of the total extracellular DNA circulating in maternal blood, even though its concentration tends to gradually increase during gestation3 and to change in the presence of some maternal or foetal conditions, such as preeclampsia, increased maternal weight or foetal aneuploidies.4, 5, 6, 7 cffDNA is rapidly cleared after delivery8 and thus does not interfere with the cffDNA released during subsequent pregnancies. Some genomic regions of cffDNA show different methylation patterns than circulating maternal DNA, a characteristic that can be used to quantify and determine the presence of foetal DNA during pregnancy, irrespective of foetal sex.9, 10 In the last decade, the discovery that all foetal chromosomes are equally represented in maternal blood has encouraged the development of non-invasive prenatal diagnostic tests worldwide. The goal of these tests is to reduce the risk of foetal loss, which is still associated with the use of invasive diagnostic procedures.11 Currently, the most common foetal aneuploidies can be detected with a high level of sensitivity and specificity through the application of next-generation sequencing (NGS) technologies coupled with dedicated bioinformatic tools.11 In addition, the characterization of genetic traits absent from the mother and paternally inherited by the foetus (ie, the RhD gene, Y chromosome, and de novo mutations) can be obtained through the application of simpler, non-invasive tests that are widely performed.12, 13, 14, 15
Despite these examples of successful translation into clinical practice, the non-invasive prenatal diagnosis (NIPD) of monogenic recessive disorders still faces several technical challenges, particularly when both parents are carriers of the same variant. The extensive contamination of cffDNA samples with maternal DNA represents one of the major obstacles to overcome for the correct identification of the foetal disease-related genotype. Nevertheless, several proof-of-concept studies have demonstrated that it is possible to infer both parental haplotypes inherited by the foetus and thus ascertain the foetal genotype through the deep sequencing of cffDNA in the genomic region containing either the disease-causing gene or the surrounding single nucleotide polymorphisms (SNPs). The feasibility of the haplotype-based approach was first described by Lo et al.2 for the non-invasive diagnosis of β-thalassemia.16 In particular, the study demonstrated that the application of relative haplotype dosage analysis (RHDO) could be a powerful strategy for inferring the foetal SNP alleles shared by both the foetus and the mother. Further studies have established that the same approach also has a high rate of success in the non-invasive diagnosis of other monogenic disorders, such as DMD,17 congenital adrenal hyperplasia,18, 19 and congenital deafness.20
Here, we describe for the first time the application of the haplotyping-based approach and semiconductor sequencing to the NIPD of β-thalassemia in 37 couples at risk for the same HBB c.118C>T variant. β-Thalassemia is the most common autosomal recessive single-gene disorder in Sardinia, where approximately 10.3% (1 out of 9 people) of the population is a carrier. The HBB c.118C>T variant (rs11549407), better known as the nonsense β°39 variant, is the most common variant, accounting for 95.7% of the β-thalassemia variants with an allele frequency of 4.8%. The platform is based on the targeted sequencing of cffDNA samples at informative SNPs spread throughout the β-globin gene cluster that have been previously selected in parental DNA. The allele counts observed at the informative loci are analyzed through an automated pipeline, which, using a hidden Markov model (HMM), predicts the two haplotypes most likely inherited by the foetus. The foetal haplotypes and the HBB alleles predicted in the cffDNA samples are finally compared with the results obtained in the corresponding foetal DNA samples with villocentesis. The results described herein show that semiconductor sequencing coupled with a dedicated bioinformatic pipeline can provide valuable results for the non invasive prenatal diagnosis of a monogenic disease, even in couples at risk for the same variant. Further improvements are needed in order to increase the detection rate of the maternally inherited haplotype.
Materials and methods
Sample collection
After ethical committee approval (Prot.no 155/CE/08, 17 December 2008), appropriate counselling and written informed consent, 20 ml of peripheral blood was collected from couples who underwent prenatal diagnosis because they were carriers of the c.118C>T variant in the HBB gene and therefore at risk for β-thalassemia. Prior to villocentesis, maternal blood samples were collected at a gestational age of 7 weeks to 14 weeks+3 days (mean 9 weeks+6 days) in EDTA-containing tubes. Plasma samples were separated after whole blood centrifugation at 1600 g for 10 min and at 16 000 g for 10 min, aliquoted into 1.5 ml tubes and finally frozen at −80 °C until cffDNA extraction.
cffDNA and genomic DNA extraction
To test the performance of the sequencing platform in the three groups of samples (wild type, heterozygous or homozygous for the c.118C>T variant), 37 plasma samples were selected and extracted retrospectively after completion of invasive prenatal diagnosis. Of them, 14 samples were wild type, 15 were heterozygous, and 8 were homozygous (Table 1). cffDNA samples were isolated from 8 ml of thawed plasma using the QIAamp Circulating Nucleic Acid Kit from Qiagen with a Qiagen vacuum manifold, following the manufacturer’s protocol (Qiagen GmbH, Hilden, Germany). Final DNA was eluted into 150 μl of AVE buffer. Parental genomic DNA was extracted from 500 μl of whole blood with a DiaSorin Blood DNA 500 extraction kit (DIASORIN S.P.A., SALUGGIA (VC), Italy) and NorDiag Arrow System (ISOGEN Life Science, Utrecht, The Netherlands). The corresponding trophoblast DNA samples were obtained by villocentesis at 11–14 weeks of gestation. After maternal decidual tissue dissection, DNA was extracted from the trophoblast tissue samples with the salting-out method. The c.118C>T variant was detected both in trophoblast DNA and in parental DNA using the Nuclear Laser Medicine Beta Globin Test kit (Nuclear Laser Medicine s.r.l, Italy).
Study design
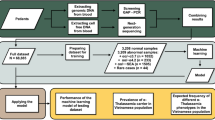
The principle of our approach is to use the cffDNA samples to infer the parental haplotypes most likely inherited by the foetus and establish the foetal HBB c.118C>T genotype accordingly. As shown in Figure 1, the analysis of each cffDNA sample is preceded by the semiconductor sequencing of the corresponding trio of familial DNA samples (maternal, paternal and trophoblast) in a 62.7 kb target region of the β-globin gene cluster (NC_000011.9 chr11: 5230230-5293230, GRCh37/hg19, UCSC Genome Browser) that contains both the HBB c.118C>T variant and SNPs that are potentially useful in determining the parental haplotype structure. In cffDNA analysis, the SNPs are considered informative when both the mother and the father are homozygous for different alleles or when at least one parent is heterozygous (Supplementary Figure 1). Once selected, the short regions (80–120 bp) containing the informative SNPs and the c.118C>T variant are individually amplified in the cffDNA samples and then pooled for library construction and semiconductor sequencing. In our study, the parental haplotypes in the target region were obtained using two different procedures. In the first step of the workflow, which was aimed at creating a unique Reference Haplotype Panel, haplotypes were constructed using the trio of sequencing data (parental and trophoblast DNA), the 1000 Genomes Project Phase3 v5 haplotypes21 (2504 individuals from 26 populations) and SHAPEIT.22, 23 Conversely, within the cffDNA pipeline, parental haplotypes were obtained by processing the parental DNA sequencing data and the previously generated Reference Haplotype Panel (Figure 2). The four parental haplotypes were finally used as references to process the sequencing data obtained from the plasma samples and to predict the haplotypes most likely inherited by the foetus.

General workflow of molecular analysis and data processing for the non-invasive prenatal diagnosis of β-thalassemia.

Flowchart of bioinformatics data analysis with a dedicated pipeline.
Amplicon library preparation in parental and trophoblast DNA
Five overlapping long (7–17.7 kb) PCRs were performed with either parental or trophoblast DNA to select the 62.7 kb region of the β-globin gene cluster (Supplementary Figure 2). The PCR conditions and the primers sequences are reported in Supplementary Note 1 and Supplementary Table 1, respectively. The five PCR products were then pooled, purified with the Agencourt AMPure XP Reagent (Beckman Coulter, Brea, CA, USA), and quantified using the Qubit dsDNA BR Assay Kit (Life Technologies, Carlsbad, CA, USA). One hundred nanograms of purified pools was fragmented to a length of 200–300 bp via enzymatic digestion, blunt ended, and ligated to Ion adapters with the Ion Xpress Plus Library kit (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s protocol. The adapter-ligated library was size selected (330 bp) by E-gel electrophoresis, amplified for 8 cycles, purified and quantified with a High Sensitivity DNA kit and 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA).
Amplicon library preparation with cffDNA samples
For each cffDNA sample, short regions (80–120 bp) containing the informative SNPs and the HBB c.118C>T variant were individually amplified. Further details of the PCR reactions and the sequences of the primers used are reported in Supplementary Note 2 and Supplementary Table 2, respectively. After pooling, the short amplicons were purified using Agencourt AMPure XP Reagent (Beckman Coulter, Brea, CA, USA), quantified with a Qubit dsDNA HS Assay Kit (Life Technologies, Carlsbad, CA, USA) and subjected to library preparation as described in the Ion Xpress Plus Library Kit (Life Technologies) protocol. The adapter-ligated library was quantified using a High Sensitivity DNA kit and a 2100 Bioanalyzer (Agilent).
Emulsion PCR and semiconductor sequencing
One hundred picomoles of the ligated pooled libraries was subjected to template preparation with the Ion OneTouch Template Kit and Ion OneTouch System v2 (Life Technologies, Carlsbad, CA, USA). Semiconductor sequencing was performed in 314/316 chips using the Ion PGM Sequencing 200 Kit v2 (Life Technologies) in a PGM system at 500 flows, in accordance with the manufacturer’s protocol.
Data analysis
The variant sites and genotypes of the genomic DNA samples were automatically detected after each sequencing run by launching the Torrent Variant Caller (TVC) plug-in with the default high-stringency settings for germline variants. The presence of these nucleotide variations was further confirmed by visual inspection of the mapped reads in Integrative Genomics Viewer (IGV).24, 25
Reference panel
The haplotype reference panel was created by integrating the 1000 Genomes Project Phase3 v5 haplotype set with the haplotypes of the 37 investigated trios and of further two trios whose corresponding cffDNA samples were not processed in this study. The FASTQ files for each trio were first aligned with the BWA program26 and analyzed with the SAMtools program27 to generate three unphased VCF files. The VCF files were filtered for quality >10 and positions (1000 G), merged with the 1000G Phase3 v5 haplotype set and then phased as a trio with the 1000G Phase3 v5 haplotype set by using SHAPEIT and information from the PIRs (phase informative reads).28
cffDNA pipeline
The cffDNA sequencing data were analyzed with an automated pipeline that, starting with the paternal, maternal and cffDNA BAM files, predicts the most likely haplotypes inherited by the foetus and validates the results with those determined by sequencing the trophoblast DNA obtained via chorionic villous sampling (CVS). The cffDNA analysis workflow is extensively described in Supplementary Note 3 and Figure 2.
Results
We tested the feasibility of semiconductor sequencing for the NIPD of β-thalassemia in 37 cffDNA samples at risk for the HBB c.118C>T variant. The plasma samples were selected retrospectively based on the results obtained in the corresponding CVS to ensure that all three genotypes were represented.
To increase the probability of discriminating the foetal DNA from the overwhelming amount of maternal DNA contaminating the cffDNA samples, we decided to use a haplotype-based approach. Accordingly, the sequencing of each cffDNA sample was preceded by the semiconductor sequencing of the corresponding trio (father/mother/CVS DNA samples) with the aim of selecting informative SNPs in a 62.7 kb target region in the β globin gene cluster. Data management was carried out through an automated pipeline; this workflow is shown in Figure 1. Namely, the pipeline first constructs the four parental haplotypes using SHAPEIT and a reference haplotype dataset without CVS DNA information. Then, it continues processing the sequencing data for the cffDNA sample and predicts the foetal HBB genotype. The final validation is completed by comparing the genotype predicted from the cffDNA sample with that established by CVS sampling. As shown in Table 1, semiconductor sequencing of cffDNA produced a median of 69462.35 reads per sample, with a mean depth of 7538X and 100% coverage in the target region. The total number of informative SNPs identified in the processed parental DNA varied greatly and ranged from 40 to 165, with a mean value of 110 (IQR 34.5; Table 2). However, on average, only 52.7% of the SNPs could be effectively sequenced and analyzed in the corresponding plasma samples. Not all the potentially informative SNPs could be effectively investigated in the cffDNA samples. In fact, the presence of highly homologous genes, such as HBG1 and HBG2, and an L1 repeat element located 3′ to the β-globin gene greatly hindered the design of specific primers that could yield amplicons shorter than 120 bp. The remaining excluded SNPs, which were mostly clustered in the same regions, were excluded from the analysis, as we observed uneven amplicon coverage caused by the unspecific overlapping of reads belonging to different amplicons in previous studies. The total number of SNPs actually used for final haplotype inference (Table 2) was further reduced by 50% within the pipeline, which automatically excludes those SNPs with coverage lower than 1000 × (paternal-only SNPs) or 1500X (maternal-only SNPs) and the heterozygous-only SNPs that show an allelic imbalance greater than 54%. These threshold values were established during the pipeline validation as they provided the highest detection rate of the foetal genotypes. For instance, 1000 × coverage was found to be deep enough for the detection of the paternal alleles that were not shared with the maternal DNA. Analysis through the automated pipeline has been completed in 30 out of 37 cffDNA samples. The pipeline was unable to proceed with the downstream data processing in the remaining seven samples because of a lack of either paternal-only SNPs (two samples) or informative SNPs useful in calculating the foetal fraction (five samples). As the paternal haplotype represents the scaffold used in the pipeline to evaluate the presence or absence of statistically relevant allelic imbalances at maternal heterozygous sites, maternal haplotype prediction could not be completed in the two cffDNA samples that lacked the paternal haplotype prediction. Conversely, the absence of a known foetal fraction prevented the inference of the paternal and the maternal haplotypes in the remaining five plasma samples. The overall data regarding the number of SNPs used to infer the maternally and paternally inherited haplotypes and the number of SNPs sequenced but ultimately excluded during the pipeline analysis are summarized in Table 3. The final results obtained for the 30 samples that completed the workflow analysis through the pipeline are reported in Table 1. In particular, the table shows the predicted and the expected foetal HBB genotypes, the number of reads, the mean depth of semiconductor sequencing and the foetal fraction calculated in each cffDNA sample. The foetal fraction values ranged from 3.7 to 12.6%, with a mean value of 6.96 (IQR 3.55), and were generated from the mean fractional read depth calculated in each plasma sample at the paternal-only and homozygous-only SNPs.
The foetal HBB genotype was correctly identified in 24 samples (82% sensibility and 77% specificity), while only the paternal haplotype prediction was correctly determined in the remaining six. As expected, we had a higher detection rate of the paternally inherited haplotypes (Table 1, Supplementary Figure 3), which were correctly inferred in the thirty samples that completed the pipeline workflow. In fact, the detection of paternal alleles absent from the maternal genome was improved by the high-depth coverage obtained by amplicon sequencing. Conversely, the maternal haplotypes were incorrectly predicted in six cffDNA samples even when the inheritance of the maternal variant was assessed by measuring the allelic imbalances observed in a medium-to-high number of heterozygous maternal SNPs (mean 18.1, IQR 7.5). In these six samples, the incorrect allelic imbalances measured at these sites caused an incorrect HMM correction and thus the prediction of the non-inherited maternal haplotype. The foetal fraction, the read depth and the time of blood draw for five of these samples were in line with those observed in the 24 correctly diagnosed samples (Table 1), and only one sample exhibited a read depth lower than the mean value obtained for the twenty-nine samples as a whole (sample 11, Table 1). The average accuracy of SNP genotyping, achieved via both sequencing and HMM inference, was found to be 99.8 and 94.5% in the cffDNA samples in which the foetal HBB genotype was correctly or incorrectly deduced, respectively.
Discussion
The ability to study foetal DNA circulating in the maternal blood during gestation is rapidly changing the approaches used worldwide in the prenatal diagnosis of the most common aneuploidies and, to a lesser extent, some autosomal recessive disorders.
Most NIPD studies published in the field of haemoglobinopathies have focused on confirming or ruling out the presence of the mutated paternal allele in couples who are carriers of different variants,29 while the successful identification of both alleles inherited by the foetus has been reported in a limited number of studies.2, 16 Among them, one report has shown the feasibility of NIPD in a large number of couples at risk for the same variant, namely, sickle cell anaemia, with an 80% success rate.30
In this study, we have explored the feasibility of the NIPD of β-thalassemia in 37 couples in which both partners were carriers of the HBB c.118C>T variant. In fact, β-thalassemia occurs at a markedly high frequency in the isolated Sardinian population, in which this is the most common variant. Therefore, in most cases, prenatal diagnosis is aimed at ascertaining the foetal genotype at a single nucleotide. This relative simplicity of invasive molecular testing represents a major obstacle in NIPD, as the technical and bioinformatic tools must be able to ascertain the presence or absence of the same maternally and/or paternally inherited foetal point variant in samples with extensive maternal DNA contamination. For this reason, the haplotype-based approach represents a valuable resource, as it provides additional information for constructing the foetal haplotypes and detecting the disease-associated genotype through the qualitative and quantitative investigation of polymorphic sites spread throughout the genomic region containing the variant of interest. Our study was thus oriented in this direction, with the aim of constructing the foetal haplotypes in a 62.7 kb region of the β-globin gene cluster by using target amplicon sequencing (PGM Ion Torrent) and an automated pipeline for data analysis. A major challenge in our work was developing a strategy for constructing the parental haplotypes. The phasing process can be accomplished in several ways that can require the analysis of parental, first-degree relative and/or proband DNA or, alternatively, the use of molecular or bioinformatics-dedicated applications.
In our work, we opted for a dual strategy. Specifically, we first created a targeted reference panel of haplotypes adapted for the NIPD of β-thalassemia by utilizing the 1000 Genomes Project Phase3 v5 haplotype set along with the haplotypes of 39 mother-father-CVS trios sequenced in the NIPD project. For all the haplotypes, we selected only the 62.7 kb region encompassing the β-globin cluster (NC_000011.9, chromosome 11: 5230230-5293230 GR37/hg19) investigated via the targeted sequencing of the DNA for each trio. The inclusion of these additional 234 haplotypes carrying the c.118C>T variant or the wild-type HBB sequence served to increase the representation of the Sardinian haplotypes in the reference panel, as only a single haplotype in the 1000G set contained this nonsense variant. Furthermore, the selection of short haplotypes did not require extended processing time during the assembly of the reference dataset. Second, we developed an automated pipeline that, with the support of a targeted reference panel, is able to process the parental and cffDNA sequencing data and provide both the haplotypes and a final report with the predicted foetal genotype within 3 h. The pipeline has been specifically designed for application to β-thalassemia; however, its structure can be easily adapted to other monogenic disorders with support from a dedicated reference dataset of haplotypes. Data analysis requires a single instance of manual intervention at the beginning of the workflow to upload the parental and cffDNA BAM files, which are then processed automatically. This simple procedure can be performed by non-skilled bioinformatics personnel. Parental phasing represents the first and most crucial step in the entire bioinformatic analysis, as the construction of the four ‘reference haplotypes’ needed for the subsequent processing of cffDNA data depends on this step. The adopted procedure is highly accurate, as the parental haplotypes constructed through the pipeline with the support of a reference panel, but not CVS information, show close to 100% accuracy for both the paternal and maternal haplotypes. Furthermore, the implementation of the 1000G reference panel with Sardinian haplotypes linked to the c.118C>T variant or the wild-type sequence has greatly improved the phasing processing. cffDNA data analysis is completed only when one of the two following conditions is met: a known foetal fraction and/or the inference of the paternal haplotype, which then triggers the detection of maternal haplotype inherited by the foetus. With this approach, the foetal genotype could be established in 30 out of 37 cffDNA samples, while failing to meet one of these conditions prevented the completion of data processing in the remaining seven samples.
The paternally inherited haplotype was correctly determined in all 30 samples. In fact, the accurate reconstruction of the parental haplotypes obtained through the pipeline coupled with the higher sequencing depth (>1000 ×) obtained for the cffDNA samples via amplicon sequencing greatly improved the identification of paternal alleles not shared with maternal DNA and, accordingly, the assigning of the correct paternal haplotype inherited by the foetus. The amplification of short regions surrounding the informative SNPs, which is more compatible with the fragmented nature of cffDNA, has proven to be not only highly sensitive in detecting low-fraction alleles but also cheaper than other target-enrichment procedures in generating cffDNA libraries. Conversely, in six out of the 30 genotyped samples, the maternal haplotype was incorrectly predicted. The overwhelming amount of maternal DNA contaminating the cffDNA is, in fact, one of the major limits that hinders the identification of the alleles shared by mother and foetus. In our samples, the maternally inherited haplotype was assigned by measuring, site by site, the allelic imbalances observed at informative SNPs (heterozygous mother and homozygous or heterozygous father) and using the HMM method. A threshold of 54% was selected for calling reliable allelic imbalances based on the observation that greater imbalances were most likely generated when one allele exhibited a different amplification efficiency than the other. In five out of six cases, the incorrect prediction of the inherited maternal haplotype was caused by the presence of unexpected allelic imbalances at a number of sites sequenced in the cffDNA samples, which resulted in an incorrect HMM inference. We suppose that the presence of a vanishing twin or PCR bias could have generated allelic imbalances different from those expected at different SNP positions, thus affecting the inference of the correct haplotype. Conversely, the incorrect construction of the maternal haplotypes with the pipeline occurred as a consequence of erroneous maternal haplotype inference in the sixth cffDNA sample. The presence of highly homologous regions spread across the beta globin cluster has greatly hampered the investigation of several potentially informative sites in cffDNA samples.
In particular, it was extremely difficult to design short amplicons for several SNPs located in the Aγ and Gγ genes and therefore ascertain their allelic status in the foetal DNA. Despite this limit, cffDNA data analysis was completed in most of the samples, even if the number of sites was extremely reduced. Furthermore, the correct haplotype prediction was not correlated with the number of informative SNP used for the analysis. In fact, the correct paternal and maternal haplotypes were detected in samples where the number of SNPs finally used for the prediction ranged from 1 to 34 (mean 13.1, IQR 10.75) and 1 to 33 (mean 17.3, IQR 11.5), respectively.
In conclusion, we have demonstrated for the first time the application of semiconductor sequencing in the NIPD of a large number of cffDNA samples at risk for β-thalassemia. We have further confirmed that this haplotyping-based approach represents a valuable resource, as it improves the detection of both parental haplotypes inherited by the foetus. However, further improvements are needed. In particular, from a technical point of view, extending the region sequenced in the parental DNA could help in recovering a higher number of informative SNPs to investigate in the cffDNA samples. Concerning the bioinformatic analysis, separating maternal haplotype prediction from the paternal prediction could increase the total number of haplotypes inferred through the pipeline since, in our approach, maternal prediction starts only when the paternal haplotype has been determined. In general, our results are encouraging, as we have proven that NIPD could be feasible in couples who are at risk for a monogenic disorder and share the same point variant.
References
Lo YM, Corbetta N, Chamberlain PF et al: Presence of fetal DNA in maternal plasma and serum. Lancet 1997; 350: 485–487.
Lo YM, Chan KC, Sun H et al: Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus. Sci Transl Med 2010; 2: 61ra91.
Wang E, Batey A, Struble C, Musci T, Song K, Oliphant A : Gestational age and maternal weight effects on fetal cell-free DNA in maternal plasma. Prenat Diagn 2013; 33: 662–666.
Hahn S, Rusterholz C, Hösli I, Lapaire O : Cell-free nucleic acids as potential markers for preeclampsia. Placenta 2011; 32 (Suppl): S17–S20.
Canick JA, Palomaki GE, Kloza EM, Lambert-Messerlian GM, Haddow JE : The impact of maternal plasma DNA fetal fraction on next generation sequencing tests for common fetal aneuploidies. Prenat Diagn 2013; 33: 667–674.
Lo YM, Zhang J, Leung TN, Lau TK, Chang AM, Hjelm NM : Rapid clearance of fetal DNA from maternal plasma. Am J Hum Genet 1999; 64: 218–224.
Rava RP, Srinivasan A, Sehnert AJ, Bianchi DW : Circulating fetal cell-free DNA fractions differ in autosomal aneuploidies and monosomy X. Clin Chem 2014; 60: 243–250.
Yu SC, Lee SW, Jiang P et al: High-resolution profiling of fetal DNA clearance from maternal plasma by massively parallel sequencing. Clin Chem 2013; 59: 1228–1237.
Poon LL, Leung TN, Lau TK, Chow KC, Lo YM : Differential DNA methylation between fetus and mother as a strategy for detecting fetal DNA in maternal plasma. Clin Chem 2002; 48: 35–41.
Chim SS, Tong YK, Chiu RW et al: Detection of the placental epigenetic signature of the maspin gene in maternal plasma. Proc Natl Acad Sci USA 2005; 102: 14753–14758.
Wong AI, Lo YM : Noninvasive fetal genomic, methylomic, and transcriptomic analyses using maternal plasma and clinical implications. Trends Mol Med 2015; 21: 98–108.
Hill M, Finning K, Martin P et al: Non-invasive prenatal determination of fetal sex: translating research into clinical practice. Clin Genet 2011; 80: 68–75.
Chitty LS, Finning K, Wade A et al: Diagnostic accuracy of routine antenatal determination of fetal RHD status across gestation: population based cohort study. BMJ 2014; 349: g5243.
Lench N, Barrett A, Fielding S et al: The clinical implementation of non-invasive prenatal diagnosis for single-gene disorders: challenges and progress made. Prenat Diagn 2013; 33: 555–562.
Chitty LS, Khalil A, Barrett AN, Pajkrt E, Griffin DR, Cole TJ : Safe, accurate, prenatal diagnosis of thanatophoric dysplasia using ultrasound and free fetal DNA. Prenat Diagn 2013; 33: 416–423.
Lam KW, Jiang P, Liao GJ et al: Noninvasive prenatal diagnosis of monogenic diseases by targeted massively parallel sequencing of maternal plasma: application to β-thalassemia. Clin Chem 2012; 58: 1467–1475.
Parks M, Court S, Cleary S et al: Non-invasive prenatal diagnosis of duchenne and Becker muscular dystrophies by relative haplotype dosage. Prenat Diagn 2016; 36: 312–320.
New MI, Tong YK, Yuen T et al: Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab 2014; 99: E1022–E1030.
Ma D, Ge H, Li X et al: Haplotype-based approach for noninvasive prenatal diagnosis of congenital adrenal hyperplasia by maternal plasma DNA sequencing. Gene 2014; 544: 252–258.
Meng M, Li X, Ge H et al: Noninvasive prenatal testing for autosomal recessive conditions by maternal plasma sequencing in a case of congenital deafness. Genet Med 2014; 16: 972–976.
The 1000 Genomes Project Consortium, Auton A The 1000 Genomes Project Consortium, Brooks LD The 1000 Genomes Project Consortium, Durbin RM et alThe 1000 Genomes Project Consortium: A global reference for human genetic variation. Nature 2015; 526: 68–74.
Delaneau O, Marchini J, Zagury JF : A linear complexity phasing method for thousands of genomes. Nat Methods 2012; 9: 179–181.
Delaneau O, Zagury JF, Marchini J : Improved whole chromosome phasing for disease and population genetic studies. Nat Methods 2013; 10: 5–6.
Robinson JT, Thorvaldsdóttir H, Winckler W et al: Integrative Genomics Viewer. Nature Biotech 2011; 29: 24–26.
Thorvaldsdóttir H, Robinson JT, Mesirov JP : Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013; 14: 178–192.
Li H, Durbin R : Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009; 25: 1754–1760.
Li H, Handsaker B, Wysoker A et al: The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 2009; 25: 2078–2079.
Hu X, Yuan J, Shi Y et al: pIRS: Profile-based Illumina pair-end reads simulator. Bioinformatics 2012; 28: 1533–1535.
Papasavva T, van Ijcken WF, Kockx CE et al: Next generation sequencing of SNPs for non-invasive prenatal diagnosis: challenges and feasibility as illustrated by an application to β-thalassaemia. Eur J Hum Genet 2013; 21: 1403–1410.
Barrett AN, McDonnell TC, Chan KC, Chitty LS : Digital PCR analysis of maternal plasma for noninvasive detection of sickle cell anemia. Clin Chem 2012; 58: 1026–1032.
Acknowledgements
This work was funded by Regione Autonoma della Sardegna. Ex Legge Regionale n. 11 (anno 2014) ‘Sostegno ricerca sulla β-Talassemia’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on European Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article

Cite this article
Saba, L., Masala, M., Capponi, V. et al. Non-invasive prenatal diagnosis of beta-thalassemia by semiconductor sequencing: a feasibility study in the sardinian population. Eur J Hum Genet 25, 600–607 (2017). https://doi.org/10.1038/ejhg.2017.26
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2017.26
This article is cited by
-
Cell therapy for the treatment of reproductive diseases and infertility: an overview from the mechanism to the clinic alongside diagnostic methods
Frontiers of Medicine (2022)
-
Noninvasive prenatal paternity determination using microhaplotypes: a pilot study
BMC Medical Genomics (2020)
-
The evolving role of genetic tests in reproductive medicine
Journal of Translational Medicine (2019)
