
Abstract
Astrocytes are implicated in information processing, signal transmission, and regulation of synaptic plasticity. Aquaporin-4 (AQP4) is the major water channel in adult brain and is primarily expressed in astrocytes. A growing body of evidence indicates that AQP4 is a potential molecular target for the regulation of astrocytic function. However, little is known about the role of AQP4 in synaptic plasticity in the amygdala. Therefore, we evaluated long-term potentiation (LTP) in the lateral amygdala (LA) and associative fear memory of AQP4 knockout (KO) and wild-type mice. We found that AQP4 deficiency impaired LTP in the thalamo-LA pathway and associative fear memory. Furthermore, AQP4 deficiency significantly downregulated glutamate transporter-1 (GLT-1) expression and selectively increased NMDA receptor (NMDAR)-mediated EPSCs in the LA. However, low concentration of NMDAR antagonist reversed the impairment of LTP in KO mice. Upregulating GLT-1 expression by chronic treatment with ceftriaxone also reversed the impairment of LTP and fear memory in KO mice. These findings imply a role for AQP4 in synaptic plasticity and associative fear memory in the amygdala by regulating GLT-1 expression.
Similar content being viewed by others


INTRODUCTION
Water homeostasis of the central nervous system (CNS) is important in both physiological and pathological conditions. Aquaporins (AQPs) are a family of membrane channels that play a significant role in regulating water homeostasis (Verkman, 2005). Aquaporin-4 (AQP4), a predominant water channel in adult brain, is widely expressed in astrocytes (Nielsen et al, 1997) and plays important roles in the regulation of extravascular brain water, brain volume homeostasis, and cerebrospinal fluid production (Amiry-Moghaddam and Ottersen, 2003). A lot of evidence shows that AQP4 is involved in the pathophysiology of cerebral disorders, including stroke, cerebral edema, traumatic brain injury, Parkinson's disease, epilepsy, and depression, and so on (Binder et al, 2006; Chi et al, 2011; Ding et al, 2009; Friedman et al, 2009; Kong et al, 2009; Thrane et al, 2011). Recent studies demonstrate that AQP4 knockout (KO) inhibits the formation of glial cell-derived neurotrophic factor and increases microglial inflammatory responses in MPTP-treated mouse model of Parkinson's disease (Chi et al, 2011; Fan et al, 2008). Moreover, AQP4 deficiency alters basal levels of amino-acid neurotransmitters, and downregulates glutamate uptake and glutamate transporter-1 (GLT-1) expression in astrocytes (Fan et al, 2005; Zeng et al, 2007). These findings strongly indicate that AQP4 plays a vital role in modulating astrocytic function. Therefore, AQP4 is not only a water channel, but also an important signal molecule in several physiological or pathophysiological procedures.
Astrocytes have been generally believed to have a mainly supportive function for neurons in CNS (Kettenmann and Ransom, 2005). However, increasing evidence suggests that astrocytes also play important roles in information processing, signal transmission, and regulation of neural and synaptic plasticity (Haydon et al, 2009; Henneberger et al, 2010; Perea et al, 2009). For example, astrocytic GLTs, lactate transporters, and glial-derived neuromodulator D-serine are required for synaptic plasticity and memory (Henneberger et al, 2010; Mothet et al, 2006; Suzuki et al, 2011). Although astrocytes play an important role in synaptic plasticity and AQP4 plays a vital role in modulating astrocytic function, little is known about the potential role of AQP4 in synaptic plasticity in the amygdala.
Glutamate is the predominant excitatory neurotransmitter in the mammalian CNS, and is critical for essentially all physiological processes, including excitatory synaptic transmission, synaptic plasticity, and neuronal development, and so on (Mayer and Westbrook, 1987). However, the excessive accumulation of glutamate in the synaptic cleft may result in neuronal overactivation and neurodegenerative diseases, such as amyotrophic lateral sclerosis, ischemia, and epilepsy. Therefore, the maintenance of physiological level of extracellular glutamate is crucial to normal synaptic transmission and prevention of excitotoxicity. GLTs, which widely localized in astrocytes, are responsible for clearing synaptically released glutamate at excitatory synapses. GLTs reduce spillover, thus ensuring a spatially restricted action of glutamate and preventing its accumulation in the extracellular space (ECS) (Amara and Fontana, 2002; Danbolt, 2001), thus contributing to the sculpture of excitatory postsynaptic currents (EPSCs) and modulation of synaptic plasticity (Tzingounis and Wadiche, 2007). GLT-1, an isoform of GLTs, exhibits the highest level of expression and is responsible for the largest proportion of glutamate transport in the hippocampus (Danbolt, 2001). GLT-1 KO or downregulation increases extracellular glutamate levels, which in turn cause excitotoxicity and impairment of long-term potentiation (LTP) (Danbolt, 2001; Katagiri et al, 2001; Wang et al, 2006), a well-established form of synaptic plasticity and the most intensely studied cellular model for memory (Malenka, 2003; Malenka and Nicoll, 1999). Conversely, an elevated expression of GLT-1 may be neuroprotective (Rothstein et al, 2005). As AQP4 deficiency downregulates GLT-1 expression and reduces glutamate uptake in astrocytes (Zeng et al, 2007), we hypothesized that AQP4 deficiency could result in the impairment of LTP and memory.
In this present study, we evaluated LTP in the lateral amygdala (LA) and associative fear memory of AQP4 KO and wild-type (WT) mice. We found that AQP4 deficiency impaired LTP in the LA and associative fear memory in KO mice. The expression of GLT-1 in the LA was downregulated in KO mice. However, pharmacological upregulation of GLT-1 expression reversed the impairment of LTP and fear memory in KO mice. These findings imply that AQP4 plays a role in synaptic plasticity and associative fear memory in the amygdala by regulating GLT-1 expression.
MATERIALS AND METHODS
Experimental Animals
Both AQP4 KO mice and WT mice were provided by Dr Gang Hu (Nanjing Medical University, Jiangsu, China). The KO mice were generated by targeted gene disruption as described (Fan et al, 2005), using an identical strategy as described previously (Ma et al, 1997). Mice were identified by PCR analyses of tail samples at 5 days of age, and kept under environmentally controlled conditions (ambient temperature, 22 °C; humidity, 40%) on a 12 h light/dark cycle with food and water ad libitum. All experiments were performed on age- and weight-matched littermates produced by intercrossing of CD1 heterozygotes. Male 6- to 8-week-old mice were used in this study. Investigators were blinded to the genotype for all experiments. All experiments were approved by IACUC (Institutional Animal Care and Use Committee of Huazhong University of Science and Technology).
Tissue Preparation and Western Blot Analysis
The amygdala tissue was prepared for the assay of protein expression. Mice were decapitated and the brain was quickly removed. Coronal brain slices (400 μm thick) containing the amygdala were cut using a vibratome (VT 1000S; Leica, Wetzlar, Germany), and LA was dissected thoroughly under the microscope using a special blade. The tissue was washed twice with ice-cold PBS and then lysed on ice in extraction buffer containing 50 mM Tris-base (pH 7.4), 100 mM NaCl, 1% NP-40, 10 mM EDTA, 20 mM NaF, 1 mM PMSF, 3 mM Na3VO4, and protease inhibitors. The homogenates were centrifuged at 12 000 g for 15 min at 4 °C. The supernatant was separated and protein concentration was estimated by Coomassie blue protein-binding assay (Nanjing Jiancheng Institute of Biological Engineering, Nanjing, China). Thereafter, the protein samples were mixed with sodium dodecyl sulfate (SDS) sample buffer, heated at 95 °C for 5 min, and stored at −80 °C until electrophoresis. Samples (20 μg) were separated by 10% SDS-polyacrylamide gel and then transferred to nitrocellulose membranes (Schleicher and Schuell, Keene, NH, USA). After blocking with 5% non-fat milk in Tris-buffered saline containing 0.1% Tween-20 (TBST) for 1 h at room temperature, transferred membranes were incubated overnight at 4 °C with different primary antibodies against β-actin (1 : 3000 dilution) (Upstate Biotechnology, Lake Placid, NY, USA), anti-AQP4 (1 : 500 dilution) (Millipore, Billerica, MA, USA), anti-glutamate/aspartate transporter (GLAST) (1 : 500 dilution), and anti-GLT-1 (1 : 300 dilution) (Santa Cruz Biotechnology, Santa Cruz, CA, USA), respectively. Following three washes with TBST buffer, membranes were then incubated with horseradish peroxidase-conjugated secondary antibodies (1 : 5000) in TBST with 1% non-fat milk for 1 h at room temperature. After repeated washes, membranes were reacted with enhanced chemiluminescence reagents (Super Signal West Pico; Pierce Chemical, Rockford, IL, USA) and visualized with Micro Chemi (DNR Bio-Imaging Systems, Jerusalem, Israel) or X-ray films (Kodak X-Omat, Rochester, NY, USA). The optical densities were scanned and the optical densities of detected bands were quantified using the NIH Image J software. The results were normalized to the quantity of β-actin in each sample lane. All assays were performed at least three times.
Reverse Transcriptase-Polymerase Chain Reaction
Total RNA was isolated from the LA tissue of brain slices using Trizol reagent (Invitrogen, Carlsbad, CA, USA), and the amount of RNA obtained was determined by spectrophotometry. Reverse transcription was performed with oligo (dT), AMV reverse transcription (Promega, Madison, WI, USA), RNase inhibitor (Promega), and dNTP mix in a final volume of 20 μl. Reverse transcriptase-polymerase chain reaction (RT-PCR) was carried out with an automatic thermocycler. The following specific primers were used: PAQP4-sense, 5′-ACCATAAACTGGGGTGGCTCAG-3′; PAQP4-antisense, 5′-TAGAGGATGCCGGCTCCAATGA-3′. GAPDH was used as an internal control. PCR products were analyzed by 2% agarose gel electrophoresis.
Electrophysiological Recordings
Mice were anesthetized and decapitated, and the brains were rapidly removed and placed in ice-cold artificial CSF (aCSF) of the following composition (in mM): 119 NaCl, 3.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose, pH 7.4 (bubbled with 95% O2/5% CO2). Coronal brain slices (400 μm thick) containing the amygdala were cut in the same solution using a vibratome (VT 1000S; Leica) and retained 1–2 h in a storage chamber for recovery. Thereafter, an individual slice was transferred into a perfusion-type recording chamber superfused with oxygenated aCSF (3–4 ml/min) at 30 °C. Field EPSPs (fEPSPs) were recorded in thalamic inputs of experimental animals using a 3 M NaCl-filled glass electrode (3–5 MΩ) placed within the LA. fEPSPs were evoked by a constant stimulation with a bipolar concentric tungsten electrode (FHC, Bowdoinham, ME, USA) placed in the internal capsule (the thalamo-LA pathway). Stimulation intensities were adjusted to produce an fEPSP with 1/3–1/2 of maximal amplitude. Stimulation frequency was 0.033 Hz. The input–output relationship (IOR) for synaptic transmission was constructed by varying the intensity of single-pulse stimulation. Paired stimuli (25, 50, 75, and 100 ms interval) were delivered to lateral nucleus and the paired-pulse facilitation (PPF) was calculated as the ratio between the mean slope of the second fEPSP (fEPSP2) over the first one (fEPSP1). After 20 min of baseline recording, LTP was induced by high-frequency stimulation (HFS) (five trains at 100 Hz for 1 s with 90 s interval between trains). Responses were recorded for 60 min following tetanus. The fEPSP slope in each time point was measured in middle 1/3 of descending branch of the fEPSP and normalized as a percentage of the averaged slope during the baseline recording period before HFS. EPSCs of principal neurons in the dorsolateral division of the LA were recorded in the whole-cell voltage-clamp mode. Recording electrodes (3–5 MΩ resistance) were filled with a solution containing (in mM): 122.5 Cs-gluconate, 17.5 CsCl, 0.2 EGTA, 10 HEPES, 8 NaCl, 2 Mg-ATP, 0.3 Na-GTP, and 5 QX314 (pH 7.2, osmolality 280–290 mOsm). Evoked EPSCs were obtained by stimulating fibers of the internal capsule. The AMPA receptor (AMPAR)-mediated component was isolated by holding the membrane potential at −70 mV. NMDA receptor (NMDAR)-mediated EPSCs were recorded at the same stimulus strength in the presence of CNQX (20 mM), a non-NMDAR antagonist, and the membrane potential was clamped at +40 mV to remove the voltage-dependent Mg2+ block of NMDAR (Katagiri et al, 2001). Ratios of NMDAR/AMPAR-mediated currents were obtained by measuring peak amplitudes of NMDAR- and AMPAR-mediated EPSCs. All experiments were performed in the presence of 50 μM picrotoxin (Sigma, St Louis, MO, USA).
Fear Conditioning Test
The experiments were performed in two contexts: the conditioning chamber (12 × 18 × 30 cm3) with a metal grid shock floor and the test context differing in shape and odor. All chambers were cleaned thoroughly after each trial. Mice were handled in the room where conditioning took place for 5 min per day for 3 days. For light fear conditioning, mice were placed into the conditioning chamber and left to explore for 3 min, and then a light conditioned stimulus (CS) was presented for 30 s that co-terminated with a single electric foot shock (0.7 mA, 1 s). This conditioning trial was repeated three times with 60 s intertrial intervals. The light was produced by an 8 W white light bulb located 10 cm behind the cage. Light intensity of the CS across contexts was equivalent (approximately 16–17 lx). Mice were returned to their home cages 1 min later. To test light-cued fear memory, different experimental mice were placed into the test context 2 and 24 h after conditioning, respectively. Freezing behavior of the corresponding animals in the test chamber was monitored for 3 min with presentation of the light. Conditioned freezing was defined as immobility, except for respiratory movements. The total freezing time in the test period was represented as a percentage. All trials were performed in darkness, except those in which the light was used as a CS.
Open Field Test
Mice were placed individually into the activity chamber (40 × 40 × 40 cm3) monitored by a video motility system. The spontaneous motor activity including total distance and average speed of mice movement during 10 min intervals was monitored and assessed.
Elevated Plus Maze Test
The elevated plus maze (EPM) consisted of a plus-shaped platform with two open (30 × 5 cm2) and two closed (30 × 5 × 15 cm3) arms and a connecting central zone (5 × 5 cm2). The apparatus was elevated 40 cm above the floor. Mice were transferred to the test room and were allowed to acclimate to the environment for 24 h before starting the experiment. The animal was placed quickly onto the central compartment facing a closed arm. During a 5 min exposure, the number of full entries into open and closed arms and the time spent on open and closed arms were recorded and calculated. Entry was defined as all four paws being positioned within one arm.
Pain Threshold Measurement
Animals were placed individually into the conditioning chamber. After a 3 min exposure, electric foot shocks (1 s) were applied starting with an intensity of 0.1 mA. The intensity was increased stepwise by 0.05 mA every 30 s between successive stimuli until the animal showed the first signs of pain (vocalizing and jumping), and the corresponding value was taken as the pain threshold.
Analysis and Statistics
Data are expressed as mean±SEM and analyzed by employing the SPSS 10.0 software. For estimation of LTP, the fEPSP slope in each time point was normalized to the averaged baseline response before tetanization and evaluated using two-way repeated-measure-ANOVA followed by Student's t-test. The results from behavioral studies and quantitative immunoblot analyses were statistically evaluated using Student's t-test and/or ANOVA, followed by Newman–Keuls post hoc test. A probability level of P<0.05 was considered as statistically significant.
RESULTS
AQP4 Deficiency Impairs LTP in the Thalamo-LA Pathway Without Affecting Basal Synaptic Transmission
To investigate the influence of AQP4 on synaptic plasticity in mice amygdala, we initially identified the expression of AQP4 in the LA with RT-PCR and western blot analyses. As expected, the Aqp4 mRNA and AQP4 protein appeared as a monomeric band in WT mice (n=3) and disappeared in KO mice (n=3; Figure 1a), suggesting that AQP4 is expressed in the amygdala of WT mice and the gene KO in KO mice is successful. We next evaluated HFS-induced LTP in the thalamo-LA pathway in brain slices from WT and KO mice (Figure 1b). At 60 min after LTP induction, the slope of fEPSPs occurring at the thalamic inputs was potentiated to 137.9±6.6% of baseline in WT mice (n=11 slices from 6 mice), but remained at only 111.1±5.1% of baseline in KO mice (n=13 slices from 7 mice; P<0.01 vs WT; Figure 1c and d). These results suggest that KO mice show a marked reduction of LTP in the thalamo-LA pathway compared with WT mice.

Aquaporin-4 (AQP4) deficiency impairs long-term potentiation (LTP) in the thalamo-LA pathway with no effect on basal synaptic transmission. (a) Expression of AQP4 in the LA from wild-type (WT) (AQP4+/+) and knockout (KO) (AQP4−/−) mice. Reverse transcriptase-polymerase chain reaction (RT-PCR) (upper) and western blot analysis (lower) revealed that the expression of Aqp4 mRNA and AQP4 protein was readily detected in LA of WT mice (n=3) but not KO mice (n=3). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and β-actin were used as internal control. (b) Schematic representation of an amygdala slice showed location of recording and stimulation electrodes in the thalamo-LA pathway. LA, lateral amygdala; BLA, basolateral amygdala; CeA, central amygdala; IC, internal capsule; EC, external capsule. (c) Time course of the field excitatory postsynaptic potential (fEPSP) evoked by stimulation of thalamic inputs recorded in amygdala slices from WT (n=11 slices from 6 mice) and KO mice (n=13 slices from 7 mice). (Inset) Schematic representation of fEPSP recorded in individual slices before (1) and 60 min after (2) the LTP-inducing stimulation in either WT (left) or KO (right) mice. (d) The histogram showed the level of LTP 60 min after high-frequency stimulation (HFS) (five trains at 100 Hz for 1 s with 90 s interval between trains) in the thalamo-LA pathway in WT and KO mice. Each point was the normalized mean±SEM of slices. **P<0.01 vs WT. (e) Typically superimposed fEPSP recorded in the thalamo-LA pathway in WT (left) and KO (right) mice with gradually increased stimulation intensity. (f) Typical fEPSP recorded in the thalamo-LA pathway from individual experiment at 50 ms interpulse interval before HFS stimulation. (g) Input–output curves in the thalamo-LA pathway illustrating the relationship between the stimulation intensity and evoked response for fEPSP recorded in brain slices from WT (n=10 slices from 5 mice) and KO mice (n=11 slices from 6 mice). No significant differences were observed between the two genotypes. (h) Paired-pulse facilitation in the thalamo-LA pathway was measured by varying the intervals (25, 50, 75, and 100 ms) between pairs of stimuli before HFS stimulation. No significant differences were observed between WT (n=10 slices from 5 mice) and KO mice (n=11 slices from 6 mice).
To determine whether the impairment of synaptic plasticity observed in AQP4 KO mice result from a general defect in synaptic transmission, we analyzed the characteristics of basal excitatory synaptic transmission in the thalamo-LA pathway in WT and KO mice. The IOR, which reflects the efficacy of synaptic transmission and assessed by the fEPSP amplitude, was not significantly altered in the thalamo-LA pathway in KO mice compared with WT mice (n=11 slices from 6 mice; Figure 1e and g). The PPF represents another important parameter of synaptic transmission likely primarily related to presynaptic function (Zucker, 1989). The analyses of fEPSPs revealed no difference in PPF between KO and WT mice (n=11 slices from 6 mice; Figure 1f and h). These results indicate that AQP4 deficiency does not alter the probability of presynaptic transmitter release. Therefore, the effects of AQP4 deficiency on LTP likely result from the changes in postsynaptic response to glutamate.
AQP4 Deficiency Impairs the Associative Fear Memory of Mice
Neuronal plasticity in the LA is believed to underlie the acquisition and retention of Pavlovian fear conditioning (Davis, 1992; LeDoux, 2000). Our finding that AQP4 deficiency impaired synaptic plasticity in amygdala raised the possibility that loss of AQP4 might alter amygdala-dependent learning behavior. To test this hypothesis, we next performed a comparative analysis of Pavlovian fear conditioning (Figure 2a). AQP4 KO and WT mice exhibited similar baseline behavior before CS. The freezing behavior of both KO and WT mice increased without any significant difference during the training of fear conditioning (Figure 2b), indicating that KO mice exhibit normal acquisition of cued fear memory. AQP4 KO mice exhibited reduced freezing 24 h after training compared with WT mice (WT: 50.8±3.7%, n=8; KO: 35.5±1.8%, n=8; P<0.01; Figure 2b), whereas they exhibited unaltered freezing 2 h after training (WT: 52.9±3.3%, n=8; KO: 51.1±4.7%, n=8; p>0.05; Figure 2b), indicating that AQP4 deficiency impairs the consolidation of associative fear memory.

Aquaporin-4 (AQP4) deficiency impairs the associative fear memory of mice. (a) Experimental design of fear conditioning tasks. (b) Freezing percentage before conditioned stimulus (pre-CS), during conditioning (training) and 24 h after conditioning (test). Wild-type (WT) and knockout (KO) mice exhibited similar freezing in conditioning chamber before CS and during conditioning training. However, freezing behavior was significantly reduced in KO mice (n=8) 24 h after conditioning training compared with WT mice (n=8). The data are expressed by mean±SEM. **P<0.01 vs WT. (c) Total distance animals moved during 10 min intervals in open field. No difference was observed between KO (n=8) and WT (n=8) mice. (d) Average speed animals moved during 10 min intervals in open field. No difference was observed between KO (n=8) and WT (n=8) mice. (e) The number of animal entries to the open arm as the percentage of the total number of arm entries in the elevated plus maze. No difference was observed between KO (n=8) and WT (n=8) mice. (f) The total number of animal entries to open and closed arms in the elevated plus maze. No difference was observed between KO (n=8) and WT (n=8) mice. (g) Thresholds of shock intensities for vocalizing and jumping are shown. No difference was detected between KO (n=8) and WT (n=7) mice.
Although both groups showed similar freezing responses during the acquisition phase, it cannot be ruled out that the impairment in fear memory retention originated from a decrease in freezing behavior due to unknown reasons (eg, differences in the motor behavior and/or anxiety-related behavior), rather than the fear-associated learning process. To assess possible general behavioral defects in KO mice, we initially performed a 10 min analysis of locomotive behavior in the open field. There were no differences in the locomotor activity between WT and KO mice. As shown in Figure 2c and d, KO mice displayed normal general locomotor activity assessed by the total distance traveled (WT, 14.6±1.5 m, n=8; KO, 12.2±3.0 m, n=8; Figure 2c) and the average movement speed (WT, 5.2±0.5 cm/s; KO, 4.9±0.6 cm/s; Figure 2d), which ruled out the possibility that freezing changes are due to hyperlocomotive activity in KO mice.
Furthermore, we investigated the anxiety-related behavior of WT and KO mice in the EPM. It was also found that there was no difference between WT and KO mice in the EPM. The total number of open arm entries was not significantly altered in KO mice (18.5±5.0% of overall entries, n=8) compared with WT mice (20.3±4.0% of overall entries, n=8; Figure 2e). Accordingly, the total number of entries (WT: 11.3±3.2; KO: 10.4±2.9; Figure 2f) and the number of entries to the closed arms (data not shown) were similar for the two genotypes. Taken together, these data argue against altered level of innate fear and anxiety in KO mice.
To exclude the possibility that nociception is altered in KO mice, the pain thresholds of WT and KO mice to the electric footshock were examined. There was no difference between WT and KO mice in vocalization and jump in response to gradually increased intensity of electric shock (WT, 0.33±0.02 mA for vocalizing and 0.38±0.02 mA for jumping, n=7; KO, 0.31±0.04 mA for vocalizing and 0.36±0.03 mA for jumping, n=8; Figure 2g), which indicates that the impairment of fear memory is not due to pain sensitivity in KO mice.
Collectively, these results suggest that the observed differences in fear conditioning were not likely to have been the result of a sensory or motor deficit.
AQP4 Deficiency Downregulates GLT-1 Expression Without Altering GLAST Level in the LA
It was reported that AQP4 deficiency reduced glutamate uptake and GLT-1 expression in primary cultured cortical astrocytes (Zeng et al, 2007). We presumed that AQP4 deficiency would influence the expression of GLTs in amygdala tissue. As GLT-1 and GLAST are mostly found in astrocytes (Rothstein et al, 1994), we investigated the regulatory role of AQP4 on GLT-1 and GLAST in amygdala tissue from AQP4 WT and KO mice using western blotting analysis. In amygdala tissue from AQP4 KO and WT mice, the bulk of the GLAST and GLT-1 proteins appeared as a monomeric band at the apparent molecular weights of 65 and 70 kDa, respectively (Figure 3a and b). Densitometric analysis showed that no significant differences in the expression of GLAST were observed between the two genotypes (n=6; Figure 3a and c). However, the expression of GLT-1 (GLT-1/β-actin) was dramatically reduced in AQP4 KO mice (51.6±4.1% of WT) compared with those in WT mice (n=6, P<0.01; Figure 3b and d). These results suggest that AQP4 deficiency downregulates GLT-1 expression without altering GLAST level in the amygdala.

Aquaporin-4 (AQP4) deficiency downregulates glutamate transporter-1 (GLT-1) expression without altering GLAST level in the lateral amygdala (LA). (a) Representative images of immunoblots using antibodies against GLAST-1 and β-actin. (b) Representative images of immunoblots using antibodies against GLT-1 and β-actin. (c) Quantitative analyses of GLAST by densitometry are summarized. Data were expressed as a percentage of value of wild-type (WT). No significant differences in GLAST expression were observed between AQP4 knockout (KO) and WT mice. (d) Quantitative analyses of GLT-1 by densitometry are summarized. Data were expressed as percentage of value of WT. The expression of GLT-1 was dramatically reduced in AQP4 KO mice compared with those in WT mice (n=6, **P<0.01 vs WT).
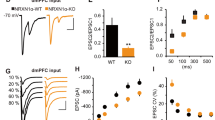
AQP4 Deficiency Selectively Increases NMDAR-Mediated Currents in the LA
It is well established that NMDARs have a higher affinity for glutamate than AMPARs (Patneau and Mayer, 1990). GLT-1 is responsible for the largest proportion of glutamate transport in the brain (Danbolt, 2001). The deficiency or downregulation of GLT-1 cause the excessive accumulation of glutamate in the synaptic cleft, which in turn result in excessive activation of neuronal NMDAR (Danbolt, 2001; Katagiri et al, 2001). Considering that AQP4 deficiency downregulates GLT-1 expression, we next assessed synaptic strength by measuring the ratio of NMDAR- to AMPAR-mediated EPSC amplitudes (NMDAR/AMPAR), and examined whether NMDAR-mediated EPSCs in AQP4 KO mice was affected by the mutation. The NMDAR/AMPAR ratio has been used reliably to compare synaptic strengths in different brain slices, because it is independent of experimental conditions of slice recording such as the positioning of the electrodes and differences in slice preparation (Saal et al, 2003). We observed that the NMDAR/AMPAR ratio was significantly larger in AQP4 KO mice (0.95±0.09, n=4 cells from 4 mice) than that in WT mice (0.59±0.06, n=4 cells from 4 mice, P<0.05; Figure 4a and b). To test whether this significant increase in NMDAR/AMPAR ratio could be due to alteration(s) of either NMDAR- or AMPAR-mediated transmission or both, we compared NMDAR- and AMPAR-mediated EPSCs. When the stimulus strength was adjusted to give a similar range of amplitudes of AMPAR-mediated EPSCs, we recorded NMDAR-mediated EPSCs without changing the stimulus strength. There was no difference in amplitudes of AMPAR-mediated EPSCs from LA principal neurons between WT and KO mice (WT, 126.4±20.8 pA; KO, 131.1±15.4 pA; Figure 4c). However, the amplitudes of NMDAR-mediated EPSCs from LA principal neurons of KO mice were significantly increased compared with those of WT mice (WT, 68.5±16.0 pA; KO, 119.1±23.4 pA; P<0.05; Figure 4d). These findings indicate that depletion of AQP4 causes a selective increase in NMDAR-dependent EPSCs, but had little effect on AMPAR-dependent EPSCs. These results indicate that NMDARs are preferentially activated than AMPARs in the LA of KO mice, due to the increased glutamate in the synaptic cleft caused by downregulation of GLT-1.

Aquaporin-4 (AQP4) deficiency selectively increases N-methyl-D-aspartate receptor (NMDAR)-mediated currents in the lateral amygdala (LA). (a) Representative traces of AMPA receptor (AMPAR) (lower traces)- and NMDAR (upper traces)-mediated excitatory postsynaptic currents (EPSCs) in AQP4 wild-type (WT) and knockout (KO) mice. Stimulus artifacts were omitted for clarity. (b) Summary histogram for the ratio of NMDAR- to AMPAR-mediated EPSC amplitudes in WT and KO mice. The NMDAR/AMPAR ratio was significantly larger in KO mice (0.95±0.09, n=4 cells from 4 mice) than in WT mice (0.59±0.06, n=4 cells from 4 mice), *P<0.05 vs WT. (c) Summary histograms for AMPAR-mediated EPSCs in WT and KO mice. The mean amplitudes of AMPAR-mediated EPSCs were not significantly different among groups. (d) Summary histograms for NMDAR-mediated EPSCs in WT and KO mice. The mean amplitudes of NMDAR-mediated EPSCs of KO mice (119.1±23.4 pA) were significantly increased compared with those in WT mice (68.5±16.0 pA). *P<0.05 vs WT.
Excessive Activation of NMDAR Contributes to the Impairment of LTP in KO Mice
It has been shown that LTP induction in the thalamo-amygdala pathway is dependent on the activation of postsynaptic NMDARs (Jung et al, 2010), and NMDAR is activated excessively by the increased glutamate in GLT-1 KO mice (Katagiri et al, 2001). As AQP4 deficiency downregulates GLT-1 expression in the amygdala and selectively increases NMDAR-mediated currents, we presumed that NMDARs were activated excessively by the increased glutamate in the synaptic cleft during HFS, which may result in the impairment of LTP in the thalamo-LA pathway of KO mice. To test this possibility, we examined the effect of low concentration of D-APV, an NMDAR antagonist, on LTP induction in the thalamo-LA pathway of WT and KO mice. The electrophysiological studies showed that HFS induced a normal LTP in WT mice (137.9±6.6%; n=11 slices from 6 mice), but an obviously impaired LTP in the presence of 1 μM D-APV (116.1±5.5%; n=8 slices from 4 mice; P<0.05 vs WT; Figure 5a and b). However, pre-incubation of amygdala slices with D-APV (1 μM) for 10 min reversed LTP deficits in KO mice (KO: 111.1±5.1%, n=13 slices from 7 mice; KO+D-APV: 141.2±4.3%, n=8 slices from four mice; P<0.05; Figure 5c and d). These results suggest that the excessive activation of NMDARs contributes to the impairment of LTP in AQP4 KO mice.

Excessive activation of N-methyl-D-aspartate receptor (NMDAR) contributes to the impairment of long-term potentiation (LTP) in knockout (KO) mice. (a) Time course of the field excitatory postsynaptic potential (fEPSP) evoked by stimulation at thalamo-lateral amygdala (LA) pathway of wild-type (WT) mice in the absence (n=11 slices from 6 mice) or presence (n=8 slices from 4 mice) of 1 μM D-2-amino-5-phosphonovaleric acid (D-APV), an NMDAR antagonist. (Inset) Schematic representation of fEPSP recorded in individual slices before (1) and 60 min after (2) the high-frequency stimulation (HFS) (five trains at 100 Hz for 1 s with 90 s interval between trains) in the absence (left) or presence (right) of D-APV. (b) The histogram showed the level of LTP 60 min after HFS at thalamo-LA pathway in WT mice in the absence or presence of 1 μM D-APV. *P<0.05 vs WT. (c) Time course of the fEPSP evoked by stimulation at thalamo-LA pathway in aquaporin-4 (AQP4) KO mice in the absence (n=13 slices from 7 animals) or presence (n=8 slices from 4 animals) of 1 μM D-APV. (Inset) Schematic representation of fEPSP recorded in individual slices before (1) and 60 min after (2) the HFS in the absence (left) or presence (right) of D-APV. (d) The histogram showed the level of LTP 60 min after HFS at thalamo-LA pathway in AQP4 KO mice in the absence or presence of 1 μM D-APV. Each point was the normalized mean±SEM of slices. ##P<0.01 vs KO.
Upregulation of GLT-1 Reverses the Impairment of LTP and Fear Memory in AQP4 KO Mice
Ceftriaxone (Cef) is known to increase both the expression and activity of GLT-1 (Rothstein et al, 2005). The KO mice were treated with Cef (Sigma) to upregulate GLT-1 and determine whether the impairment of LTP and fear memory could be reversed. AQP4 KO mice received a daily intraperitoneal (i.p.) injection of saline or Cef (200 mg/kg/day dissolved in saline) for 5 days. At 24 h after the final injection, mice were used for western blotting analysis, electrophysiological studies, and fear conditioning test, respectively (Figure 6a). Western blotting analysis showed that Cef treatment significantly increased GLT-1 expression by 49.9±8.2% in LA tissue of AQP4 KO mice (n=6, P<0.01 vs KO; Figure 6b). The electrophysiological studies showed that the relative slope of fEPSP 60 min after HFS was 110.5±5.3% of baseline in saline-treated KO mice (n=9 slices from 5 mice), but potentiated to 139.8±4.4% in Cef-treated KO mice (n=10 slices from 5 mice; P<0.01; Figure 6c and d). Fear conditioning test showed that Cef- and saline-treated KO mice exhibited similar baseline behaviors before CS. However, Cef-treated KO mice exhibited significantly increase in freezing behavior 24 h after conditioning training (44.7±4.6%, n=8) compared with saline-treated KO mice (31.5±3.5%, n=8, p<0.05; Figure 6e). Taken together, these results indicate that chronic treatment with Cef reverses the impairment of LTP and fear memory in KO mice by upregulating GLT-1 expression.

Ceftriaxone (Cef) treatment (intraperitoneally (i.p.), 200 mg/kg/day for 5 days) reverses the impairment of long-term potentiation (LTP) and fear memory in aquaporin-4 knockout (AQP4 KO) mice. (a) Experimental design of electrophysiological studies and fear conditioning tasks. (b) The expression of glutamate transporter-1 (GLT-1) in amygdala tissues of saline- and Cef-treated KO mice. (Above) Representative images of immunoblots using antibodies against GLT-1 and β-actin. (Below) Quantitative analyses of GLT-1 by densitometry are summarized. Data were expressed as percentage of value of KO. Cef treatment (i.p. 200 mg/kg/day for 5 days) increased GLT-1 expression in KO mice (n=6, ##P<0.01 vs KO). (c) Time course of the field excitatory postsynaptic potential (fEPSP) evoked by stimulation at thalamo-lateral amygdala (LA) pathway in saline- and Cef-treated KO mice. (Inset) Schematic representation of fEPSP recorded in individual slices before (1) and 60 min after (2) the LTP-inducing stimulation in saline- (left) and Cef-treated (right) KO mice. Cef treatment reversed the impairment of LTP in KO mice. (d) The histogram showed the level of LTP 60 min after high-frequency stimulation (HFS) in saline- (n=9 slices from 5 mice) and Cef-treated KO mice (n=10 slices from 5 mice). Each point was the normalized mean±SEM of slices. ##P<0.01 vs saline-treated KO mice. (e) Total percentage of freezing in saline (n=8) and Cef-treated KO mice (n=8) when tested before conditioned stimulus (pre-CS) and 24 h after conditioning training (test), respectively. Data are expressed by mean±SEM. #P<0.05 vs saline-treated KO mice.
DISCUSSION
These studies demonstrated a new role for AQP4 in synaptic plasticity and memory. We found that AQP4 deficiency impaired LTP in the thalamo-LA pathway and amygdala-dependent fear memory of adult mice. The impairment was involved in the downregulation of GLT-1 expression, which in turn resulted in the redundant glutamate in the synaptic cleft and excessive activation of NMDARs. Interestingly, upregulation of GLT-1 expression reversed the impairment. These findings support an unanticipated role for AQP4 in synaptic plasticity and associative fear memory by regulating GLT-1 expression. The results also provide new evidence for astrocytic function in the regulation of synaptic plasticity.
AQP4 is mostly expressed in astrocytes (Amiry-Moghaddam and Ottersen, 2003), and plays a significant role in regulating water homeostasis and astrocytic function (Verkman, 2005; Zeng et al, 2007). A growing body of evidence indicates that astrocyte function is implicated in synaptic plasticity (Henneberger et al, 2010). We found that AQP4 was also expressed in LA. Therefore, it is likely that AQP4 deficiency influence synaptic plasticity in the amygdala. These studies confirmed that AQP4 deficiency impaired the LTP induced by HFS in the thalamo-LA pathway. LTP reflects a persistent enhancement in synaptic strength in which both presynaptic and postsynaptic mechanisms might be involved (Malenka and Nicoll, 1999). IOR and PPF are two important parameters of synaptic transmission likely primarily related to presynaptic function. It is worth noting that AQP4 deficiency had no effect on IOR and PPF in the thalamo-LA pathway, arguing against a critical function of AQP4 for presynaptic transmitter release. Therefore, the impairment of LTP in AQP4 KO mice is due to the changes in postsynaptic response to glutamate. Notably, Skucas et al (2011) recently reported that AQP4 KO impaired the TBS-induced LTP, but exhibited the normal LTP induced by HFS in hippocampal slices. In contrast, we found that AQP4 deficiency impaired the HFS-induced LTP in the thalamo-LA pathway. The possible reasons are that different brain regions (hippocampus and amygdala) have different response to tetanic stimulation in AQP4 KO mice, and the experimental conditions, such as the parameter of HFS and the composition of artificial CSF, are different in Skucas’ and our studies.
LTP is widely considered to be one of the major mechanisms by which the brain ‘acquires’ and ‘stores’ information (Citri and Malenka, 2008; Neves et al, 2008). The amygdala is a key component of the neural circuitry underlying both innate and acquired fear behavior (Barad et al, 2006; Kim and Jung, 2006). Therefore, the effect of AQP4 deficiency on synaptic plasticity may result in the impairment of associative fear memory. Fear conditioning task was performed to examine the behavioral consequences of the depletion of AQP4 at the intact animal level. Considering that the hearing of AQP4 KO mice is impaired (Li and Verkman, 2001), we used light but not tone as CS in fear conditioning test. In Pavlovian fear conditioning, memory acquisition refers to the process mediating the initial learning of the CS–US association during the training of fear conditioning, whereas memory consolidation refers to the process converting short-term memories into stable long-term memories (Abel and Lattal, 2001; Makkar et al, 2010). Cued fear memory encoded in the amygdala has been shown to be consolidated within 24 h and short-term fear memory is known to be maintained for up to 4 h (Schafe and LeDoux, 2000). Therefore, if there is an impairment in the consolidation of cued fear memory, memory retention assessed 24 h after conditioning would be impaired, whereas memory retention assessed within 4 h after conditioning would be intact (Lee et al, 2011). In this study, we found that the freezing behavior of AQP4 KO mice significantly reduced 24 h after training without any significant difference during the training and 2 h after training when compared with that of WT mice, indicating that AQP4 deficiency impairs the consolidation of associative fear memory without altering the normal acquisition of fear memory. Interestingly, it was reported that AQP4 deficiency had no effect on contextual fear memory (Skucas et al, 2011), which seems inconsistent with our result. In fact, these results are not contradictory. Contextual fear memory is hippocampus-dependent, whereas cued fear memory is amygdala-dependent. The mechanisms of the two fear memories are not identical, and the impairment of cued fear memory is not necessarily associated with contextual fear memory. Therefore, it is not surprising that AQP4 KO mice exhibited impairment on cued fear memory, but had a negligible functional impact on contextual fear memory.
It is likely that AQP4 may alter associative fear learning by modulating elementary behavioral aspects. To address this issue, we also examined locomotor activity and anxiety-related behavior. None of these tests yielded a significant difference between WT and KO mice, ruling out the possibility that the observed differences in fear conditioning is due to a motor deficit. Moreover, it is important to exclude the possibility that the impairment of fear memory is due to the alteration of pain threshold, Bao et al (2010) recently reported that pain thresholds of KO mice were increased with thermal and chemical stimulation, but not altered with mechanical stimulation. However, we found the pain threshold of KO mice did not alter with electric foot shocks. The possible reason for this is that the pain response of KO mice to different stimulation is not identical, but its mechanism is unclear. Even if the pain threshold of KO mice is increased with electric foot shocks, there is little impact on the test of fear memory, because the stimulation intensity (0.7 mA) used in training of fear conditioning is much higher than pain threshold of KO mice (0.36±0.03 mA) in this study. Therefore, it is impossible that the impairment of fear memory is due to the alteration of pain threshold. Taken together, these data that the expression pattern of AQP4 in the amygdala acts in concert with the impairment of synaptic plasticity in the LA of AQP4-deficient mice reinforce the view that AQP4 plays a role in cued fear memory.
Glutamate is the principal excitatory neurotransmitter in the CNS. During neural activity, glutamate rapidly diffuses into synaptic cleft and is quickly uptaken by GLTs in astrocytes (Clements, 1996). GLT-1 is responsible for more than 90% glutamate uptake of astrocytes (Danbolt, 2001; Rothstein et al, 1996). AQP4 is prominently present in astrocytes where GLTs are localized (Nielsen et al, 1997). The interaction between the two protein molecules is speculated to be an intimate spatial relationship and be parts of a same supramolecular complex. Therefore, it is presumed that AQP4 KO disturbed the direct physical contact between AQP4 and GLTs, as well as their signaling transduction and the functions of GLTs. Previous studies have proved that AQP4 deficiency downregulates GLT-1 expression and reduces glutamate uptake in astrocytes (Zeng et al, 2007). Consistent with the previous report, we also showed that GLT-1 expression was decreased in LA tissue from AQP4 KO mice. These results provide direct evidence for the interaction between AQP4 and GLTs. Thus, it is possible that AQP4 deficiency reduces glutamate uptake in LA and results in excessive accumulation of glutamate in the synaptic cleft. Moreover, reducing GLT-1 function can also result in the increase of extracellular glutamate concentrations, but it is unclear whether the AQP4 deficiency reduces GLT-1 function. In general, increasing glutamate release or reducing glutamate uptake can alter basal synaptic transmission. However, we found that AQP4 deficiency reduced GLT-1 expression, which accordingly reduced glutamate uptake, but failed to have a major effect on basal synaptic transmission, as reported by Skucas et al (2011). One possible reason for this is that AQP4 deficiency reduces neuronal excitability by expanding ECS volume (Binder et al, 2004, 2006), which has been demonstrated to play an important role in cell–cell communication and extracellular buffering of ions such as K+ and glutamate. Therefore, the activation of glutamate receptors in KO mice requires much more glutamate than that in WT mice. When recording basal fEPSP induced by single stimulus in amygdala slices, increased glutamate in the synaptic cleft in KO mice likely produced the similar effect on basal synaptic transmission with that in WT mice. Therefore, it is possible that AQP4 deficiency reduces GLT-1 expression without affecting basal synaptic transmission.
Appropriate glutamate concentration in the synaptic cleft is required for LTP induction. However, the excessive accumulation of glutamate in the synaptic cleft may result in neuronal overactivation and neurodegenerative diseases. Therefore, the maintenance of physiological level of extracellular glutamate is crucial to normal synaptic transmission and prevention of excitotoxicity. Increasing evidence shows that deficiency of GLT-1 causes excitotoxicity and excessive activation of NMDAR, and then impaired LTP and fear memory (Danbolt, 2001; Katagiri et al, 2001; Rothstein et al, 1996; Tzingounis and Wadiche, 2007; Wang et al, 2006). In this study, we found that AQP4 deficiency increased the NMDAR-dependent EPSCs without altering AMPAR-dependent EPSCs in LA. One reasonable interpretation of this result is that the downregulation of GLT-1 increased the glutamate concentration in the synaptic cleft, which in turn excessively activated NMDARs. As astrocytes may release glutamate from volume-sensitive channels (Takano et al, 2005), there is the possibility that reduced water transport via AQP4 alter glutamate release from volume-sensitive channels in astrocytes. Thus, increased activation of NMDAR in AQP4 KO mice may also result from altered glutamate release and uptake in astrocytes. It is well known that LTP induction in the thalamo-amygdala pathway is dependent on activation of postsynaptic NMDARs. However, the excessive activation of NMDARs during HFS could result in the impairment of LTP (Katagiri et al, 2001). We found that low concentration of NMDAR antagonist reversed the impairment of LTP in KO mice, suggesting that the excessive activation of NMDARs blocks LTP induction in AQP4 KO mice. In fact, even in normal animals, excessive activation of NMDARs could impair LTP induction in conditions such as ischemia or brain damage during which a large amount of glutamate is released, and the impairment could be prevented by partial blockade of NMDARs in these conditions. It has been reported that extrasynaptic NMDAR is involved in neurodegenerative disorders (Hardingham and Bading, 2010) and exerts different functions from synaptic NMDAR in many physiological processes (Gladding and Raymond, 2011). Moreover, increasing extracellular glutamate levels excessively activate extrasynaptic NR2B-containing NMDAR and inhibit LTP (Li et al, 2011). Therefore, we presume that the impairment of LTP in AQP4 KO mice may involve the excessive activation of extrasynaptic NR2B-containing NMDAR, which requires to be further investigated in the future.
As the downregulation of GLT-1 impairs LTP in KO mice, it is possible that the upregulation of GLT-1 may reverse this impairment. It is well known that Cef, one of the β-lactam antibiotics, increases expression and activity of GLT-1 (Rothstein et al, 2005). Previous studies have shown that single daily injection of Cef (200 mg/kg) for 5–7 days increases the GLT-1 expression and glutamate uptake in both the cortex and the striatum, as well as in the nucleus accumbens (Knackstedt et al, 2010; Miller et al, 2008; Sari et al, 2010). Therefore, the Cef-induced increase in GLT-1 expression has a direct effect on glutamate function. Consistent with this view, we found that chronic treatment with Cef (i.p., 200 mg/kg) not only increased GLT-1 expression in LA but also reversed the impairment of LTP and fear memory in AQP4 KO mice. Our behavioral test indicated that Cef did not alter locomotor activity or pain thresholds of mice (data not shown), which was consistent with previous reports (Miller et al, 2008; Rothstein et al, 2005). Thus, accelerated clearance of extracellular glutamate appears to underlie the ability of Cef to reverse the impairment of LTP and fear memory. Taken together, these findings confirm that impairment of LTP and fear memory in AQP4 KO mice is due to the downregulation of GLT-1 expression, which in turn results in excessive accumulation of glutamate in the synaptic cleft and excessive activation of NMDARs.
In general, the induction of LTP is related to neuronal activity that is sensitive to the extracellular K+ concentration. There have been some studies about the impact of AQP4 deficiency on K+ channels and extracellular K+ concentration (Binder et al, 2006; Nagelhus et al, 2004; Zhang and Verkman, 2008), but the concept is inconsistent. Recently, Strohschein et al (2011) reported that stimulation in the alveus evoked smaller increases and slower recovery of [K+]o in the stratum pyramidal of AQP4-deficient mice when compared with WT mice. These results suggest that the impairment of LTP in AQP4-deficient mice may also involve the alteration of extracellular K+ concentration. In the study of Strohschein et al (2011), the paired-pulse stimulation and strains stimulation (10 s, 20 Hz) but not HFS (1 s, 100 Hz) was used to evoke the increase in [K+]o, whether AQP4 deficiency affects the increase in [K+]o induced by HFS is still unclear. Hence, the correlation of HFS-induced LTP and K+ buffering in AQP4-deficient mice is uncertain.
In conclusion, we demonstrate that AQP4 regulates synaptic plasticity in the LA and associative fear memory by regulating GLT-1 expression. Considering that amygdala is important in emotional and fear memories and related to many psychiatric disorders, our novel findings imply that the regulation of emotional memory by modulating AQP4 expression may represent a target for the treatment of cognitive or psychiatric disorders such as post-traumatic stress disorder and anxiety disorder.
References
Abel T, Lattal KM (2001). Molecular mechanisms of memory acquisition, consolidation and retrieval. Curr Opin Neurobiol 11: 180–187.
Amara SG, Fontana AC (2002). Excitatory amino acid transporters: keeping up with glutamate. Neurochem Int 41: 313–318.
Amiry-Moghaddam M, Ottersen OP (2003). The molecular basis of water transport in the brain. Nat Rev Neurosci 4: 991–1001.
Bao F, Chen M, Zhang Y, Zhao Z (2010). Hypoalgesia in mice lacking aquaporin-4 water channels. Brain Res Bull 83: 298–303.
Barad M, Gean PW, Lutz B (2006). The role of the amygdala in the extinction of conditioned fear. Biol Psychiatry 60: 322–328.
Binder DK, Papadopoulos MC, Haggie PM, Verkman AS (2004). In vivo measurement of brain extracellular space diffusion by cortical surface photobleaching. J Neurosci 24: 8049–8056.
Binder DK, Yao X, Zador Z, Sick TJ, Verkman AS, Manley GT (2006). Increased seizure duration and slowed potassium kinetics in mice lacking aquaporin-4 water channels. Glia 53: 631–636.
Chi Y, Fan Y, He L, Liu W, Wen X, Zhou S et al (2011). Novel role of aquaporin-4 in CD4+ CD25+ T regulatory cell development and severity of Parkinson's disease. Aging Cell 10: 368–382.
Citri A, Malenka RC (2008). Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33: 18–41.
Clements JD (1996). Transmitter timecourse in the synaptic cleft: its role in central synaptic function. Trends Neurosci 19: 163–171.
Danbolt NC (2001). Glutamate uptake. Prog Neurobiol 65: 1–105.
Davis M (1992). The role of the amygdala in fear and anxiety. Annu Rev Neurosci 15: 353–375.
Ding JY, Kreipke CW, Speirs SL, Schafer P, Schafer S, Rafols JA (2009). Hypoxia-inducible factor-1alpha signaling in aquaporin upregulation after traumatic brain injury. Neurosci Lett 453: 68–72.
Fan Y, Kong H, Shi X, Sun X, Ding J, Wu J et al (2008). Hypersensitivity of aquaporin 4-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine and astrocytic modulation. Neurobiol Aging 29: 1226–1236.
Fan Y, Zhang J, Sun XL, Gao L, Zeng XN, Ding JH et al (2005). Sex- and region-specific alterations of basal amino acid and monoamine metabolism in the brain of aquaporin-4 knockout mice. J Neurosci Res 82: 458–464.
Friedman B, Schachtrup C, Tsai PS, Shih AY, Akassoglou K, Kleinfeld D et al (2009). Acute vascular disruption and aquaporin 4 loss after stroke. Stroke 40: 2182–2190.
Gladding CM, Raymond LA (2011). Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci 48: 308–320.
Hardingham GE, Bading H (2010). Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11: 682–696.
Haydon PG, Blendy J, Moss SJ, Rob Jackson F (2009). Astrocytic control of synaptic transmission and plasticity: a target for drugs of abuse? Neuropharmacology 56 (Suppl 1): 83–90.
Henneberger C, Papouin T, Oliet SH, Rusakov DA (2010). Long-term potentiation depends on release of D-serine from astrocytes. Nature 463: 232–236.
Jung SY, Kim J, Kwon OB, Jung JH, An K, Jeong AY et al (2010). Input-specific synaptic plasticity in the amygdala is regulated by neuroligin-1 via postsynaptic NMDA receptors. Proc Natl Acad Sci USA 107: 4710–4715.
Katagiri H, Tanaka K, Manabe T (2001). Requirement of appropriate glutamate concentrations in the synaptic cleft for hippocampal LTP induction. Eur J Neurosci 14: 547–553.
Kettenmann H, Ransom BR (2005). Neuroglia, 2nd edn. Oxford University Press: New York, p. xix, 601pp.
Kim JJ, Jung MW (2006). Neural circuits and mechanisms involved in Pavlovian fear conditioning: a critical review. Neurosci Biobehav Rev 30: 188–202.
Knackstedt LA, Melendez RI, Kalivas PW (2010). Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry 67: 81–84.
Kong H, Sha LL, Fan Y, Xiao M, Ding JH, Wu J et al (2009). Requirement of AQP4 for antidepressive efficiency of fluoxetine: implication in adult hippocampal neurogenesis. Neuropsychopharmacology 34: 1263–1276.
LeDoux JE (2000). Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184.
Lee EJ, Son GH, Chung S, Lee S, Kim J, Choi S et al (2011). Impairment of fear memory consolidation in maternally stressed male mouse offspring: evidence for nongenomic glucocorticoid action on the amygdala. J Neurosci 31: 7131–7140.
Li J, Verkman AS (2001). Impaired hearing in mice lacking aquaporin-4 water channels. J Biol Chem 276: 31233–31237.
Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011). Soluble Abeta oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31: 6627–6638.
Ma T, Yang B, Gillespie A, Carlson EJ, Epstein CJ, Verkman AS (1997). Generation and phenotype of a transgenic knockout mouse lacking the mercurial-insensitive water channel aquaporin-4. J Clin Invest 100: 957–962.
Makkar SR, Zhang SQ, Cranney J (2010). Behavioral and neural analysis of GABA in the acquisition, consolidation, reconsolidation, and extinction of fear memory. Neuropsychopharmacology 35: 1625–1652.
Malenka RC (2003). The long-term potential of LTP. Nat Rev Neurosci 4: 923–926.
Malenka RC, Nicoll RA (1999). Long-term potentiation—a decade of progress? Science 285: 1870–1874.
Mayer ML, Westbrook GL (1987). The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol 28: 197–276.
Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR et al (2008). Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience 153: 329–337.
Mothet JP, Rouaud E, Sinet PM, Potier B, Jouvenceau A, Dutar P et al (2006). A critical role for the glial-derived neuromodulator D-serine in the age-related deficits of cellular mechanisms of learning and memory. Aging Cell 5: 267–274.
Nagelhus EA, Mathiisen TM, Ottersen OP (2004). Aquaporin-4 in the central nervous system: cellular and subcellular distribution and coexpression with KIR4.1. Neuroscience 129: 905–913.
Neves G, Cooke SF, Bliss TV (2008). Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci 9: 65–75.
Nielsen S, Nagelhus EA, Amiry-Moghaddam M, Bourque C, Agre P, Ottersen OP (1997). Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J Neurosci 17: 171–180.
Patneau DK, Mayer ML (1990). Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J Neurosci 10: 2385–2399.
Perea G, Navarrete M, Araque A (2009). Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32: 421–431.
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW et al (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675–686.
Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D et al (1994). Localization of neuronal and glial glutamate transporters. Neuron 13: 713–725.
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE et al (2005). Beta-lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature 433: 73–77.
Saal D, Dong Y, Bonci A, Malenka RC (2003). Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577–582.
Sari Y, Prieto AL, Barton SJ, Miller BR, Rebec GV (2010). Ceftriaxone-induced up-regulation of cortical and striatal GLT1 in the R6/2 model of Huntington's disease. J Biomed Sci 17: 62.
Schafe GE, LeDoux JE (2000). Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci 20: RC96.
Skucas VA, Mathews IB, Yang J, Cheng Q, Treister A, Duffy AM et al (2011). Impairment of select forms of spatial memory and neurotrophin-dependent synaptic plasticity by deletion of glial aquaporin-4. J Neurosci 31: 6392–6397.
Strohschein S, Huttmann K, Gabriel S, Binder DK, Heinemann U, Steinhauser C (2011). Impact of aquaporin-4 channels on K+ buffering and gap junction coupling in the hippocampus. Glia 59: 973–980.
Suzuki A, Stern SA, Bozdagi O, Huntley GW, Walker RH, Magistretti PJ et al (2011). Astrocyte-neuron lactate transport is required for long-term memory formation. Cell 144: 810–823.
Takano T, Kang J, Jaiswal JK, Simon SM, Lin JH, Yu Y et al (2005). Receptor-mediated glutamate release from volume sensitive channels in astrocytes. Proc Natl Acad Sci USA 102: 16466–16471.
Thrane AS, Rappold PM, Fujita T, Torres A, Bekar LK, Takano T et al (2011). Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci USA 108: 846–851.
Tzingounis AV, Wadiche JI (2007). Glutamate transporters: confining runaway excitation by shaping synaptic transmission. Nat Rev Neurosci 8: 935–947.
Verkman AS (2005). More than just water channels: unexpected cellular roles of aquaporins. J Cell Sci 118: 3225–3232.
Wang ZY, Zhang YQ, Zhao ZQ (2006). Inhibition of tetanically sciatic stimulation-induced LTP of spinal neurons and Fos expression by disrupting glutamate transporter GLT-1. Neuropharmacology 51: 764–772.
Zeng XN, Sun XL, Gao L, Fan Y, Ding JH, Hu G (2007). Aquaporin-4 deficiency down-regulates glutamate uptake and GLT-1 expression in astrocytes. Mol Cell Neurosci 34: 34–39.
Zhang H, Verkman AS (2008). Aquaporin-4 independent Kir4.1 K+ channel function in brain glial cells. Mol Cell Neurosci 37: 1–10.
Zucker RS (1989). Short-term synaptic plasticity. Annu Rev Neurosci 12: 13–31.
Acknowledgements
We are grateful to Dr Gang Hu from Nanjing Medical University for supplying mice. This work was supported by grants from the Key Project of National Natural Science Foundation of China (NSFC No. 30930104) and NSFC grant (No. 81171258) to Dr JG Chen. It was also supported by the Program for New Century Excellent Talents in Universities of China (NCET-08-0225) and NSFC Grant (No. 81173039) to Dr F Wang.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Li, YK., Wang, F., Wang, W. et al. Aquaporin-4 Deficiency Impairs Synaptic Plasticity and Associative Fear Memory in the Lateral Amygdala: Involvement of Downregulation of Glutamate Transporter-1 Expression. Neuropsychopharmacol 37, 1867–1878 (2012). https://doi.org/10.1038/npp.2012.34
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/npp.2012.34
Keywords
This article is cited by
-
Maternal e-cigarette use can disrupt postnatal blood-brain barrier (BBB) integrity and deteriorates motor, learning and memory function: influence of sex and age
Fluids and Barriers of the CNS (2023)
-
Sleep fragmentation affects glymphatic system through the different expression of AQP4 in wild type and 5xFAD mouse models
Acta Neuropathologica Communications (2023)
-
Aquaporin-4 Deficiency is Associated with Cognitive Impairment and Alterations in astrocyte-neuron Lactate Shuttle
Molecular Neurobiology (2023)
-
Inhibition of mGluR5 alters BDNF/TrkB and GLT-1 expression in the prefrontal cortex and hippocampus and ameliorates PTSD-like behavior in rats
Psychopharmacology (2023)
-
Drug development in targeting ion channels for brain edema
Acta Pharmacologica Sinica (2020)
