
Abstract
Xeroderma pigmentosum (XP, OMIM 278700–278780) is a group of autosomal recessive diseases characterized by hypersensitivity to UV rays. There are seven complementation groups of XP (XPA to XPG) and XPV. Among them, the XP group C (XP-C) is the most prevalent type in Western Europe and in the United States. We report here on the clinical and genetic investigation of XP-C patients in 14 Tunisian families. As the XPC V548A fs X572 mutation has been identified in Algerian and Moroccan populations, Tunisian patients were first screened for this mutation by a direct sequencing of exon 9 of the XPC gene. All patients with a severe clinical form had this mutation, thus showing the homogeneity of the mutational spectrum of XPC in Tunisia. A potential founder effect was searched and confirmed by haplotype analysis. Taking into account the similarity of the genetic background, we propose a direct screening of this mutation as a rapid and cost-effective tool for the diagnosis of XP-C in North Africa.
Similar content being viewed by others
Introduction
Xeroderma pigmentosum (XP) is a rare genodermatosis, characterized by an extremely high sun sensitivity of the skin, by ocular abnormalities and by a high incidence of skin tumors.1 XP patients have been identified throughout the world in all ethnic groups.2 This disease is rare worldwide, but is relatively frequent in Tunisia, with an estimated prevalence of 1 patient per 10 000.3 This might be explained by the high rate of consanguineous marriage and endogamy.
XP is genetically heterogeneous: eight genes (XPA to XPG and XPV) have been identified so far. The XP complementation groups differ in frequency and geographical distribution. Among its seven complementation groups, the XP group C (XP-C) is the most prevalent type in Western Europe and in the United States.4 The XPC protein is involved in the first step of the nucleotide excision repair (NER); it is important in the recognition of target lesions and in the recruitment of the incision complex.5 This human protein forms a strong heterotrimeric complex with Rad23B and centrin 2, and is involved in the recognition of UV ray-induced DNA lesions during global genome repair but not during transcription-coupled repair.6, 7, 8 In vitro experiments have shown that the XPC complex might contribute toward cancer prevention by participating in the base excision repair of 8-OH-Gua and other oxidative DNA lesions.9
Among the studied populations, no hot-spot mutation in the XPC gene was identified. The V548A fsX572 mutation was previously described in three unrelated families from Italy, Algeria and Morocco, and also with a compound heterozygous XPC mutation in Honduras and USA patients.10, 11, 12, 13
To elucidate the spectrum of XPC gene mutations, 20 Tunisian patients with an XP severe clinical form were analyzed. We report here the relatively homogeneous mutation spectrum of the XPC gene in Tunisian patients and discuss its implication for molecular diagnosis of XP-C in North Africa.
Materials and methods
Patients
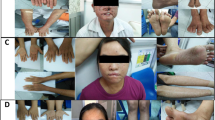
Twenty XP-C patients (9 male and 11 female individuals) and their family members were examined at the Tunisian reference XP department (Dermatology Department of the Habib Thameur Hospital in Tunis). Classification of these patients as XPC was on the basis of clinical features, such as early onset of the disease, which could start at the first month of birth, and severity. Chronic photophobia is the first sign, followed by photosensitivity marked by persistent erythema, pigmentosum macula and skin malignancy developed before 10 years of age. The diagnosis of skin cancer, suspected during clinical monitoring, was confirmed by dermoscopical and histological investigation.
Molecular investigation
After signing an informed consent form, all families were interviewed using a structured questionnaire, including a pedigree drawing, to collect information about consanguinity, affected members, associated diseases and family history. DNA was extracted from peripheral blood leucocytes by the classical salting-out method.14 To screen for the V548A fsX572 XPC mutation, a region of 609 bp length of exon 9 of the XPC gene was amplified from genomic DNA using the primers F: 5′-CCAGGGTGTCTTATAAAGAGG-3′ and R: 5′-CAAGGCCTTACCTCCAAG-3′ (3′-UTR). The PCR amplification reaction was carried out in a volume of 25 μl containing 10 ng of DNA, 10 × PCR buffer, 0.2 mM of each dNTP, 1.5 mM MgCl2, 0.5 μM of one of each primer and 0.5 U Amplitaq DNA polymerase (Invitrogen, Foster City, CA, USA). DNA was denatured for 5 min at 96°C, followed by 40 cycles of denaturation at 94°C for 30 s, annealing at 55°C for 35 s, elongation at 72°C for 40 s and terminal elongation for 10 min at 72°C. The PCR products were purified by using the QIA quick gel extraction purification kit (Qiagen, Hilden, Germany) and sequenced with the Big Dye terminator kit (Applied Biosystems, Foster City, CA, USA) using one of the PCR primers on an ABI prism 3130 DNA sequencer (Applied Biosystems) in accordance with the manufacturer's recommendations.
To seek for a potential founder effect, available family members were genotyped using three microsatellite markers spanning a 1.4 Mb interval flanking the XPC locus (cen-D3S3602, D3S1585 (XPC) and D3S3542-tel). Microsatellite markers were selected from the genetic maps available on NCBI (http://www.ncbi.nih.gov/) and Ensembl (http://www.ensembl.org/) genome browsers on the basis of their heterozygosity percentage and closeness to the XPC gene. Genotyping was performed as described elsewhere.15
Results
In this study, we report on 20 XPC cases belonging to 14 families who originated from various geographical locations in Tunisia, mainly from coastal regions. Twelve of the 14 families (85%) were consanguineous and the most common form is first-cousin marriages (57%). The average age of the patients is 11 years, with an allocation from 2 to 24 years. Clinical features are summarized in Table 1. For patients who were referred to our reference XP department before the age of 1 year, the diagnosis was based on the presence of the specific pigmentosum macula (association between achromia spots and badly limited multiple pigmented macula). No patient had neurological abnormalities.
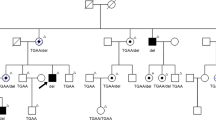
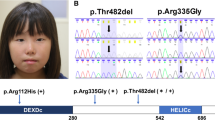
Mutation screening showed that all studied patients presented the homozygous deletion of two bases TG in exon 9 at position 1744–1745 (NC_000003.10). This deletion leads to a frameshift mutation and a premature termination of the encoded protein, V548A fsX572 XPC (Figure 1). To investigate whether a common ancestral chromosome accounted for the recurrence of the identified mutation, patients and their family, whenever available, were genotyped with three selected microsatellite markers flanking the XPC gene, and the mutation-associated haplotypes were constructed by visual inspection. Six XP-C patients from four unrelated families were homozygous for the same haplotype, thus suggesting a founder effect (Figure 2).

Genomic DNA sequence showing the TG deletion V548A fsX572 in exon 9 of the XPC gene in a homozygous state (a) or in a heterozygous parent (b), which is compared with the wild-type sequence of an unrelated control (c).

Pedigrees and haplotype analysis for four investigated XP-C Tunisian families, with microsatellite markers flanking the XPC gene. The disease haplotype is indicated by shading. m, mutant allele; N, normal allele; nd, not determined.
Discussion
XP is among the most severe and even life-threatening genodermatosis. It is a highly heterogeneous disease at the clinical and genetic level. In Tunisia, a preliminary investigation showed that approximately 40% of collected cases had a severe clinical form and they are suspected to be XP-C.3 We reported here on the clinical and mutational investigation of XP-C in Tunisian patients to introduce carrier detection, genetic counseling and prenatal diagnosis.
Clinical investigation showed that all studied patients correspond to a severe clinical form. Ninety percent of them developed pre-cancerous lesions or cancer: basal-cell carcinoma (BCC) (70%, the average age of onset of the first BCC is 8.2 years) or squamous-cell carcinoma (SCC) (55%, the average age of onset of the first SCC is 7.3 years), and some patients developed several melanomas (MM) (20%, the average age of onset of the first MM is 11.6 years). The severity of this disease among Tunisian patients is similar to that described previously in North African, Italian, Turkish and Ashkenazi–Jewish Israeli patients.16, 17, 18
The V548A fs X572 mutation affects the C-ter of the XPC protein residues 492–940. These residues had critical interactions with DNA, RAD23B, CETN2 and TFIIH. The XPC–RAD23B–CETN2 complex participates in the first fundamental step of GG-NER, DNA damage recognition, and sets the stage for the following two steps: DNA damage excision and grap-filling synthesis and ligation.19 Therefore, it is expected that the XPC V548A fs X572 mutation reduces the ligand-binding capacity, explaining the severe phenotype in the affected patients.
As previous studies of XP-C showed that Moroccan, Algerian and Italian patients had the V548A fsX572 XPC mutation,10, 11, 12, 13, 18 patients were screened for this mutation. All investigated patients bear this mutation. The identification of the prevalent mutation opened the way for molecular diagnosis. Indeed, premarital counseling was proposed to one couple (family XPC30MO) and the results showed that the father was heterozygous for the mutation, whereas the mother did not carry this mutation (homozygous normal). Consequently, this couple has the same risk of having an affected child as the general population. To identify a potential founder effect, haplotype analysis was performed. All available patients from unrelated families shared the same haplotype, thus suggesting a common ancestor. A similar figure should be found in other North African countries as they share a common ethnic background and consequently the same mutational spectrum as already shown for other genodermatosis.20, 21
In our study, clinical similarity correlates with genetic homogeneity; in fact, all patients with a severe clinical form of XP bear the V548A fsX572 XPC mutation. The recurrence of this mutation will greatly simplify the molecular diagnosis of XPC to propose genetic counseling, prenatal diagnosis and to allow an early management of affected children.
References
Kraemer, K. H., Patronas, N. J., Schiffmann, R., Brooks, B. P., Tamura, D. & DiGiovanna, J. J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype–phenotype relationship. Neuroscience 4, 1388–1396 (2007).
Stary, A. & Sarasin, A. The genetics of the hereditary xeroderma pigmentosum syndrome. Biochimie 84, 49–60 (2002).
Zghal, M., Fazaa, B. & Kamoun, M. R. Xeroderma pigmentosum. EMC (Elsevier SAS, Paris) Dermatologie 10, 98–660 (2006).
Kleijer, W. J., Laugelc, V., Berneburgd, M., Nardoe, T., Fawcett, H., Gratchev, A. et al. DNA Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair 5, 983–989 (2008).
Venema, J., van Hoffen, A., Karcagi, V., Natarajan, A. T., van Zeeland, A. A. & Mullenders, L. H. Xeroderma pigmentosum complementation group C cells remove pyrimidine dimmers selectively from the transcribed strand of active genes. Mol. Cell Biol. 11, 4128–4134 (1991).
Sugasawa, K., Ng, J. M., Masutani, C., Iwai, S., van der Spek, P. J., Eker, A. P. et al. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol. Cell 2, 223–232 (1998).
Araki, M., Masutani, C., Takemura, M., Uchida, A., Sugasawa, K., Kondoh, J. et al. Centrosome protein centrin 2/caltractin 1 is part of the xeroderma pigmentosum group C complex that initiates global genome nucleotide excision repair. J Biol Chem 276, 18665–18672 (2001).
Maillard, O., Solyom, S. & Naegeli, H. An aromatic sensor with aversion to damaged strands confers versatility to DNA repair. PLoS Biol. 5, 717–728 (2007).
D'Errico, M., Parlanti, E., Teson, M., de Jesus, B. M., Degan, P., Calcagnile, A. et al. New functions of XPC in the protection of human skin cells from oxidative damage. EMBO J. 25, 4305–4315 (2006).
Khan, S. G., Oh, K. S., Shahlavi, T., Ueda, T., Busch, D. B., Inui, H. et al. Reduced XPC DNA repair gene mRNA levels in clinically normal parents of xeroderma pigmentosum patients. Carcinogenesis 27, 84–94 (2006).
Chavanne, F., Broughton, B. C., Pietra, D., Nardo, T., Browitt, A., Lehmann, A. R. et al. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell, protein, and transcript levels. Cancer Res. 60, 1974–1982 (2000).
Li, L., Bales, E. S., Peterson, C. A. & Legerski, R. J. Characterization of molecular defect in xeroderma pigmentosum group C. Nat. Genet. 5, 413–417 (1993).
Daya-Grosjean, L., James, M., Drougard, C. & Sarasin, A. An immortalized xeroderma pigmentosum, group C, cell line which replicates SV40 shuttle vectors. Mutat. Res. 183, 185–196 (1987).
Miller, S. A., Dykes, D. D. & Polesky, H. F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 3, 1215 (1988).
Bouchlaka, C., Abdelhak, S., Amouri, A., Ben Abid, H., Hadiji, S., Frikha, M. et al. High prevalence of group A and identification of new FANCA mutations. J. Hum. Genet. 48, 352–361 (2003).
Gozukara, E. M., Khan, S. G., Metin, A., Emmert, S., Busch, D. B., Shahlavi, T. et al. A stop codon in xeroderma pigmentosum group C families in Turkey and Italy: molecular genetic evidence for a common ancestor. J. Invest. Dermatol. 117, 197–204 (2001).
Slor, H., Batko, S., Khan, S. G., Sobe, T., Emmert, S., Khadavi, A. et al. Clinical, cellular, and molecular features of an Israeli Xeroderma pigmentosum family with a frameshift mutation in the XPC gene: sun protection prolongs life. J. Invest. Dermatol. 115, 974–980 (2000).
Mahindra, P., DiGiovanna, J. J., Tamura, D., Brahim, J. S., Hornyak, T. J., Stern, J. B. et al. Skin cancers, blindness, and anterior tongue mass in African brothers. J. Am. Acad. Dermatol. 5, 881–886 (2008).
Bunick, C. G., Miller, M. R., Fuller, B. E., Fanning, E. & Chazin, W. J. Biochemical and structural domain analysis of xeroderma pigmentosum complementation group C protein. Biochemistry 50, 14965–14979 (2006).
Charfeddine, C., Mokni, M., Ben Mousli, R., Elkares, R., Bouchlaka, C., Boubaker, S. et al. A novel missense mutation in the gene encoding SLURP-1 in patients with Mal de Meleda from northern Tunisia. Br. J. Dermatol. 149, 1108–1115 (2003).
Lefévre, C., Audebert, S., Jobard, F., Bouadjar, B., Lakhdar, H. & Boughdene-Stambouli, O. et al. Mutations in the transporter ABCA12 are associated with lamellar ichthyosis type 2. Hum. Mol. Genet. 18, 2369–2378 (2003).
Acknowledgements
We thank the patients and their families for their collaboration and understanding. This work was supported by the Tunisian Ministry of Higher Education and Scientific Research and the Tunisian Ministry of Health (Research Unit on ‘Molecular Investigation of Genetic Orphan Diseases’ UR 04/SP03).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ben Rekaya, M., Messaoud, O., Talmoudi, F. et al. High frequency of the V548A fs X572 XPC mutation in Tunisia: implication for molecular diagnosis. J Hum Genet 54, 426–429 (2009). https://doi.org/10.1038/jhg.2009.50
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2009.50
Keywords
This article is cited by
-
Xeroderma pigmentosum and acute myeloid leukemia: a case report
Journal of Medical Case Reports (2021)
-
A novel frameshift mutation in the XPC gene in a Moroccan patient: a case report
Journal of Medical Case Reports (2017)
-
Genetic investigation of XPA gene: high frequency of the c.682C>T mutation in Moroccan XP patients with moderate clinical profile
BMC Research Notes (2017)
-
c.1643_1644delTG XPC mutation is more frequent in Moroccan patients with xeroderma pigmentosum
Archives of Dermatological Research (2013)
-
Founder mutations in Tunisia: implications for diagnosis in North Africa and Middle East
Orphanet Journal of Rare Diseases (2012)
