
Abstract
MicroRNAs (miRNAs) negatively regulate target gene expression by binding to 3′-untranslated region of target mRNA. Single-nucleotide polymorphisms (SNPs) that reside in the miRNA target sites can affect the bindings of miRNAs to mRNAs. Compelling evidence has shown that miRNAs contribute to the etiology of type 2 diabetes (T2DM). We hypothesized that SNPs in diabetes-related miRNA target-binding sites could be associated with the risk of T2DM. We selected 10 SNPs on miRNA-binding sites by using bioinformatics software. Genotypes of T2DM patients (n=1017) and normal controls (n=1059) were analyzed by TaqMan assay. The variant genotypes rs1366600CC and TC/CC in the insulin receptor (INSR) gene, rs2292899GA in the acyl-CoA synthetase 1 (ACSL1) gene and rs11724758AA in the fatty-acid-binding protein 2 (FABP2) gene were associated with T2DM (adjusted odds ratios (ORs) (95% confidence intervals)=2.03 (1.02–4.01), 1.28 (1.04–1.57), 1.22 (1.004–1.49) and 0.76 (0.58–0.997), respectively). The analysis stratified by age, gender, waist circumstance and living habits also revealed these genotypes’ effect. Furthermore, crossover analysis indicated those who exposed to both environmental factor and putative risk genotypes did have the highest risk of T2DM. A cumulative effect of SNPs rs1366600, rs2292899 and rs11724758 was observed with individuals carrying 2, 3 and 4–6 risk alleles having a gradually increased risk of T2DM (OR=1.52, 1.81 and 2.28, P for trend <0.001). Our result suggested that INSR rs1366600, ACSL1 rs2292899 and FABP2 rs11724758 could influence the susceptibility to T2DM in Chinese Han population, most likely through their effects on the specific miRNA-binding sites and functional characterizations of three genes are needed.
Similar content being viewed by others
Introduction
Diabetes mellitus is the most common metabolic disorder worldwide. In developed countries, diabetes is already the leading cause of blindness, renal failure and lower limb amputations, and is a major risk factor for cardiovascular disease and stroke.1 The prevalence of diabetes has grown rapidly during the past 10 years in China and reached 9.7%. It is currently affecting 92.4 million adults in our country, including type 2 diabetes (T2DM) accounting for 90% of cases.2 T2DM is caused by defects in insulin action and insulin production. There are strong evidences that an adverse genetic background together with environmental factors could impair insulin sensitivity in target tissues and insulin secretion from pancreatic β-cells.1
The recent breakthrough discovery of a new family of naturally endogenous, small (∼22 nucleotides), non-coding but functional RNAs have been proven to have significant roles in a wide spectrum of diseases including T2DM.3 Considerable data suggest that miRNAs has a direct role in insulin secretion and production, pancreatic islet development, insulin action and indirectly control glucose and lipid metabolism.4 Generally, miRNA exerts its function through binding to mRNA 3′-untranslated region (3′-UTR) and negatively regulating gene expression by cleavage or translational repression of mRNA.3 Single-nucleotide polymorphisms (SNPs) that reside in the microRNA (miRNA) target site can affect the binding of miRNA to mRNA, which can either create illegitimate-binding sites or abolish existing-binding sites.5 Recent studies have indicated that polymorphisms in miRNA target sites can affect gene and protein expression and lead to influence the risk of certain human diseases, including Tourette’s syndrome and cancers.6, 7, 8 However, gene polymorphisms in miRNA-binding sites have not been reported to be associated with susceptibility of T2DM. In the present case–control study, we investigated the effect of 10 potentially functional polymorphisms within the eight diabetes-related miRNAs target sites on T2DM risk.
Materials and methods
Subjects
This study included 1017 T2DM patients and 1059 T2DM-free controls. All subjects were biologically unrelated ethnic Han Chinese from Nanjing City and surrounding regions in Jiangsu Province, China. The patients were consecutively recruited between March 2008 and August 2010 from the diabetes inpatient or outpatient clinic of three affiliated hospitals of Nanjing Medical University (The Affiliated Changzhou Second Hospital of NJMU, the Third Affiliated Hospital of NJMU and the Affiliated Nanjing First Hospital of NJMU) without the restrictions of age and sex. A diagnosis of T2DM required either a fasting plasma glucose ⩾7.0 mmol l−1 (126 mg dl−1) or a 2-h glucose of ⩾11.1 mmol l−1 (200 mg dl−1) after an oral glucose tolerance test. All the patients were tested by glutamic acid decarboxylase autoantibodies (GAD) and islet cell antibodies (ICA512) (performed by the clinical laboratories in the three affiliated hospitals) to exclude patients with type 1 diabetes. T2DM-free controls were randomly selected from the physical examination center within the same geographical area and the period when the cases were recruited. Controls were non-diabetic as determined by an oral glucose tolerance test, which was performed according to World Health Organization criteria. These control subjects were frequency-matched to the cases on the basis of age (±5 years), sex and residential area. After informed consent was obtained, each subject was personally interviewed by trained interviewers using a standard questionnaire to collect information on demographic data and environmental exposure history, including tobacco smoking and drinking. Those who reporting at least one cigarette per day were defined as current cigarette smokers, otherwise they were considered as non-smoker. Total alcohol intake was expressed as the sum of millimeters of alcohol per week from wine, beer, cider and spirits. An approximately 5 ml venous blood sample was collected from each subject after the interview.
Measurements
Weight, height and waist circumstance (WC) were measured by trained personnel and body mass index (in kg m−2) was calculated. Blood pressure was measured on the right arm after a 10-min rest using a standard sphygmomanometer of appropriate cuff size. After an overnight fast, venous blood samples were drawn and promptly centrifuged, and the serum was stored at −20 °C. All samples were run in the same assay. Blood sugar was measured in the laboratories in the three affiliated hospitals of NJMU with the glucose oxidase method. Total cholesterol, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol and triglycerides were determined in the three affiliated hospitals by an enzymatic colorimetric method (Au5400, Olympus, Tokyo, Japan). DNA was extracted from peripheral blood by the use of proteinase K and phenol/chloroform. This study was approved by the Research Ethics Committee of Nanjing Medical University.
SNP selection
We first obtained 21 diabetes-related miRNAs (miR-375,9, 10 miR-96,11 miR-9,12 miR-30d,10, 13 miR-34a,10, 14 miR-146a,10 miR-126,15 miR-let-7b,16 miR-7,17 miR-21,15 miR-29a,10, 18 miR-143,19 miR-99a,20 miR-132,20 miR-24,15 miR-20b,15 miR-130a,21 miR-410,21 miR-200a,21 miR-124a,10, 11, 22 miR-17-5P20) through an extensive mining of bioinformatics according to previously reported literatures and miRBase database (http://www.mirbase.org/search.shtml). The Patrocles database (http://www.patrocles.org/Patrocles_targets.htm)23 and PolymiRTS database (http://compbio.uthsc.edu/miRSNP/)24 were first used to predict and identify potentially functional polymorphisms within miRNA-binding sites. Then we used the TargetScan25 and miRanda26 algorithms to further identify putative miRNA-binding sites within the 3′-UTR of each candidate gene and used the ensemble genome browser 64 (http://asia.ensembl.org/index.html) to search for the SNPs in the miRNA-binding seed region.The SNPs in miRNA target sites that appeared in at least two of above four prediction algorithm were selected. All SNPs were filtered by using the criteria that minor allele frequency of ⩾0.05 in the Chinese Han population identified from the National Center for Biotechnology Information dbSNPs database (http://www.ncbi.nlm.nih.gov/), and the candidate genes where the selected SNPs located in were connected with the molecular mechanisms of T2DM according to the literature. Eventually, 10 SNPs (Table 1) were selected for subsequent association analyses.
Genotyping assay
Genomic sequences were obtained from the Hapmap database (http://www.hapmap.org/). A 5′-Nuclease TaqMan assay (Applied BioSystems, Foster City, CA, USA) was used to genotype the polymorphisms in 384-well plates on ABI PRISM 7900HT Sequence Detection system (Applied BioSystems). The primers and probes of TaqMan assays were designed using Primer Express Oligo Design software v2.0 (Applied BioSystems) and were available upon request as TaqMan Pre-Designed SNP Genotyping Assays. PCR reactions were performed in a 5-μl reaction mixture containing 5 ng DNA, 2.5 μl 2* TaqMan Universal PCR Master Mix and 0.083 μl 40* Assay Mix. The primer sequences used were shown in (Supplementary Table S1). The PCR conditions were: 50 °C for 2 min, 95 °C for 10 min, 95 °C for 15 s and then 60 °C for 1 min; forty cycles of real-time PCR were performed. Individual genotype identification was performed by SDS software 2.0 (ABI). Each plate contained two samples from the same individual as positive controls and two blank samples as negative controls for the genotyping quality confirmation. There was 100% consistency in a 5% sample of duplicate testing. All the genotyping assays were done without knowing the subjects’ case and control status.
Statistical analysis
Differences between cases and controls in demographic characteristics, risk factors and genotype frequencies were evaluated by two-sided χ2-square test (for categorical variables) or Student’s t-test (for continuous variables). Hardy–Weinberg equilibrium was assessed by a goodness-of-fit χ2 test to compare the observed genotype frequencies with the expected ones among the controls. Associations between gene genotypes and risk of T2DM were estimated by computing odds ratios (ORs) and their 95% confidence intervals (CIs) from unconditional logistic regression analyses with adjustment for age, gender and WC. We carefully controlled the potential influence of multiple comparisons using the thousand times permutation test by using Stata11.0 software (version 9.2; StataCorp LP, College Station, TX, USA). The gene–environmental interaction was assessed with crossover analysis by using Stata11.0 software. WC⩾85 cm for men or WC⩾80 cm for women was defined as abdominal obesity. All of the statistical analyses were performed with SPSS18.0 software (Version 18.0, SPSS Inc., Chicago, IL, USA). All tests were two sided and the criterion for significance was set at P<0.05.
Results
Basic characteristics
The characteristics of the cases and controls enrolled in this study are shown in Table 2. There was no significant difference in the distribution of age and gender between the cases and controls. Of the selected characteristics, significant differences existed between cases and controls for blood pressure, waist circumference, body mass index, total cholesterol, triglycerides, high-density lipoprotein cholesterol, low-density lipoprotein cholesterol and fasting plasma glucose.
Logistic regression analysis for the association between SNP genotypes in miRNA target sites and T2DM
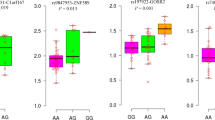
The genotype distribution for all the SNPs did not show any deviation from the Hardy–Weinberg equilibrium (all P>0.05 in controls). The logistic regression analysis revealed that rs1366600CC homozygote was associated with a significant risk effect for T2DM, compared with the rs1366600TT wide-type homozygote (adjusted OR=2.03; 95% CIs=(1.02–4.01)). When assuming a co-dominant allele effect, the combined TC/CC variant genotype was associated with a significantly increased risk of T2DM compared with GG genotype (adjusted OR=1.28; 95% CIs=(1.04–1.57)). Likewise, compared with rs2292899GG wild-type genotype, the variant genotype GA was associated with an increased risk of T2DM (adjusted OR=1.22; 95% CIs=(1.004–1.49)). However, the variant genotype AA of rs11724758 was associated with a significantly reduced risk of T2DM compared with the GG genotype with adjusted OR and 95% CIs of 0.76 (0.58–0.997). After inspection by the thousand times permutation test, the frequency distribution of those three sites genotyped between the case group and control group still contained statistical differences (Table 3). The remaining seven SNPs did not show significant difference in the associations of T2DM (Supplementary Table S2).
Stratified analysis of miRNA target SNP genotypes with T2DM susceptibility
In stratification analyses, we found the association between rs1366600CC genotype and T2DM risk was significant among the elderly (age >55) (adjusted OR=2.54; 95% CI=(1.06–6.07)) and non-drinkers (adjusted OR=2.36; 95% CIs=(1.08–5.16)). Similarly, rs1366600CT increased the risk of T2DM in the elderly (>55 years old) and abdominal obesity people (adjusted ORs=1.35, 1.59; 95% CIs=(1.02–1.79), (1.15–2.19), respectively). We only observed a higher risk of T2DM in the normal WC people who carried the rs2292899GA genotype (P=0.025; adjusted OR=1.38; 95% CIs=(1.04–1.82)). There was more pronounced decrease in T2DM risks among males, elderly (age >55), smokers and drinkers carrying rs11724758AA genotype (adjusted ORs=0.64, 0.57, 0.54, 0.43; 95% CIs=(0.44–0.93), (0.39–0.82), (0.33–0.90), (0.23–0.78), respectively) (Table 4). There was no significant difference in the magnitude of the associations between the remaining seven SNPs and T2DM in subjects with different age, gender, abdominal obesity, smoking, drinking (data not shown).
Crossover analysis in assessing gene–environmental interaction
Rs1366600, rs2292899 and rs11724758 which had significant associations with the risk of T2DM in the single-locus analysis were, respectively, assessed the gene–environmental interaction by crossover analysis. The dominant models of rs1366600, rs2292899 and rs11724758 were defined as G+. Rs1366600TT, rs2292899GG, rs11724758GG were defined as G−. Abdominal obesity was defined as the E+ and the normal WC was defined as E−. In the analysis of rs1366600 and rs2292899, subjects who only carried the environmental risk factor or putative risk genotype had the increased risk of T2DM (adjusted ORs=2.17, 1.32, 2.73; 95% CIs=(1.75–2.68), (1.01–1.72), (2.04–3.67), respectively), while those who exposed to both environmental factor and genotype factors (putative risk genotypes) had the highest risk of T2DM (adjusted ORs=3.56, 3.04; 95% CIs=(2.61–4.87), (2.31–3.99), respectively). Nevertheless, the subjects with abdominal obesity exposed to rs11724758GA/AA variant genotype (putative protective genotype) had an increased risk of T2DM (adjusted OR=2.24; 95% CI=(1.71–2.95)), while the abdominally obese subjects exposed to rs11724758GG genotype (putative risk genotype) had the highest risk for T2DM (adjusted OR=2.62; 95%CI=(1.92–3.58)) (Table 5).
Assessing gene–gene combined effect on T2DM risk
We then analyzed the cumulative effect of three SNPs (rs1366600, rs2292899 and rs11724758) alleles and risk of T2DM. These three loci were evaluated together by the number (0–6) of putative risk alleles (rs1366600 C, rs2292899 A and rs11724758 G). Compared with the reference group (0–1), a significant increased risk of T2DM was detected as the number of risk alleles increased (P for trend <0.001, Table 6). Furthermore, compared with the reference group (0–1), individuals carrying two or more risk alleles still had a 1.70-fold increased risk for T2DM.
Discussion
Recent studies1, 4 demonstrated that SNPs located within miRNA-binding sites are likely to affect the expression of the miRNA targets that may contribute to the susceptibility to human diseases. In short, an SNP may either abolish or weaken a miRNAs target or create a perfect sequence match to the seed of a miRNAs.5 Abelson et al.6 showed that the var321 in the human SLITRK1 gene 3′-UTR replaces a G: U wobble base pair with an A: U Watson–Crick pairing and strengthens an existing miR-189 target site, further amplifying the down regulation of SLITRK1, which leads to Tourette syndrome. Mishra et al.7 demonstrated that a 3′-UTR 829C→T in the human dihydrofolate reductase leads to a decrease in the miR-24-binding site resulting in dihydrofolate reductase overexpression and methotrexate resistance. We used different algorithms to predict SNPs in diabetes-related miRNA target sites located on 3′-UTRs of the genes involved in the development and progression of T2DM. In this study, 10 SNPs were eventually selected and genotyped, and we found significant associations between three SNPs (rs1366600, rs2292899 and rs11724758) and the risk of T2DM in Chinese Han population with a moderate sample size of 1017 T2DM cases and 1059 T2DM-free controls.
In the single-locus analyses, rs1366600CC, TC/CC of insulin receptor (INSR) gene contributed an independently increased risk for T2DM compared with rs1366600TT (adjusted P=0.031, 0.011, respectively). The association was more evident in elderly subjects (carrying CC, TC/CC genotypes), non-drinkers (carrying CC genotype) and the abdominally obese (carrying TC genotype). The SNP rs1366600 was located on 3′-UTR of the INSR gene. The INSR gene encodes protein having direct role in insulin signaling pathway and is implicated in the insulin signal transduction and insulin sensitivity modulation. Some polymorphisms localized mainly on coding region of the INSR gene had shown correlation with insulin resistance (IR) and T2DM.27 Rare evidence showed the correlation of SNPs on 3′-UTR of the INSR gene with increased risk to IR or T2DM. For example, Malodobra et al.27 showed that G/G genotype of rs3745551 on 3′-UTR of the INSR gene dominated in IR diabetic patients and had effect on insulin resistant phenotype development. Thus far, there has been little focus on the association between the SNP rs1366600 and the risk of T2DM. The SNP rs1366600 of INSR gene was predicted to be located on binding site of miR-20b seed. Zampetaki et al.15 observed lower plasma level of miR-20b in diabetic subjects of the Bruneck study, indicating the important role of miR-20b in diabetes. In present study, we predicted that the variant allele C of rs1366600 in 3′-UTR of the INSR gene may cause a loss of the binding site for the miR-20b by algorithms and identified that it is associated with higher diabetes risks. However, how the miRNA regulates the expression of target gene INSR and the exact biological mechanism of the effect that the SNP has on miRNA binding needs further research. We hypothesized that the SNP rs1366600 in miR-20b target site can have an effect on INSR gene expression, further to change the function of the receptor. Although normal insulin secretion continues, the receptor cannot react to the existing insulin, ultimately leading to IR or T2DM.
We also found that the acyl-CoA synthetase 1 (ACSL1) rs2292899GA heterozygote genotype was associated with a significantly increased risk of T2DM. The association between higher risk of T2DM and rs2292899GA was more evident in the normal WC subjects (Table 4). The association between T2DM and rs2292899AA homozygotes was not showed in present study, but the A was considered as risk allele. This was due to the insufficient sample size and rs2292899AA contributed an increased risk for T2DM compared with rs2292899GG (adjusted OR=1.13, despite P=0.416). The SNP rs2292899 was located on 3′-UTR of the ACSL1 gene. ACSL1, the major acyl-CoA synthetase of adipocytes, is involved in peroxisome proliferator-activated receptor signaling pathway. Peroxisome proliferator-activated receptors have been identified as the key regulators of fatty acid and lipoprotein metabolism, glucose homeostasis, cellular proliferation/differentiation and the immune response. Although ACSL1 clearly has an important role relevant to lipid metabolism and fatty-acid-induced insulin resistant, rare studies to date have investigated the association between genetic variants of ACSL1 and T2DM.28 The SNP rs2292899 of ACSL1 gene was predicted to be located on binding site of miR-34a seed. Expression of miR-34a increased in the β-cell line MIN6B1 or pancreatic islets after exposed to palmitate in islets of diabetic db/db mice. Elevated miR-34a resulted in sensitization to apoptosis and impaired nutrient-induced insulin secretion.14 Kong et al.10 showed that hypertriglyceridemia as one of the characteristic blood lipid abnormalities in diabetes could favor the onset of diabetes via higher serum level of miR-34a. The variant allele A of rs2292899 on 3′-UTR of the ACSL1 gene was predicted to cause a loss of the binding site for the miR-34a by algorithms and identified that it could cause higher diabetes risks. How the SNP located on miRNA binding affects the target gene ACSL1 expression eventually leading to T2DM also needs to study further.
In this study, we found the variant genotype AA of rs11724758 compared with the GG genotype was associated with a significantly decreased risk of T2DM (adjusted P=0.035). In the stratified analyses, we similarly found that there was pronounced decrease in T2DM risk in males, elderly (age >55), smokers and drinkers carrying rs11724758AA genotype. The SNP rs11724758 was on 3′-UTR of the fatty-acid-binding protein 2 (FABP2) gene. The FABP2 involved in peroxisome proliferator-activated receptor signaling pathway has an important role in intracellular transportation of dietary long-chain fatty acids and fat absorption.29 It has been reported that a polymorphism at codon 54 of the FABP2 gene, changing G to A (Ala54Thr), is associated with high IR, fasting insulin concentrations and involved in the development of T2DM.29 But no evidence indicates any association between rs11724758 and T2DM. We predicted rs11724758 located in binding site of miR-132 seed. Klöting et al.20 performed a global miRNA gene expression assay in different fat depots of overweight and obese individuals and found that the expression of miR-132 had a role in the link between adipose tissue dysfunction and the development of obesity-associated disorders including T2DM. We hypothesize that the variant allele A of rs11724758 on 3′-UTR of the FABP2 gene may cause loss of the binding site for the miR-132. Likewise, further functional studies are needed to validate the hypothetically functional mechanism.
The crossover analysis of the genetic polymorphisms of three genes and environmental risk factor (abdominal obesity) indicated that, subjects who exposed to both the abdominal obesity and putative risk genotypes (rs1366600TC/CC and rs2292899GA/AA) had higher risk of T2DM than those who exposed to single environmental factor or genotype factor. This may be attributed to the gene–environmental interaction that the risk effect of these two putative risk genotypes could be strengthened by the abdominal obesity. In addition, we found the carriers with potential protective genotype (rs11724758GA/AA), who did expose to the abdominal obesity, had a decreased risk effect for T2DM compared with those who exposed to the abdominal obesity and potential risk genotype. This may be ascribed to that the potential protective genotype could decrease the effect of abdominal obesity factor on risk of T2DM.
Importantly, a cumulative effect was observed when the genotypes of rs1366600, rs2292899 and rs11724758 were combined to be analyzed with T2DM risk. Although in different signaling pathway, mutations in the INSR, ACSL1 and FABP2 genes are connected with IR and have a significant role in the pathogenesis of T2DM. Results from the study of Ning et al. demonstrated that insulin and insulin signaling induced IR through interactions with fat, which was in turn likely caused by the excessive accumulation of activated long-chain acyl-CoAs in mitochondria. It indicated that fat may not be able to induce IR without insulin,30 while ACSL1 and FABP2 genes have a major role in fatty acid and lipoprotein metabolism.
In conclusion, our results suggest the rs1366600 of INSR gene, rs2292899 of ACSL1 gene and rs11724758 of FABP2 gene in miRNA-binding sites had significant associations with T2DM risk in Chinese Han population. The mechanism which caused T2DM susceptibility by subtle gene regulation is still not fully appreciated and clear. Further functional studies are necessary to verify whether the miRNA regulates the expression of target gene by binding to predicted target sequences.
References
Guay, C., Roggli, E., Nesca, V., Jacovetti, C. & Regazzi, R. Diabetes mellitus, a microRNA-related disease? Transl. Res. 157, 253–264 (2011).
Yang, W., Lu, J., Weng, J., Jia, W., Ji, L., Xiao, J. et al. Prevalence of diabetes among men and women in China. New Engl. J. Med. 362, 1090–1101 (2010).
Lynn, F. C. Meta-regulation: microRNA regulation of glucose and lipid metabolism. Trends Endocrinol. Metab. 20, 452–459 (2009).
Dehwah, M. A., Xu, A. & Huang, Q. MicroRNAs and type 2 diabetes/obesity. J. Genet. Genomics 39, 11–18 (2012).
Chen, K., Song, F., Calin, G. A., Wei, Q., Hao, X. & Zhang, W. Polymorphisms in microRNA targets: a gold mine for molecular epidemiology. Carcinogenesis 29, 1306–1311 (2008).
Abelson, J. F., Kwan, K. Y., O'Roak, B. J., Baek, D. Y., Stillman, A. A., Morgan, T. M. et al. Sequence variants in SLITRK1 are associated with Tourette's syndrome. Science 310, 317–320 (2005).
Mishra, P. J., Humeniuk, R., Mishra, P. J., Longo-Sorbello, G. S., Banerjee, D. & Bertino, J. R. A miR-24 microRNA binding-site polymorphism in dihydrofolate reductase gene leads to methotrexate resistance. Proc. Natl Acad. Sci. USA 104, 13513–13518 (2007).
Chin, L. J., Ratner, E., Leng, S., Zhai, R., Nallur, S., Babar, I. et al. A SNP in a let-7 microRNA complementary site in the KRAS 3’untranslated region increases Non–small cell lung cancer risk. Cancer Res. 68, 8535–8540 (2008).
Poy, M. N., Eliasson, L., Krutzfeldt, J., Kuwajima, S., Ma, X., Macdonald, P. E. et al. A pancreatic islet-specific microRNA regulates insulin secretion. Nature 432, 226–230 (2004).
Kong, L., Zhu, J., Han, W., Jiang, X., Xu, M., Zhao, Y. et al. Significance of serum microRNAs in pre-diabetes and newly diagnosed type 2 diabetes: a clinical study. Acta Diabetol. 48, 61–69 (2010).
Lovis, P., Gattesco, S. & Regazzi, R. Regulation of the expression of components of the exocytotic machinery of insulin-secreting cells by microRNAs. Biol. Chem. 389, 305–312 (2008).
Ramachandran, D., Roy, U., Garg, S., Ghosh, S., Pathak, S. & Kolthur-Seetharam, U. Sirt1 and mir-9 expression is regulated during glucose-stimulated insulin secretion in pancreatic β-islets. FEBS J. 278, 1167–1174 (2011).
Tang, X., Muniappan, L., Tang, G. & Ozcan, S. Identification of glucose-regulated miRNAs from pancreatic {beta} cells reveals a role for miR-30d in insulin transcription. RNA 15, 287–293 (2009).
Lovis, P., Roggli, E., Laybutt, D. R., Gattesco, S., Yang, J. Y., Widmann, C. et al. Alterations in microRNA expression contribute to fatty acid-induced pancreatic beta-cell dysfunction. Diabetes 57, 2728–2736 (2008).
Zampetaki, A., Kiechl, S., Drozdov, I., Willeit, P., Mayr, U., Prokopi, M. et al. Plasma microRNA profiling reveals loss of endothelial miR-126 and other microRNAs in type 2 diabetes. Circ. Res. 107, 810–817 (2010).
Krek, A., Grun, D., Poy, M. N., Wolf, R., Rosenberg, L., Epstein, E. J. et al. Combinatorial microRNA target predictions. Nat. Genet. 37, 495–500 (2005).
Bravo-Egana, V., Rosero, S., Molano, R. D., Pileggi, A., Ricordi, C., Dominguez-Bendala, J. et al. Quantitative differential expression analysis reveals miR-7 as major islet microRNA. Biochem. Biophys. Res. Commun. 366, 922–926 (2008).
He, A., Zhu, L., Gupta, N., Chang, Y. & Fang, F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol. Endocrinol. 21, 2785–2794 (2007).
Esau, C., Kang, X., Peralta, E., Hanson, E., Marcusson, E. G., Ravichandran, L. V. et al. MicroRNA-143 regulates adipocyte differentiation. J. Biol. Chem. 279, 52361–52365 (2004).
Klöting, N., Berthold, S., Kovacs, P., Schon, M. R., Fasshauer, M., Ruschke, K. et al. MicroRNA expression in human omental and subcutaneous adipose tissue. PLoS ONE 4, e4699 (2009).
Hennessy, E., Clynes, M., Jeppesen, P. B. & O’Driscoll, L. Identification of microRNAs with a role in glucose stimulated insulin secretion by expression profiling of MIN6 cells. Biochem. Bioph. Res. Commun. 396, 457–462 (2010).
Baroukh, N., Ravier, M. A., Loder, M. K., Hill, E. V., Bounacer, A., Scharfmann, R. et al. MicroRNA-124a regulates Foxa2 expression and intracellular signaling in pancreatic beta-cell lines. J. Biol. Chem. 282, 19575–19588 (2007).
Hiard, S., Charlier, C., Coppieters, W., Georges, M. & Baurain, D. Patrocles: a database of polymorphic miRNA-mediated gene regulation in vertebrates. Nucleic Acids Res. 38, D640–D651 (2010).
Ziebarth, J. D., Bhattacharya, A., Chen, A. & Cui, Y. PolymiRTS database 2.0: linking polymorphisms in microRNA target sites with human diseases and complex traits. Nucleic Acids Res. 40, D216–D221 (2012).
Lewis, B. P., Burge, C. B. & Bartel, D. P. Conserved Seed pairing, often flanked by adenosines, indicates that thousands of human genes are MicroRNA targets. Cell 120, 15–20 (2005).
Betel, D., Wilson, M., Gabow, A., Marks, D. S. & Sander, C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 36, D149–D153 (2008).
Malodobra, M., Pilecka, A., Gworys, B. & Adamiec, R. Single nucleotide polymorphisms within functional regions of genes implicated in insulin action and association with the insulin resistant phenotype. Mol. Cell Biochem. 349, 187–193 (2011).
Lobo, S., Wiczer, B. M. & Bernlohr, D. A. Functional analysis of long-chain acyl-CoA synthetase 1 in 3T3-L1 adipocytes. J. Biol. Chem. 284, 18347–18356 (2009).
Baier, L. J., Sacchettini, J. C., Knowler, W. C., Eads, J., Paolisso, G., Tataranni, P. A. et al. An amino acid substitution in the human intestinal fatty acid binding protein is associated with increased fatty acid binding, increased fat oxidation, and insulin resistance. J. Clin. Invest. 95, 1281–1287 (1995).
Ning, J., Hong, T., Yang, X., Mei, S., Liu, Z., Liu, H. Y. et al. Insulin and insulin signaling play a critical role in fat induction of insulin resistance in mouse. Am. J. Physiol. Endocrinol. Metab. 301, E391–E401 (2011).
Acknowledgements
We would like to thank Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Supplementary information
Rights and permissions
About this article
Cite this article
Zhao, X., Ye, Q., Xu, K. et al. Single-nucleotide polymorphisms inside microRNA target sites influence the susceptibility to type 2 diabetes. J Hum Genet 58, 135–141 (2013). https://doi.org/10.1038/jhg.2012.146
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2012.146
Keywords
This article is cited by
-
Associations between genetic variations in microRNA and myocardial infarction susceptibility: a meta-analysis and systematic review
Herz (2022)
-
MicroRNAs and Synaptic Plasticity: From Their Molecular Roles to Response to Therapy
Molecular Neurobiology (2022)
-
Polymorphisms in miRNA binding sites involved in metabolic diseases in mice and humans
Scientific Reports (2020)
-
Investigation of miRNA-binding site variants and risk of gestational diabetes mellitus in Chinese pregnant women
Acta Diabetologica (2017)
-
The pre-mir-27a variant rs895819 may contribute to type 2 diabetes mellitus susceptibility in an Iranian cohort
Journal of Endocrinological Investigation (2016)
