
Abstract
Pressure overload enhances salt-induced sympathoexcitation through hypothalamic mineralocorticoid receptor (MR)-epithelial Na channel activation. Pressure overload also increases hypothalamic angiotensin type 1 receptors (AT1R). However, the role of AT1R in pressure overload-induced MR activation and salt-induced sympathoexcitation remains unknown. Therefore, the aim of the present study was to address this question. We performed aortic banding (AB) on mice from the Institute of Cancer Research. The expression of hypothalamic MR, serum/glucocorticoid-induced protein kinase-1 (SGK-1) and AT1R increased independently of plasma renin activity at 2 or 4 weeks after AB. Next, we performed AB in AT1aR-knockout (KO) mice and c57BL6/J wild-type (WT) mice. Sham-operated (Sham) mice were used as a control. Four weeks after AB (AB-KO or AB-WT), the expression of hypothalamic MR and SGK-1 increased in both AB-WT and AB-KO compared with Sham-WT and Sham-KO, respectively. The expression of AT1R was also greater in AB-WT than in Sham-WT. In addition, mice were fed a high-salt (8%) diet for an additional 4 weeks (ABH-KO and ABH-WT). High salt loading increased the urinary excretion of norepinephrine, a marker of sympathetic activity in ABH-WT, concomitant with hypothalamic MR activation, but not in ABH-KO. These results indicate that pressure overload activated hypothalamic MR independently of AT1R. After salt intake, however, AT1R was necessary to maintain hypothalamic MR activation and salt-induced sympathoexcitation.
Similar content being viewed by others


Introduction
High salt intake causes sympathoexcitation and blood pressure elevation, particularly in salt-sensitive models.1, 2 It is well known that the central nervous system has an important role in salt-induced sympathoexcitation and hypertension.3, 4, 5 Intracerebroventricular infusion of high-Na artificial CSF enhances sympathetic activity and increases blood pressure in both the salt-sensitive and salt-resistant models. The degree of increase, however, is greater in the salt-sensitive models than in the salt-resistant models.6 In addition, we confirmed that there was also a greater blood pressure elevation in response to high-Na intracarotid artery infusion in the salt-sensitive model than in the salt-resistant model.7 Therefore, an inappropriate response to high Na levels in the brain is suggested to be a key factor in salt-sensitive hypertension.
We recently reported that mice with pressure overload acquired salt sensitivity for sympathetic activity.8 In mice with pressure overload, but not in Sham mice, high salt intake causes sympathoexcitation. Intracerebroventricular infusion of high-Na artificial CSF causes sympathoexcitation both in mice with pressure overload and in Sham mice. However, the degree of induction of sympathetic nerve activation is greater in mice with pressure overload than in Sham mice. We also confirmed an increased Na concentration in the brains of mice with pressure overload after salt intake.8 These results suggest that in mice with pressure overload, the responses of sympathetic nerve activation to high Na levels in the brain is enhanced inappropriately. Furthermore, we confirmed that in mice with pressure overload, the hypothalamic mineralocorticoid receptor (MR)-epithelial Na channel (ENaC) pathway contributes to the acquisition of salt sensitivity for sympathetic activity.9 In this model, the expression levels of hypothalamic angiotensin type 1 receptors (AT1R) also increased. Brain AT1R is known to be involved in sympathoexcitation in animal models of hypertension and heart failure.10, 11, 12, 13 However, the role of hypothalamic AT1R in pressure overload-induced MR activation and salt-induced sympathoexcitation remains unknown. Therefore, the aim of the present study was to answer this question. To this end, we performed aortic banding (AB) to create a pressure overload model using Institute of Cancer Research (ICR) or AT1aR-knockout (KO) mice and measured the plasma renin activity (PRA), plasma aldosterone concentration (PAC), sympathetic activity by urinary norepinephrine (uNE) excretion in response to a high-salt diet, MR expression, serum/glucocorticoid-induced protein kinase-1 (SGK-1, a marker of MR activity) expression, ENaC expression and AT1R expression in the hypothalamus.
Methods
Animals
The study protocol was reviewed and approved by the Committee on Ethics of Animal Experiments, Kyushu University Graduate School of Medical Sciences, and conducted according to the Guidelines for Animal Experiments of Kyushu University and the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH publication No. 85-23, revised 1996). Ten-week-old male ICR mice (SLC, Hamamatsu, Japan), AT1aR KO mice (Jackson Laboratory, Bar Harbor, ME, USA; B6.129P2-Agtr1atm1Unc/J), and C57 black6/J (WT mice; SLC) were used.
Mouse pressure overload model preparation
The suprarenal abdominal aorta was banded in mice (AB mice) under sodium pentobarbital (25–35 mg kg−1 intraperitoneally anesthesia, and the depth of anesthesia was maintained by isoflurane inhalation (1.5–2.0%). The abdominal aorta was constricted at the suprarenal level with 5-0 silk sutures guided by a blunted 27-gauge needle, which was withdrawn as quickly as possible.10, 11 In the case of ICR mice, the mice were assigned 2 or 4 weeks later as AB2 or AB4 mice, and Sham mice served as control (Sham2 mice or Sham4 mice, respectively). In the case of the KO/WT mice, the mice were classified 4 weeks later as AB-KO mice or AB-WT mice, and Sham mice served as the control (Sham-KO or Sham-WT). Furthermore, the AB-KO mice, AB-WT mice and Sham-WT mice were fed with a high-salt (8% NaCl) diet for an additional 4 weeks (ABH-KO mice, ABH-WT mice, and ShamH-WT mice).
Evaluation of left ventricle systolic function
The left ventricle (LV) systolic function was evaluated by echocardiography. Serial M-mode echocardiography was performed on mice under light sodium pentobarbital anesthesia with spontaneous respiration. We used an echocardiography system (SSD5000; Aloka, Tokyo, Japan) with a dynamically focused 10-MHz linear array transducer. M-mode tracings were recorded from the short-axis view at the level of the papillary muscle. The LV end-diastolic diameter (LVDD), LV end-systolic diameter (LVSD) and LV wall thickness (LVWT) were measured. The LVWT was calculated as the mean thickness of the interventricular septum and the posterior LV wall. The per cent fractional shortening (%FS) was calculated as follows: %FS=(LVDD−LVSD)/LVDD × 100.
Measurement of PRA and PAC
To measure PRA and PAC, within minutes after the mice were injected with an overdose of sodium pentobarbital, a blood sample was collected from the right ventricle into a standard EDTA-containing syringe. The plasma obtained by centrifugation for 10 min at 6000 r.p.m. was used for the PRA and PAC measurements. PRA was measured by radioimmunoassay using a kit for animal samples (SRL Inc., Tokyo, Japan), and PAC was measured using an enzyme-linked immunosorbent assay.
Evaluation of sympathetic activity and confirmation of salt sensitivity
Sympathetic activity was evaluated by measuring 24-hour uNE excretion using high-performance liquid chromatography (SRL Inc.).8, 9
The AB2, AB4, Sham2 and Sham4 mice were fed a high-salt diet (8%) or a regular-salt diet for 5 days. Sympathetic activity in response to a 5-day high-salt diet was evaluated by 24-hour uNE excretion to confirm the acquired salt sensitivity.
Measurement of blood pressure and heart rate
Under isoflurane anesthesia (1.5–2.0%), a catheter (stretched PE50 tubing) was inserted into the right carotid artery. PE50 tubing was tunneled subcutaneously from the neck incision to the back of the neck. PE50 tubing was filled with heparinized saline.
After the animals recovered from anesthesia (almost 3 h after surgical procedure), their conscious-state blood pressure and heart rate were measured.
Measurement of organ weight
After completion of the experiments, the mice were killed with an overdose of sodium pentobarbital, and the heart and lungs were removed and weighed.
Western blot analysis
The animals were killed with an overdose of sodium pentobarbital, and the hypothalamus, including circumventricular tissues, was obtained. The tissues were homogenized in a lysing buffer containing 40 mmol l−1 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1% Triton X-100, 10% glycerol, 1 mmol l−1 sodium orthovanadate and 1 mmol l−1 phenylmethylsulfonyl fluoride. The protein concentration was determined using a bicinchoninic acid protein assay kit (Pierce Chemical Co., Rockford, IL, USA). A 15-μg aliquot of protein from each sample was separated on a polyacrylamide gel with 10% sodium dodecyl sulfate. The proteins were subsequently transferred onto polyvinylidene difluoride membranes (Immobilon-P membranes; Millipore, Billerica, MA, USA). The membranes were incubated in immunoreaction enhancer solution (Can Get Signal; Toyobo, Osaka, Japan) with a rabbit immunoglobulin G polyclonal antibody against MR (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA), a rabbit polyclonal antibody against SGK-1 (1:1000; Abcam, Cambridge, UK), a rabbit immunoglobulin G monoclonal antibody against AT1R (1:1000; Santa Cruz Biotechnology), a goat immunoglobulin G polyclonal antibody against αENaC, a rabbit polyclonal antibody against βENaC or a rabbit polyclonal antibody against γENaC (1:1000; Santa Cruz Biotechnology). The membranes were then incubated with a horseradish peroxidase-conjugated horse anti-rabbit or anti-goat immunoglobulin G antibody (1:10 000). Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control for the brain tissues. Immunoreactivity was detected by enhanced chemiluminescence autoradiography (ECL Western Blotting Detection Kit; Amersham Pharmacia Biotech, Uppsala, Sweden), and the positive bands on the film were analyzed using the public domain software NIH Image (developed at the US National Institutes of Health and available on the internet at http://rsb.info.nih.gov/nih-image/).
Statistical analysis
All values are expressed as the mean±s.e. An analysis of variance was used to compare uNE excretion, organ weight, LVDD, LVWT, %FS and protein expression levels between groups. Any two mean values were compared by application of the Bonferroni procedure. Differences were considered significant when the P-value was <0.05.
Results
Acquired salt sensitivity for sympathetic activity in AB mice
The sympathetic activity, as evaluated by uNE excretion in response to the 5-day high-salt diet, was increased in AB2 and AB4 mice but not in Sham2 and Sham4 mice (Figure 1). The sympathetic activity did not change in the groups without a high-salt diet.

Urinary norepinephrine excretion in response to a 5-day high-salt diet in each group. HS indicates high-salt diet (*P<0.05 vs. Sham2, Sham2+HS, and AB2, n=6–8, #P<0.05 vs. Sham4, Sham4+HS, and AB4, n=6 for each). Sham2, 2 weeks after sham operation; Sham4, 4 weeks after sham operation; AB2, 2 weeks after aortic banding; AB4, 4 weeks after aortic banding.
PRA and PAC in AB2 and AB4 mice
Neither PRA nor PAC in the AB2 and AB4 mice differed from those of the nontreated control mice (Figure 2a).

(a) PRA and PAC in AB2 and AB4 mice. Control indicates nontreated 10-week-old ICR mice. n=5 for each. PAC, plasma aldosterone concentration; PRA, plasma renin activity. (b) Representative western blots showing hypothalamic MR, SGK-1, αENaC and AT1R expression from each group. The graph shows the means for the quantification of four or five separate experiments. The data are the ratio relative to glyceraldehyde 3-phosphate dehydrogenase expression and are expressed as a ratio relative to Sham2 or Sham4, which were assigned a value of 1. (*P<0.05 vs. Sham2, #P<0.05 vs. Sham4.) AB2, 2 weeks after aortic banding; AB4, 4 weeks after aortic banding; Sham2, 2 weeks after sham operation; Sham4, 4 weeks after sham operation.
Blood pressure and heart rate in AB2 and AB4 mice
The mean blood pressure was greater in the AB mice than in the Sham mice both 2 and 4 weeks after AB. The mean blood pressure in the AB4 mice was greater than that in the AB2 mice (Table 1).
Expression levels of hypothalamic MR, SGK-1, ENaC and AT1R in AB2 and AB4 mice
The expression levels of hypothalamic MR, SGK-1, αENaC and AT1R were greater in the AB2 and AB4 mice than in the Sham2 and Sham4 mice (Figure 2b). The expressions of hypothalamic βENaC and γENaC did not differ between the groups.
Characteristics of AB-KO and AB-WT mice
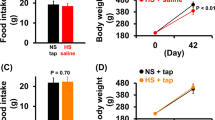
The body weight, relative heart weight and relative lung weight in the AB mice did not differ from those in the Sham mice (both WT and KO) (Figure 3a). Echocardiography revealed that LVWT was greater in the AB-WT and AB-KO mice than in the Sham-WT and Sham-KO mice. The %FS did not differ between the Sham mice and the AB mice (both WT and KO) (Figure 3b). Additionally, the sympathetic activity (measured by uNE excretion) did not differ between the Sham mice and the AB mice (both WT and KO) (Figure 3c).

(a) BW, heart weight/BW and lung weight/BW in each group (n=6 in WT, n=5 in KO, *P<0.05 vs. Sham-WT, #P<0.05 vs. Sham-KO). BW, body weight. (b) Cardiac function in each group. LVDD, left-ventricular end-diastolic diameter; LVWT, LV wall thickness; %FS, percent fractional shortening (n=5 for each, *P<0.05 vs. Sham-WT or KO, #P<0.05 vs. AB-WT or KO, $P<0.05 vs. ABH-WT). (c) Urinary norepinephrine excretion in each group (n=8 in WT, n=5 in KO, *P<0.05 vs. Sham-WT, #P<0.05 vs. others). AB, 4 weeks after aortic banding; ABH, AB mice with high salt diet for an additional 4 weeks; KO, AT1aR KO; Sham, sham-operated mice; WT, wild type c57black6/J mice.
Characteristics of ABH-KO and ABH-WT mice
The body weight and relative lung weight in the ABH mice were similar to those in the other groups in both the WT and KO mice. Compared with the AB and Sham mice, the relative heart weight in the ABH mice increased in both the WT and KO mice (Figure 3a). Echocardiography revealed that the LVWT was greater in the ABH-WT and ABH-KO mice than in the AB-WT and AB-KO mice, respectively. The %FS decreased in both the ABH-WT and ABH-KO mice compared with AB-WT and AB-KO mice, respectively. However, the extent to which %FS decreased was greater in the ABH-WT mice than in the ABH-KO mice (Figure 3b). In the WT mice, sympathetic activity (measured by uNE excretion) increased in the ABH mice compared with the AB or Sham mice but not the KO mice. Sympathetic activity in the ShamH-WT mice was higher than that in the sham-WT mice but lower than that in the ABH-WT mice (Figure 3c).
Blood pressure and heart rate in AB and ABH mice
The mean blood pressure increased in the AB-WT mice compared with that of the Sham-WT mice, and in the ABH-WT mice, the mean blood pressure was reduced to levels similar to that of the Sham-WT mice. The heart rate tended to increase in the AB-WT mice; a significant increase was observed in the ABH-WT mice compared with the Sham-WT mice (Table 1).
PAC in AB and ABH mice
In the WT mice, PAC significantly decreased in the ABH mice compared with the Sham or AB mice (Sham-WT, 443±79 pg ml−1; AB-WT, 410±68 pg ml−1; ABH-WT, 273±81 pg ml−1, n=5 for each). In the KO mice, PAC also decreased in the ABH mice compared with the Sham or AB mice (Sham-WT, 569±33 pg ml−1; AB-WT, 517±66 pg ml−1; ABH-WT, 340±67 pg ml−1, n=5 for each).
Expressions of hypothalamic MR, SGK-1, ENaC and AT1R
In the WT mice, the expression of hypothalamic MR, SGK-1 and αENaC were greater in the AB mice than in the Sham mice, and the expression of AT1R also increased in the AB mice compared with that in the Sham mice (Figure 4). In the KO mice, the expression levels of hypothalamic MR and SGK-1 also increased in the AB mice compared with that in the Sham mice (Figure 4). The extent to which the expression of SGK-1 increased in the WT mice, however, was greater than that of the KO mice (1.38±0.06 vs. 1.21±0.02, P<0.05, relative to Sham-WT or Sham-KO, respectively, which was assigned a value of 1). The enhanced expression levels of hypothalamic MR, SGK-1, αENaC and AT1R were maintained after high salt loading in the WT mice (ABH-WT). However, the expression of those proteins after high salt loading in KO mice (ABH-KO) returned to levels equal to that of the Sham mice (Figure 4). High salt loading did not alter the protein expression in WT mice (ShamH-WT) (MR, 0.96±0.09; SGK1, 0.98±0.08; αENaC, 0.94±0.11, N=3 for each, relative to Sham-WT, which was assigned a value of 1), and the expression levels of hypothalamic βENaC and γENaC did not differ among the groups.

Representative western blots showing hypothalamic MR, SGK-1, αENaC and AT1R expression from each group. The graph shows the means for the quantification of five separate experiments in WT mice and three separate experiments in KO mice. The data are the ratio relative to glyceraldehyde 3-phosphate dehydrogenase expression and are expressed as a ratio relative to Sham-WT or Sham-KO, which were assigned a value of 1. (*P<0.05 vs. Sham-WT, #P<0.05 vs. Sham-KO.) AB, 4 weeks after aortic banding; ABH, AB mice with high-salt diet for an additional 4 weeks; KO, AT1aR KO; Sham, sham-operated mice; WT, wild-type c57black6/J mice.
Discussion
Our findings indicate that mice with pressure overload experienced increased hypothalamic MR activity that was independent of AT1R, and, after salt intake, AT1R is necessary to maintain brain MR activation and salt-induced sympathoexcitation. Thus, we suggest new targets for the studies of the prevention of salt-induced sympathoexcitation in patients with pressure overload conditions, such as hypertensive heart disease.
We previously demonstrated that pressure overload activated the hypothalamic MR-ENaC pathway; thus, concomitant salt loading led to sympathoexcitation.8, 9 We also found that pressure overload increased hypothalamic AT1R, and blockage of the brain AT1R effectively prevented salt-induced sympathoecitation.8, 9 However, the relationship between AT1R and MR and the role of these receptors in salt-induced sympathoexcitation remained unclear. We induced pressure overload by AB at the suprarenal level. It is possible that activation of the circulating renin-angiotensin system due to a decrease in renal blood flow might have influenced our observations in this study, because circulating renin-angiotensin system is known to affect brain renin-angiotensin system through circumventricular organs.14 Therefore, we measured the PRA in mice with pressure overload. Both 2 and 4 weeks after AB, the PRA did not increase. In addition, we confirmed that the PAC in AB2 and AB4 mice was also at levels similar to that of the control group. On the other hand, the expressions of hypothalamic MR, SGK-1, αENaC and AT1R were greater in the AB2 and AB4 mice than in the Sham2 and Sham4 mice, respectively. These results indicate that activation of the hypothalamic MR pathway and increased AT1R do not depend on the PRA and PAC. Furthermore, we used AT1aR-KO mice in the present study. Pressure overload significantly increased hypothalamic MR and SGK-1 not only in WT mice but also in KO mice, although the degree of the pressure overload-induced increase in SGK-1 expression was greater in WT mice than in KO mice. These results indicate that the hypothalamic MR pathway is activated, at least in part, by pressure overload mechanisms that are independent of AT1aR.
Another important finding of the present study is that mice with pressure overload needed AT1aR to maintain hypothalamic MR activation after salt intake. The expression levels of hypothalamic MR, SGK-1, αENaC and AT1R were greater in the ABH-WT mice than in the Sham-WT mice, which is in agreement with sympathoexcitation. In contrast, the expression levels of hypothalamic MR, SGK-1 and αENaC in ABH-KO mice were at similar levels to those observed in the Sham-KO mice. Additionally, the sympathetic activity in the ABH-KO mice did not differ from that of the Sham-KO mice, suggesting that salt-induced sympathoexcitation in mice with pressure overload requires AT1aR.
We did not address the role of the AT1b receptor in AT1aR-KO mice in the present study. We previously confirmed that intracerebroventricular infusion of the AT1 receptor blocker failed to prevent pressure overload-induced hypothalamic MR enhancement.9 In addition, salt-induced sympathoexcitation was completely blocked in the ABH-KO mice in the present study, suggesting the requirement of AT1aR in sympathoexcitation. Expression of the brain AT1b receptor is reported to occur at similar levels in WT mice and AT1aR-KO mice.15 Based on these findings, we believe that AT1bR is not responsible for salt-induced sympathoexcitation.
The precise mechanism(s) involved in AT1R-independent hypothalamic MR activation remains unknown. Aldosterone is a major ligand of MR,16 and it has been suggested that aldosterone is synthesized locally in the brain.17, 18 Hypothalamic aldosterone synthesis is reported to increase by high salt loading in a salt-sensitive hypertensive model. In the present study, pressure overload-induced hypothalamic MR activation was caused before high salt loading; therefore, aldosterone-independent mechanisms might contribute to this mechanism. In the kidneys, small G protein Rac1 was demonstrated to activate the MR pathway in an aldosterone-independent manner.19 Therefore, it is possible that a similar mechanism might be responsible for the pressure overload-induced hypothalamic MR activation that is independent of aldosterone.
The AT1R-dependent mechanism(s) involved in hypothalamic MR activation and sympathoexcitation after high salt loading also remains unclear. In both WT and KO mice, the PAC in ABH mice decreased compared with that of the Sham or AB mice. This finding suggested that the hypothalamic MR activation was independent of systemic aldosterone. In mice with pressure overload, high salt loading is presumed to increase the Na levels in the CSF,8 which may increase brain aldosterone synthesis.18 CYP11B2, an aldosterone synthase, is activated by angiotensin II via AT1R.20 Therefore, in mice with pressure overload, high salt loading might lead to an increase in aldosterone synthesis via AT1R. Interestingly, the expression levels of hypothalamic MR-SGK-1-αENaC were lower in ABH-KO mice than in AB-KO mice but were at similar levels in Sham-KO mice. High salt loading is reported to decrease MR pathway activity.21 Therefore, in the AT1aR-absent condition, the MR pathway-inactivated mechanisms might have a significant role in ABH-KO mice. However, we did not address these issues in the present study, and further studies are warranted to clarify the mechanisms.
We failed to demonstrate the attenuation of pressure overload-induced LV hypertrophy in AB-KO and ABH-KO mice. However, it has been reported that pressure overload induced LV hypertrophy in AT1aR-KO mice.22 In the present study, we confirmed that aortic banding increased blood pressure in the WT mice. Although we did not measure blood pressure in the KO mice, the baseline blood pressure was ∼20 mm Hg lower in AT1aR-KO mice than in WT mice.22, 23, 24 However, the degree of blood pressure elevation due to aortic banding was similar to that observed in the WT mice,22 and we successfully demonstrated LV hypertrophy in AB-KO mice, which suggests that the impact of aortic banding on LV hypertrophy is similar between WT mice and KO mice. In the present study, we started to feed the mice with a high-salt diet 4 weeks after AB (ABH mice). LV function was more deteriorated in the ABH mice than in the AB mice (both KO and WT). The blood pressure in the ABH-WT mice decreased to levels similar to or lower than those observed in the Sham-WT mice, which was most likely due to LV dysfunction.8 The decrease in %FS was smaller in the ABH-KO mice than in the ABH-WT mice. The greater decreases in %FS in the ABH-WT mice than in the ABH-KO mice observed in the present study might have been caused by AT1R-mediated salt-induced sympathoexcitation.
In the present study, we evaluated three ENaC subunits (αENaC, βENaC and γENaC) and demonstrated that pressure overload increased the expression of hypothalamic αENaC. ENaC are reported to exist in the choroid plexus25 and neurons of the hypothalamus and supraoptic nucleus.26 In addition, a recent study reported that αENaC exists in both magnocellular neurons and parvocellular neurons, but βENaC and γENaC exist only in magnocellular neurons in the hypothalamus.26 In addition, we recently demonstrated that three ENaC subunits also exist in the choroid plexus and contribute to salt-sensitive hypertensive mechanisms in stroke-prone spontaneously hypertensive rats.7 As we performed western blotting using the hypothalamus including the circumventricular organs, we were not able to precisely determine the site of ENaC expression in the present study. Therefore, further studies are needed to clarify the site specificity and the pathophysiological significances of the enhanced expression of αENaC in mice with pressure overload.
Previously, in the case of ICR mice, we reported increased-αENaC expression in mice with pressure overload (AB mice). The expressions of γENaC, however, increased in mice with pressure overload after salt loading (ABH mice).9 The reasons for these discrepancies remain unclear. The genetic backgrounds of the response for salt loading differ between ICR mice and c57black/J mice. In fact, c57black/J mice are known as a salt-sensitive model.27 We also confirmed the salt-induced sympathoexcitation in ShamH-WT mice in the present study without changing the expression levels of hypothalamic MR-SGK-1-αENaC. Therefore, the differences in the genetic backgrounds between the two strains might affect the enhancement of the ENaC subunits in response to high salt loading.
In conclusion, the present findings strongly suggest that pressure overload activates hypothalamic MR independently of AT1R. After salt intake, however, AT1R is necessary to maintain hypothalamic MR activation and salt-induced sympathoexcitation.
References
Wang JM, Veerasingham SJ, Tan J, Leenen FH . Effects of high salt intake on brain AT1 receptor densities in Dahl rats. Am J Physiol Heart Circ Physiol 2003; 285: H1949–H1955.
Stocker SD, Madden CJ, Sved AF . Excess dietary salt intake alters the excitability of central sympathetic networks. Physiol Behav 2010; 100: 519–524.
Huang BS, Van Vliet BN, Leenen FH . Increases in CSF[Na+] precede the increases in blood pressure in Dahl S rats and SHR on high-salt diet. Am J Physiol Heart Circ Physiol 2004; 287: H1160–H1166.
Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TP, Van Huysse JW, Zhang J, Wier WG . How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 2012; 302: H1031–H1049.
Takahashi H, Yoshika M, Komiyama Y, Nishimura M . The central mechanism underlying hypertension: areview of the roles of sodium ions, epithelial sodium channels, the renin-angiotensin-aldosterone system, oxidative stress and endogenous digitalis in the brain. Hypertens Res 2011; 34: 1147–1160.
Huang BS, Wang H, Leenen FH . Enhanced sympathoexcitatory and pressor responses to central Na+ in Dahl salt-sensitive vs. -resistnat rats. Am J Physiol Heart Circ Physiol 2001; 281: H1881–H1889.
Nakano M, Hirooka Y, Matsukawa R, Ito K, Sunagawa K . Mineralocorticoid receptors/epithelial Na+ channels in the choroid plexus are involved in hypertensive mechanisms in stroke-prone spontaneously hypertensive rats. Hypertens Res 2013; 36: 277–284.
Ito K, Hirooka Y, Sunagawa K . Acquisition of brain Na sensitivity contributes to salt-induced sympathoexcitation and cardiac dysfunction in mice with pressure overload. Circ Res 2009; 104: 1004–1011.
Ito K, Hirooka Y, Sunagawa K . Blockade of mineralocorticoid receptors improves salt-induced left-ventricular systolic dysfunction through attenuation of enhanced sympathetic drive in mice with pressure overload. J Hypertens 2010; 28: 1449–1458.
Eshima K, Hirooka Y, Shigematsu H, Matsuo I, Koike G, Sakai K, Takeshita A . Angiotensin in the nucleus tractus solitarii contributes to neurogenic hypertension caused by chronic nitric oxide synthase inhibition. Hypertension 2000; 36: 259–263.
Shigemastu H, Hirooka Y, Eshima K, Shihara M, Tagawa T, Takeshita A . Endogenous angiotensin II in the NTS contributes to sympathetic activation in rats with aortocaval shunt. Am J Physiol Regul Integr Comp Physiol 2001; 280: R1665–R1673.
Allen AM . Role of angiotensin in the rostral ventrolateral medulla in the development and maintenance of hypertension. Curr Opin Pharmacol 2011; 11: 117–123.
Wang WZ, Gao L, Wang HJ, Zucker IH, Wang W . Interaction between cardiac sympathetic afferent reflex and chemoreflex is mediated by the NTS AT1 receptors in heart failure. Am J Physiol Heart Circ Physiol 2008; 295: H1216–H1226.
Hendel MD, Collister JP . Contribution of the subfornical organ to angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol 2005; 288: H680–H685.
Rocha MJ, Chen Y, Oliveira GR, Morris M . Physiological regulation of brain angiotensin receptor mRNA in AT1a deficient mice. Exp Neurol 2005; 195: 229–235.
Nagai Y, Miyata K, Sun GP, Rahman M, Kimura S, Miyatake A, Kiyomoto H, Kohno M, Abe Y, Yoshizumi M, Nishiyama A . Aldosterone stimulates collagen gene expression and synthesis via activation of ERK1/2 in rat renal fibroblasts. Hypertension 2005; 46: 1039–1045.
Gomez-Sanchez EP, Gomez-Sanches CM, Plonczynski M, Gomez-Sanchez CE . Aldosterone synthesis in the brain contributes to Dahl salt-sensitive rat hypertension. Exp Physiol 2009; 95: 120–130.
Huang BS, White RA, Ahmad M, Jeng AY, Leenen FH . Central infusion of aldosterone synthase inhibitor prevents sympathetic hyperactivity and hypertension by central Na+ in Wister rats. Am J Physiol 2008; 295: R166–R172.
Shibata S, Nagase M, Yoshida S, Kawarazaki W, Kurihara H, Tanaka H, Miyoshi J, Takai Y, Fujita T . Modification of mineralocorticoid receptor function by Rac1 GTPase: implication in proteinuric kidney. Nat Med 2008; 14: 1370–1376.
Lai L, Chen J, Hao CM, Lin S, Gu Y . Aldosterone promotes fibronectin production through a Smad2-dependent TGF-β1 pathway in mesangial cells. Biochem Biophys Res Commun 2006; 348: 70–75.
Lienhard D, Lauterburg M, Escher G, Frey FJ, Frey BM . High salt intake down-regulates colonic mineralocorticoid receptors, epithelial sodium channels and 11 β-Hydroxysteroid dehydrogenase type 2. PLoS One 2012; 7: e37898.
Harada K, Komuro I, Shiojima I, Hayashi D, Kudoh S, Mizuno T, Kijima K, Matsubara H, Sugaya T, Murakami K, Yazaki Y . Pressure overload induces cardiac hypertrophy in angiotensin II type 1A receptor knockout mice. Circulation 1998; 97: 1952–1959.
Chen D, Greca LL, Head GA, Walther T, Mayorov DN . Blood pressure reactivity to emotional stress is reduced in AT1A-receptor knockout mice on normal, but not high salt intake. Hypertens Res 2009; 32: 559–564.
Wichi RB, Farah V, Chen Y, Irigoyen MC, Morris M . Deficiency in angiotensin AT1a receptors prevents diabetes-induced hypertension. Am J Physiol Regul Integr Comp Physiol 2007; 292: R1184–R1189.
Amin MS, Reza E, Wang H, Leenen FH . Sodium transport in the choroid plexus and salt-sensitive hypertension. Hypertension 2009; 54: 860–867.
Teruyama R, Sakuraba M, Wilson LL, Wandrey NEJ, Armstrong WE . Epithelial Na+ sodium channels in magnocellular cells of the rat supraoptic and paraventricular nuclei. Am J Physiol Endocrinol Metab 2012; 302: E273–E285.
Escano CS, Armando I, Wang X, Asico LD, Pascua A, Yang Y, Wang Z, Lau YS, Jose PA . Renal dopaminergic defect in C57Bl/6J mice. Am J Physiol Regul Integr Comp Physiol 2009; 297: R1660–R1669.
Acknowledgements
We express our sincere thanks to Naomi Shirouzu for help with the western blot analysis. This work was supported by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion Science (21790730, 24390198).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Ito, K., Hirooka, Y., Nakano, M. et al. Role of hypothalamic angiotensin type 1 receptors in pressure overload-induced mineralocorticoid receptor activation and salt-induced sympathoexcitation. Hypertens Res 36, 513–519 (2013). https://doi.org/10.1038/hr.2012.221
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hr.2012.221
Keywords
This article is cited by
-
Review of the health benefits of habitual consumption of miso soup: focus on the effects on sympathetic nerve activity, blood pressure, and heart rate
Environmental Health and Preventive Medicine (2020)
