
Abstract
Purpose
To evaluate the effects of the duration of oral corticosteroid treatment on the recurrence of inflammation in Vogt-Koyanagi-Harada (VKH) disease.
Methods
Retrospective analysis of 35 VKH patients who received oral corticosteroid during the first attack of VKH with a minimum follow-up of 6 months. Patients were divided into two groups on the basis of the oral corticosteroid treatment duration of less than 6 months or 6 months or more. Kaplan–Meier survival and Cox-regression analyses were carried out to compare the recurrence rates of inflammation in the two groups.
Results
The mean age of onset was 42.5 years and the mean follow-up duration was 3.6 years. During the follow-up period, 10 (58.8%) of the 17 patients who received oral corticosteroid for less than 6 months compared with 2 (11.1%) of the 18 patients who had treatment for 6 months or more developed recurrence of inflammation (P=0.003). Cox-regression analysis showed that the duration of oral corticosteroid treatment for less than 6 months was the only significant risk factor for recurrence of VKH after adjustment for age, gender, and the initial dosage of oral corticosteroid treatment (adjusted odds ratio=8.8, P=0.008). Patients who received oral corticosteroid treatment for less than 6 months were also more likely to have one eye with visual acuity of 20/200 or worse (P=0.016).
Conclusions
Early withdrawal of oral corticosteroid is associated with increased risk of recurrence of VKH and worse visual prognosis. Oral corticosteroid should be tapered off slowly and maintained for at least 6 months for the treatment of acute VKH.
Similar content being viewed by others


Introduction
Vogt-Koyanagi-Harada (VKH) disease is a multisystem disorder characterized by bilateral granulomatous panuveitis. Clinical features of VKH include anterior uveitis, exudative retinal detachment, and depigmented fundal lesions, as well as the presence of neurological, auditory, or cutaneous manifestations.1, 2, 3 VKH is particularly common in certain ethnic groups with more darkly pigmented skin including Asians, Hispanics, and Indians.3, 4 It can be divided into various stages including prodromal, acute, chronic, and chronic-recurrent stages.2, 3 During the acute stage of VKH, treatment usually requires aggressive systemic corticosteroid therapy to prevent irreversible damage causing visual loss.5, 6, 7 However, recurrence of inflammation is not uncommon after cessation of corticosteroid therapy, especially when the steroid is tapered too rapidly.6 Therefore, in acute VKH, it has been recommended to maintain high dose oral corticosteroid therapy and taper it gradually over at least 3–6 months.3, 6 Prolonged duration of corticosteroid treatment is not without its side effects. On the other hand, recurrent or persistent ocular inflammation associated with chronic recurrent form of VKH may lead to irreversible visual loss with various long-term ocular complications including cataract, secondary glaucoma, optic atrophy, chronic pigmentary changes, and choroidal neovascularization.8, 9 In the literature, there has been no formal study to evaluate the effects of the duration of corticosteroid on the recurrence of inflammation in patients with VKH. In this study, we aim to analyse the effects of the duration of oral corticosteroid on the outcome of patients with acute VKH.
Patients and methods
This was a retrospective study of patients with a history of acute VKH disease who attended the medical retina and uveitis clinic at the Hong Kong Eye Hospital from January 2003 to July 2006. Patients’ presentation ranged from February 1995 to July 2006. The case notes of all patients who received oral corticosteroid treatment for acute VKH during their initial presentation periods were reviewed. A total of 40 patients were identified, and five patients with follow-up duration of less than 6 months, chronicity of uveitis of more than 4 months at presentation, previous penetrating eye injury or ocular operations, and those treated with intravenous corticosteroid or who did not receive oral steroid were excluded. The diagnosis and classification of VKH disease was established according to the criteria of the International Workshop of Vogt-Koyanagi-Harada Disease.10
Oral corticosteroid was given in the form of prednisolone, and the initial dose and tapering of corticosteroid was guided by the severity of ocular inflammation by the treating ophthalmologists. Patients were classified into two groups for analysis: (i) those who received initial oral corticosteroid treatment for less than six months and (ii) those who received oral corticosteroid for 6 months or more. Categorical variables between the two groups were compared using Fisher's exact test or χ2 test and continuous variables were compared using the non-parametric Mann–Whitney U-test. Kaplan–Meier survival analysis with log-rank test was performed to assess the recurrence of inflammation after cessation of oral corticosteroid during the follow-up period between the two duration groups. Recurrence was defined as relapse of inflammation after inactivity for 3 months or more without oral corticosteroid therapy.11 To adjust for potential confounding factors that might influence the recurrence rate, Cox-regression analysis was performed to adjust for the effects of age, gender, and the initial dosage of oral corticosteroids. Statistical analyses were carried out using SPSS 15.0, and a P-value of ⩽0.05 was considered as statistically significant.
Results
A total of 35 patients were included in the study and there were 18 (51.4%) male and 17 (48.6%) female patients. Thirty-three (94.3%) patients presented with bilateral ocular involvement, whereas two (5.7%) patients had unilateral presentation. The mean±SD age at the time of presentation was 42.5±18.4 (range, 10–72 years). The mean±SD duration of disease at presentation was 3.2±4.4 weeks (range, 1–16 weeks). The mean±SD duration of follow-up was 3.6±3.1 (range, 0.7–12.0 years).
Twenty-four (68.6%) of the 35 patients were classified as having incomplete VKH disease, whereas six (17.1%) and five (14.3%) patients had probable and complete VKH, respectively. The ocular findings at initial presentation are summarized in Table 1. The most common initial clinical signs among the patients included exudative retinal detachment (30 patients, 85.7%), anterior uveitis (28 patients, 80.0%), and optic disc swelling (21 patients, 60.0%). Five (14.3%) patients presented with sunset glow fundus with diffuse atrophy of the retinal pigment epithelium. Table 2 displays the extraocular manifestation of the patients. Headache (23 patients, 65.7%) and tinnitus (10 patients, 28.6%) were found to be the most common extraocular symptoms. In addition, six (17.1%) patients were found to have cutaneous manifestations including alopecia, poliosis, and vitiligo at the initial presentation.
For the 68 eyes affected in the 35 VKH patients, the median visual acuity at presentation was 20/50 (range, 20/20 to hand motion). Thirteen (19.1%) eyes had a visual acuity of 20/200 or worse at the initial presentation. At the last follow-up, the median visual acuity improved to 20/30 (range, 20/15 to hand motion) and the improvement was statistically significant (Wilcoxon sign-rank test, P<0.001). Figure 1 displays the baseline and final visual acuity of all eyes in the study. The final visual acuity was 20/200 or worse in nine (13.2%) eyes and the causes for poor vision included sunset glow fundus with diffuse RPE atrophy in eight eyes and glaucoma in four eyes. Four of the 13 eyes with baseline visual acuity of 20/200 or worse had vision of 20/200 or worse at the last follow-up.

Scatter plot of baseline and final visual acuity of 68 eyes of 35 patients with Vogt-Koyanagi-Harada disease.
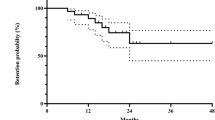
Eighteen (51.4%) of 35 patients received oral corticosteroid treatment for 6 months or more, whereas the remaining 17 (24.3%) patients received oral corticosteroid for less than 6 months. The mean initial dosage of oral prednisolone was 51.2 mg/day (range, 20–80 mg/day) and the mean total dosage of oral prednisolone given during the first 6 months was 3.68 g (range, 1.05–7.84 g). Twenty-three (65.7%) patients received oral corticosteroid of at least 1 mg/kg/day as the initial dose of treatment. The main reason for early discontinuation of oral corticosteroid treatment was apparent control of active intraocular inflammation in the patients. The baseline demographic data of patients receiving the two durations of corticosteroid treatment were shown in Table 3. Kaplan–Meier survival analysis of the recurrence of VKH disease is displayed in Figure 2. During the follow-up period, 10 (58.8%) of the 17 patients in the short duration group had recurrence of inflammation after cessation of corticosteroid therapy compared with only two (11.1%) of 18 patients in the long treatment duration group. The difference was statistically significant (log-rank test, P=0.003). Two patients in the long duration treatment group had recurrence of inflammation that required the use of second-line immunosuppressive therapy for steroid-sparing purpose, including cyclosporine A and azathioprine in one patient each. Cox-regression analysis showed that after adjustment for age, gender, and the initial dosage of oral corticosteroid treatment, short duration of oral corticosteroid treatment of less than 6 months was the only significant risk factor for recurrent inflammation in acute VKH (adjusted odds ratio=8.8, P=0.008).

Kaplan–Meier survival curve of recurrence of intraocular inflammation in Vogt-Koyanagi-Harada disease patients.
Tables 4 and 5 display the ocular complications of VKH disease among the patients during the follow-up period and at various time points, respectively. The most common ocular complications encountered during the follow-up period include sunset glow fundus (18 patients, 51.4%), RPE clumping (16 patients, 45.7%), and cataract (14 patients, 40.0%). Posterior synechiae (11 patients, 31.4%) and ocular hypertension (5 patients, 14.3%) were also among the common ocular complications. Eleven (31.4%) patients required cataract surgery during the follow-up period, whereas three (8.6%) patients had glaucoma surgery performed for secondary uveitic glaucoma. The rates of all ocular complications were similar in the short duration steroid treatment group compared with the long duration group (P>0.2). The proportion of patients with all types of ocular complications increased with longer duration of follow-up.
At the last follow-up, seven (41.2%) of 17 patients who received oral corticosteroid therapy of less than 6 months had one eye with visual acuity of 20/200 or worse compared with only one (5.6%) patient in the longer duration group (Fisher's exact test, P=0.016). Patients who developed cataract, ocular hypertension, optic atrophy, or choroidal neovascularization were more likely to have a visual acuity of 20/200 or worse in one eye compared with those without these complications (Fisher's exact test, P<0.001).
Discussion
The use of high dose systemic corticosteroid with gradual tapering for around 6 months has been recommended for the treatment of patients with acute VKH disease.3, 6 In cases with severe ocular inflammation associated with acute VKH, intravenous pulse steroid therapy has also been advocated.5, 12 A recent study by Read et al,7 however, showed no detectable difference in the visual outcome and complications in the route of corticosteroid administration, and therefore we preferred to use oral corticosteroid for the patients as they do not require hospitalization. The therapeutic effect of systemic corticosteroid lies mainly on its anti-inflammatory and immunosuppressant properties. Despite the recommendation of using a prolonged duration of oral corticosteroid therapy, there has been no formal study to compare the recurrence rate of inflammation in VKH disease with different duration of corticosteroid treatment. Long-term, high-dose systemic corticosteroid therapy can potentially result in systemic adverse effects, and therefore clinicians should balance the benefit of uveitis control and the risk of its side effects.
In this study, we evaluated the recurrence rate of inflammation in VKH patients who received two different durations of oral corticosteroid therapy during the acute phase of the disease. The baseline characteristics of the patients in both groups were similar in terms of age, duration of presentation, baseline visual acuity, length of follow-up, and initial dose of oral prednisolone. Our findings support the notion that oral corticosteroid should be continued for at least 6 months, as the recurrence rate of inflammation in patients who had oral corticosteroid of 6 months or more was significantly lower compared with those who received oral corticosteroid for less than 6 months. Moreover, we also found that patients who received oral corticosteroid for 6 months or more were also less likely to have one eye with a final visual acuity of 20/200 or worse compared with those who received treatment for less than 6 months. These findings suggest that systemic corticosteroid treatment should be tapered gradually and maintained for at least 6 months to prevent future recurrence and to optimize the visual outcome.
In terms of ocular complications, we did not find a significant difference in the complication rates between the two treatment duration groups. The most common sight-threatening complications encountered in the study were cataract, which developed in 40% of patients, followed by ocular hypertension in 14.3% of patients. The proportions of VKH patients who developed these ocular complications were comparable with other studies in the literature.3, 13, 14, 15, 16, 17 Yang et al13 showed that the duration of VKH at presentation is an important factor for the development of glaucoma and ocular hypertension. Chee et al14 also demonstrated that the use of high-dose systemic prednisolone within 2 weeks of initial presentation will result in significantly fewer ocular complications compared with those who received systemic steroid after 4 weeks. The results emphasized the importance of commencing high-dose systemic corticosteroid treatment in patients with VKH as early as possible after presentation.
The main limitation of this study was the retrospective nature of the study and only patients referred to the medical retina and uveitis clinic of our tertiary care hospital were included into the analysis. Therefore, selection bias may occur owing to the inclusion of VKH patients with more severe disease activity, as patients with a milder form of VKH might be excluded. Second, there was a lack of standardized treatment regime in the use of oral corticosteroid in the patients. Therefore, a large variation in the initial dose of oral corticosteroid existed among the patients, and some patients might not have received a sufficiently high dose of oral corticosteroid for the optimal treatment effects. Third, we have excluded patients who did not receive oral corticosteroid and those who presented with chronic VKH of more than 4 months, and the visual outcome in these patients might be different compared with those included in the study. Lastly, the duration of follow-up was variable in this retrospective study, and comparison of the visual outcome between the two steroid treatment groups might therefore introduce some bias to the analysis despite the two groups having a similar mean duration of follow-up. Nonetheless, the results of our study suggested that systemic corticosteroid treatment in VKH disease should be tapered off gradually and maintained for at least 6 months to minimize recurrence of inflammation and to optimize the visual outcome of the patients. Further prospective long-term study will be useful to determine the optimal treatment regime for treating VKH and to evaluate its effect in recurrence rates, ocular complications, and visual outcomes of patients.
References
Synder DA, Tessler HA . Vogt-Koyanagi-Harada syndrome. Am J Ophthalmol 1980; 90: 69–75.
Read RW . Vogt-Koyanagi-Harada disease. Ophthalmol Clin North Am 2002; 15: 333–341.
Moorthy RS, Inomata H, Rao NA . Vogt-Koyanagi-Harada syndrome. Surv Ophthalmol 1995; 39: 265–292.
Sugira S . Vogt-Koyanagi-Harada disease. Jpn J Ophthalmol 1978; 22: 9–35.
Samamoto Y, Ohno S, Matsuda H . Studies on corticosteroid therapy in Vogt-Koyanagi-Harada disease. Ophthalmologica 1990; 201: 162–167.
Rubsamen PE, Gass JD . Vogt-Koyanagi-Harada syndrome. Clinical course, therapy and long-term visual outcome. Arch Ophthalmol 1991; 109: 682–687.
Read RW, Yu F, Accorinti M, Bodaghi B, Chee SP, Fardeau C et al. Evaluation of the effect on outcomes of the route of administration of corticosteroids in acute Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2006; 142: 119–124.
Read RW, Rechodouni A, Butani N, Johnston R, LaBree LD, Smith RE et al. Complications and prognostic factors in Vogt-Koyanagi-Harada disease. Am J Ophthalmol 2001; 131: 599–606.
Bykhovskaya I, Thorne JE, Kempen JH, Dunn JP, Jabs DA . Vogt-Koyanagi-Harada disease: clinical outcomes. Am J Ophthalmol 2005; 140: 674–678.
Read RW, Holland GN, Rao NA, Tabbara KF, Ohno S, Arellanes-Garcia L et al. Revised diagnostic criteria for Vogt-Koyanagi-Harada disease: report of an international committee on nomenclature. Am J Ophthalmol 2001; 131: 647–652.
Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of uveitis nomenclature (SUN) working group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol 2005; 140: 509–516.
Yamanaka E, Ohguro S, Yamamoto S, Nakagawa Y, Imoto Y, Tano Y . Evaluation of pulse corticosteroid therapy for Vogt-Koyanagi-Harada disease assessed by optical coherence tomography. Am J Ophthalmol 2002; 132: 454–456.
Yang P, Ren Y, Li B, Fang W, Meng Q, Kijlstra A . Clinical characteristics of Vogt-Koyanagi-Harada syndrome in Chinese patients. Ophthalmology 2007; 114: 606–614.
Chee SP, Jap A, Bacsal K . Spectrum of Vogt-Koyanagi-Harada in Singapore. Int Ophthalmol 2007; 27: 137–142.
Khairallah M, Zaouali S, Messaoud R, Chaabane S, Attia S, Ben Yahia S et al. The spectrum of Vogt-Koyanagi-Harada disease in Tunisia, North Africa. Int Ophthalmol 2007; 27: 125–130.
Tugal-Tutkun I, Ozyazgan Y, Akova YA, Sullu Y, Akyol N, Soylu M et al. The spectrum of Vogt-Koyanagi-Harada in Turkey. Int Ophthalmol 2007; 27: 117–123.
Al-Kharashi AS, Aldibhi H, Al-Fraykh H, Kangave D, Abu El-Asrar AM . Prognostic factors in Vogt-Koyanagi-Harada disease. Int Ophthalmol 2007; 27: 201–210.
Author information
Authors and Affiliations
Corresponding author
Additional information
Presented in part as a poster at the 9th International Ocular Inflammation Society Conference in Paris, France, 17–20 September 2007Financial and proprietary interest: none
Rights and permissions
About this article
Cite this article
Lai, T., Chan, R., Chan, C. et al. Effects of the duration of initial oral corticosteroid treatment on the recurrence of inflammation in Vogt-Koyanagi-Harada disease. Eye 23, 543–548 (2009). https://doi.org/10.1038/eye.2008.89
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2008.89
Keywords
This article is cited by
-
A randomized non-inferiority trial of therapeutic strategy with immunosuppressants versus biologics for Vogt-Koyanagi-Harada disease
Nature Communications (2023)
-
Incidence and pre/post-treatment risk factors of glaucoma in Vogt-Koyanagi-Harada disease
International Ophthalmology (2023)
-
Outcomes of retinal pigment epithelial detachment in Vogt-Koyanagi-Harada disease: a longitudinal analysis
BMC Ophthalmology (2022)
-
Multicenter, retrospective, observational study for the Treatment Pattern of systemic corticoSTERoids for relapse of non-infectious uveitis accompanying Vogt-Koyanagi-Harada disease or sarcoidosis
Japanese Journal of Ophthalmology (2022)
-
Outcomes of patients with acute Vogt–Koyanagi–Harada disease treated with intravenous corticosteroid pulse followed by the slow tapering of oral corticosteroid therapy
International Ophthalmology (2022)
