
Abstract
Insight into the mode of action of newly discovered antiviral agents is now almost a prerequisite for clinical development. This protocol describes a method that provides information on the target of inhibitors of the human immunodeficiency virus (HIV); it can also be adapted to other viruses. The results from this experiment are available within 2 d. This time-based approach determines how long the addition of a compound can be postponed before losing its antiviral activity in cell culture. The target of an antiviral compound can be identified by comparing its relative position in the time scale to that of reference drugs. Therefore, it is more precise than, for example, in the case of HIV, a determination of pre- or postintegrational mode of action, and combines in one routine different assays for studying mechanisms of action.
Similar content being viewed by others

Introduction
Shortly after the isolation of HIV, an intensive search for compounds that would inhibit infectivity and replication of this virus was initiated. A major breakthrough in this search was the establishment of a rapid and automated anti-HIV screening assay1,2. The active development of new anti-HIV compounds continues, and many lead compounds still emerge from initial antiviral screens. Knowing the target of action of these new drugs is not only of extreme importance for swift progress in the development of novel anti-HIV strategies but is also essentially a prerequisite for their clinical development.
Methods for determining drug targets of action
The in vitro tests most commonly used to determine the target of action of anti-HIV drugs are assays measuring the activity of the following viral enzymes: reverse transcriptase (RT) (polymerase and RNaseH activity)3,4,5,6,7, integrase8,9 and protease10. The advantage of these enzymatic assays in the test tube is that they are straightforward to perform, do not require any special safety infrastructure and produce clear interpretable results. However, if there are no previous indications of the presumed target of a new drug, it is difficult to know with which assay to start. In that case, viral resistance is selected against the inhibitor and the target of action is then needed to be determined by sequence analysis, which is time-consuming and cumbersome. Currently, many other test-tube assays have been described for measuring the distinct interactions (protein-protein, RNA-protein) that are essential for viral replication, and new assays are being developed11,12.
However, on the basis of test-tube studies of mechanisms of action, several compounds have been mistakenly ascribed to inhibit a certain target that, upon further study in cell culture, appeared to inhibit an off-site target. For example, suramin was first found to be a very potent inhibitor of the retroviral reverse transcription process in the test tube and to block the in vitro infectivity and cytopathic effect of HIV3,13. It was not only the first antiviral drug to be shown in vitro to block HIV infection but was also the first to be proven effective in suppressing HIV replication in HIV-infected patients14. Although suramin was originally pursued for its anti-HIV potential because of its RT-inhibitory capacity, it has since become clear that, in cell culture, suramin primarily targets the viral adsorption step15 involving the viral envelope glycoprotein gp120 (ref. 16). Another example is the Tat inhibitor CGP64222 (ref. 17), shown to target viral entry in cell culture18. Yet another example is L-chicoric acid and its derivatives, which counteract HIV-1 integrase activity in vitro but in cell culture block virus entry19,20. Therefore, tools to characterize the mechanism of action of virus inhibitors in cell culture are required to define their correct targets of action21.
There are several tests for studying the target of action of antiviral compounds in cell culture. For example, by comparing the activity of a compound on acutely1 versus persistently or latently infected cells22,23,24, it can be easily determined whether an inhibitor targets a pre- or a postintegrational step of the viral replication cycle25. A drug blocking the production of a new virus from persistently infected cells presumably targets a postintegrational event because in the persistent infection system all preintegrational steps, such as virus binding, reverse transcription and integration, are omitted. Alternatively, for several steps in the HIV replication cycle (e.g., virus binding26, integration21, transcription27 and viral budding28), there exist specific tests in cells that can be performed to study the effect of a potential inhibitor of this target. For example, if one presumes that a newly discovered inhibitor interferes with virus binding, a virus-binding assay26 can be performed. However, none of these assays combine all these properties in just one test that covers a full replication cycle.
Overview of the time-of-addition method
In this protocol, we describe an approach routinely used in our lab that can be used to narrow down the mechanism/target of action of a newly discovered anti-HIV drug in cell culture by comparing its time of intervention with that of well-characterized inhibitors. This time-of-addition (TOA) approach determines how long the addition of a compound can be postponed before it loses its antiviral activity and was first used by Pauwels et al.29 to delineate the target of action of tetrahydro-imidazo[4,5,1-jk][1,4]-benzodiazepine-2-(1H)-one (TIBO) derivatives, later recognized as prototype of the NNRTIs (non-nucleoside RT inhibitors). Indeed, when an inhibitor that interferes with, for example, the viral RT enzyme, is present at the time when the reverse transcription process occurs within the viral replication cycle, it will be able to inhibit virus replication. In contrast, when this inhibitor is added once the reverse transcription is completed, the inhibitor will no longer be effective in blocking viral replication.
To replicate, HIV undergoes several major essential steps that occur in a well-established chronological order (see overview in Box 1 and ref. 30 for a review). A single round of HIV-1 replication takes ∼24 h (Fig. 1). Therefore, the standard protocol of a TOA experiment is based on the bulk infection of susceptible cells for 1 h; this is followed by removing the unbound virus and subsequent dispensing of the infected cells in a 96-well tissue culture plate and adding test compounds at 0, 1, 2, 3,..., 24 h after virus infection. Further, the extent of virus replication is monitored at 31 h after infection. To synchronize the infection in all conditions, it is a prerequisite to wash out all unbound virus at 1 h after infection. The distinct steps of the procedure are detailed in Figure 2. Key to the success of the experiment and to obtaining a maximal viral output that can be reliably measured is the use of: a highly susceptible cell line, e.g., the human T-lymphotropic virus type I (HTLV-I)-transformed MT-4 cell line; a high multiplicity of infection (0.5–1); and an optimal harvest time. For HIV, the newly produced virus is harvested 31 h after infection (Fig. 1) to ensure that only one replication cycle has been completed.

Highly susceptible MT-4 cells were infected with HIV-1 at a high multiplicity of infection (0.5). Virus was incubated with cells for 1 h and unbound virus was subsequently removed by extensive and repeated washing to synchronize the replication. Next, infected cells were incubated for different time periods as indicated, and the produced (released) virus in the supernatant was quantified by monitoring the virus-associated Gag core p24 antigen. From 26 h after infection, an exponential increase in virus released in the supernatant of the infected cells was observed, demonstrating that one round of virus replication roughly encompasses 1 d.

Corresponding PROCEDURE step numbers are included.
As other viruses also undergo major processes of replication in a chronological order, this protocol can be adapted for the identification of the target of action of inhibitors of viruses other than HIV (e.g., coronaviruses)31,32. In principle, this protocol is applicable to any virus/inhibitor combination, although in the absence of known inhibitors of the replication of the virus under study, it would be more challenging to interpret the data. However, for most viruses at least a few inhibitors with an established mode of action are known.
The advantage of this method is that a single experiment will give an indication of the possible target of interaction of an inhibitor and provide the basis for further investigations. The major limitation, however, is the availability of well-characterized inhibitors for the virus of interest. In principle, the more well-characterized inhibitors that exist for a certain virus, the more precisely a TOA experiment can reveal the target of action of a new inhibitor. A second limiting factor is that the result obtained from compounds with limited selectivity is not easily interpretable. Indeed, the use of compound concentrations of 10- to 100-fold their antiviral activity (half-maximum inhibitory concentration or IC50) are most favorable in this experiment, and compounds with limited selectivity will often be toxic at these concentrations.

Experimental design
Compound concentration. The antiviral activity (IC50) and toxicity (CC50) of a compound should be determined before a TOA experiment is performed. The concentration of compound used in the TOA experiment is 100-fold its IC50, as established in an MT-4/MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay1. It should be confirmed that the compound is not toxic to the cells at this concentration; if so, a lower concentration should be used, although we recommend not to use a concentration lower than tenfold its IC50, as this could result in an insufficient inhibition of viral replication. Moreover we recommend checking the solubility of the compound in aqueous medium at the intended concentration, as precipitation reduces the final concentration of the compound.
Virus replication capacity. Not any cell line can be used in this protocol. It is important to use a highly susceptible cell line in combination with a highly replicative virus to ensure that replication can be accurately measured 31 h after infection (in the case of HIV). A pretest should be carried out to ensure that sufficiently high virus production is measured after 31 h of infection (Fig. 1). For other viruses, the time required for one replication round should first be determined in order to assign a suitable detection window. In addition, the amount of inoculum required should be optimized to ensure that sufficient virus is released into the supernatant at the time of harvest.
Controls. If available, always include sufficient control/reference compounds that are well established in their target of interaction.
Detection of viral replication. The virus replication can be monitored in different ways. For HIV, usually the virus-associated p24 antigen in the supernatant of infected cells is quantified by ELISA (commercially available through different suppliers). Other methods such as virus-associated RT activity (assay kits are available through different suppliers) or quantitative RT-PCR33 can be used as alternatives.
Materials
REAGENTS
-
MT-4 cells. They can be obtained from the NIH AIDS Research and Reference Reagent Program (cat. no. 120)
-
Complete medium (see REAGENT SETUP)
-
RPMI-1640 medium (1× with HEPES without glutamine; Invitrogen, cat. no. 42402016)
-
Heat-inactivated FCS (Invitrogen, cat. no. 10270-106)
-
L-glutamine (Invitrogen, cat. no. 25030024)
-
Gentamicin (50 mg ml−1, liquid; Invitrogen, cat. no. 15750045)
-
Virus stock. It can be obtained from the NIH AIDS Research and Reference Reagent Program (cat. no. 398). Stocks should be prepared and titrated to determine the 50% cell culture infective dose (CCID50) as described in Box 2
Caution
All experiments using the virus should be performed in an appropriate safety laboratory (L3) by trained and skilled personnel, taking the required security precautions, especially as virus stocks with high titer are to be used in these experiments.
-
Stock solutions of test compounds (see REAGENT SETUP)
-
Triton X-100 (5% (vol/vol); Sigma, cat. no. T8787). It is used to inactivate the virus.
-
HIV-1 p24 Core Profile ELISA kit (PerkinElmer, cat. no. NEK050B001KT)

EQUIPMENT
-
Centrifuge (Multifuge 3S-R, Heraeus)
-
Centrifuge tubes (50 ml; TPP, cat. no. 91050)
-
Eppendorf high-performance microcentrifuge 5417R (Eppendorf, cat. no. 5407 000.317 and cat. no. 5490 061.004 (FA-45-24-11 fixed-angle rotor 24 × 1.5/2.0 ml, including aerosol-tight, aluminum lid))
-
Cup Eppendorf 1.5-ml safe lock (Eppendorf, cat. no. E10210)
-
Tissue culture test plates 96F (TPP, cat. no. 92696)
-
Polystyrene round-bottom tube (5 ml; BD Falcon, cat. no. 352054)
-
Microscope
-
CO2 incubator
-
Laminar Airflow class IIA (Clean Air, cat. no. EF/A6)
-
L3 safety laboratory
REAGENT SETUP
Complete medium
-
RPMI-1640 containing 20 mM HEPES buffer supplemented with 10% (vol/vol) heat-inactivated FCS, 2 mM L-glutamine and 20 μg ml−1 gentamicin. This can be stored at 4 °C for 1 month.
Preparation of stock solutions of test compounds
-
Homogeneous aliquots of the test compounds have to be prepared. These can be prepared as described earlier in reference 1. The most commonly used solvents are DMSO, water or buffer solutions. Ensure that the compounds dissolve well in the solvent used. As a rule of thumb, for long-term storage the compounds are stored at 4 °C; however, the storage conditions and the maximum storage duration depend on the compound concerned. If one presumes degradation of a compound, we advise comparing the antiviral activity of a fresh solution with that of an old solution in the antiviral MT-4/MTT assay1. During the experiment, the compound solutions need to be maintained at room temperature (21 °C), protected from light.
Procedure
Sample preparation
Timing 35 min
-
1
Prepare a solution of the test compounds at, preferentially, 1,000-fold their IC50 in complete medium. In the experiment, the final concentration of the compound will be 100-fold its IC50. Per assessed time point, 22 μl (one well of a 96-well plate) is needed for each compound. The calculation for the required reagents of a typical experiment testing seven compounds at 11 time points is described in Box 3.
-
2
Transfer 22 μl of each compound solution prepared in Step 1 to a 1.5-ml tube; this will serve as time point 0 h.
-
3
Cultivate MT-4 cells in a humidified atmosphere (≥95% humidity) at 37 °C and 5% CO2. Subcultivate the cells every 2 d (seed at 3 × 105) or 3 d (seed at 1.5 × 105 cells per ml). Count the cells (10 × 104 cells are needed per 96-well plate).
Critical Step
Choice of cell type is very important; a highly susceptible cell line is prerequisite for the success of the experiment.
-
4
Obtain the required number of cells for the experiment, pellet the cell suspension by centrifugation (5 min, 220g at room temperature) and discard the supernatant. Resuspend the cells in complete medium at a density of 5 × 105 cells per ml.

Infection
Timing 45 min, plus 1 h incubation time
-
5
To this cell suspension, add the required amount of virus to reach a multiplicity of infection of 0.5 or 7.2 × 104 CCID50 per well; this is bulk infection (See Box 2 for virus titration and Box 3 for example calculations).
Caution
All experiments using virus should be performed in an appropriate safety laboratory (L3) by trained and skilled personnel, taking the required security precautions, especially as virus stocks with high titer are to be used in these experiments.
-
6
Immediately after infection, add 200 μl of this suspension (bulk infection) to the 1.5-ml tubes containing 22 μl of each compound solution at 1,000-fold its IC50; these are the tubes from Step 2, which represent time point 0.
-
7
Place the 1.5-ml tubes and the rest of the bulk infection at 37 °C in a humidified atmosphere of 5% CO2 in air. Incubate for 1 h.
-
8
After 1 h of incubation, add 1 ml of complete medium to each 1.5-ml tube and enlarge the volume of the bulk suspension to 40 ml with complete medium.
-
9
Pellet the infected cells in the 1.5-ml tubes and in the bulk infection by centrifugation (4 min, 330g, room temperature). Remove the supernatants and resuspend the cells in 40 ml of complete medium.
-
10
Repeat Step 9 twice (a total of three centrifugations), and finally resuspend in complete medium (200 μl for the 1.5-ml tubes and 5 × 105 cells per ml for the bulk infection).
Critical Step
Appropriate washing to remove the unbound virus is necessary to minimize background.
-
11
During the washing steps (Steps 9 and 10), add 22 μl of each compound solution in the wells of the 96-well plate, which will be used for time points 0 and 1 h.
-
12
Transfer 200 μl from the bulk suspension to all wells of time point 1 h and transfer 200 μl from the 1.5-ml tubes to the 22 μl of compound solution in the plate corresponding to time point 0.
-
13
Mix the bulk suspension again and add 200 μl to each remaining well in the 96-well plate that will be used for the experiment. Place the plate at 37 °C in a humidified atmosphere of 5% CO2 in air, and store the compound solutions in the dark at room temperature.
Compound addition
Timing 5 min for each time point, with intervals of 1 h
-
14
Each hour, add 22 μl of the compound dilution to the wells corresponding to the correct time point and place the plate back at 37 °C in a humidified atmosphere of 5% CO2 in air.
-
15
At 31 h after infection, microscopically score the plate for cytotoxicity by visually monitoring the cell morphology and/or for eventual crystallization/precipitation of the compounds in solution. Next, take 110 μl supernatant from each well, inactivate by adding 12 μl Triton X-100 (5%) and store at −80 °C until you can quantify the virus-associated p24 core antigen by ELISA.
Pause point
The samples may be stored at −80 °C (for up to 6 months) without affecting the results.
-
16
Quantify the virus-associated p24 core antigen in the supernatant by ELISA; we use a commercially available kit from PerkinElmer and follow the manufacturer's instructions.
-
17
Plot the amount of p24 (pg ml−1) versus time points (see Fig. 3, panel b).
Figure 3: Schematic illustration of the HIV replication cycle and a typical result from a time-of-addition experiment. 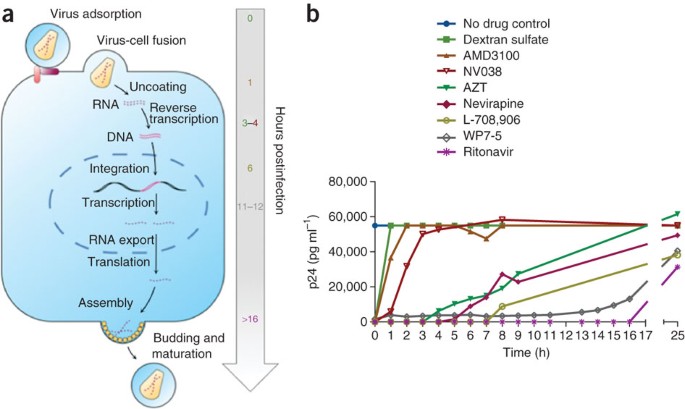
(a) Chronological representation of the essential steps in the HIV-1 viral replication cycle. (b) Example of typical results obtained with the well-characterized anti-HIV drugs: dextran sulfate (an inhibitor of the viral adsorption to the host cell)35, AMD3100 (targeting the CXCR4 coreceptor binding)36, NV038 (an NCp7 inhibitor)37, AZT (an NRTI)38, nevirapine (an NNRTI)39, L-708,906 (a strand transfer integrase inhibitor)40, WP7-5 (a transcription inhibitor)24,41 and ritonavir (a protease inhibitor)42. Viral replication is inhibited up to a time point corresponding to the occurrence of the replication process targeted by the drug.

Troubleshooting
Troubleshooting advice can be found in Table 1.
Timing
Sample preparation
Steps 1–4: 35 min
Infection
Steps 5 and 6: 10 min
Step 7: 1-h incubation
Steps 8–12: ∼30 min
Step 13: 5 min
Compound addition
Step 14: 5 min for each compound, with intervals of 1 h
Step 15: 20 min
Step 16: Time required to perform p24 ELISA is dependent on the kit and corresponding manufacturer's instructions
Step 17: 10 min
Time from start of experiment until obtaining the final results: ∼2 d
Anticipated results
This protocol enables the target of action of newly discovered antiviral drugs to be determined in cell culture. For example, the RT inhibitor AZT is able to inhibit the replication of HIV-1 when it is present within the first 3 h of infection (Fig. 3b); on the contrary, when it is added at 4 h after infection or later, it is unable to inhibit the viral replication because its target of action (the reverse transcription process) has already occurred and the target is no longer of importance for the further replication of the virus in a single round of infection. It should be pointed out that it is imperative to include control drugs with a known and well-characterized target of action in each experiment. When a drug with an unknown target of action shows a similar profile to the established anti-HIV agent, it strongly suggests that this drug targets the same process or at least targets one that is operative at the same time. Most notably, using this assay makes it possible to discriminate between an RT inhibitor of the nucleoside RT inhibitor type (NRTI) versus NNRTI type (Fig. 3). Indeed, we have consistently observed in all experiments that NRTIs (2′,3′-dideoxynucleoside (ddN) analogs) lose their antiviral activity exactly 1 h before that of any NNRTI. This is most likely because of the fact that ddN analogs need to undergo phosphorylation by cellular enzymes before becoming active, as chain terminators, in the RT reaction.
One should keep in mind that for drugs with a dual target of action, i.e., one molecule targeting two distinct steps within the replication cycle, always the last occurring target in the viral replication will be revealed by this experiment, as this is the last step at which replication can be blocked by the inhibitor. To illustrate this point, several combinations of two well-known HIV inhibitors were combined in one experiment (Fig. 4). In the case of dextran sulfate (an inhibitor of viral entry) combined with AZT (an RT inhibitor), the TOA assay only reveals reverse transcription as a target for this combination. For AZT combined with L-870,810 (an integrase inhibitor), only the integrase is revealed as a target. A special example of drugs with a dual mechanism of action are polysulfonate dendrimers, which at low concentrations in cell culture inhibit the viral adsorption but when added at higher concentrations also target a process coinciding with the viral reverse transcription (Fig. 5)34. This can be explained by both the extent of cellular uptake of the drug(s) at different concentrations and their differential potency against different processes. Therefore, in a TOA experiment, it is advised to test compounds at different concentrations.

In this experiment, two well-known HIV inhibitors were combined. When a virus-binding inhibitor (dextran sulfate) is combined with a reverse transcriptase inhibitor (AZT), only the reverse transcription is revealed as the target, as the reverse transcription occurs later in time than the virus-binding process. Similarly, when a reverse transcriptase inhibitor (AZT) is combined with an integrase inhibitor (L-870,810), only the integration is revealed as the target for this drug combination.

At low concentrations, the BRI2923 blocks virus entry, whereas at higher concentrations, it also targets the viral reverse transcription process. Dextran sulfate and AZT were included as reference compounds for virus entry and reverse transcription.
It is possible that a compound will not completely block HIV replication. In many cases this is because of the compound being present at too low a concentration to completely inhibit the virus replication or having too low an intrinsic potency. In the first case, this can be remedied by increasing the concentration of the compound, but cytotoxicity and solubility in aqueous medium of the compound will be limiting factors. We have noticed that there are compounds that are able to inhibit the viral replication and generate an IC50 value in the replication assay but that do not reach a complete (100%) block of viral replication because of a lack of intrinsic potency. Even when no complete inhibition of viral replication can be established with a certain compound, a satisfactory and reliable result could be obtained in this TOA assay. In this case, one can expect to see a residual amount of p24 at time points occurring before the target of the compound that is not zero (only reached when there is a 100% block of viral replication) but lower than in the control without the compound. It is also possible that sometimes a slight slope in loss of activity will be observed instead of a clear jump. This can be because of toxicity of the compound or because the compound is targeting a step in the viral replication that involves an iterative process that takes place for several hours.
Many variations of the TOA experiment can be envisaged, e.g., a so-called negative TOA experiment can be performed for drugs that need time to be processed by the host cell to become active in the cells. In such an experiment, the compound is added at different time points before the virus infection takes place. A similar approach can be used when the mechanism of action of a compound is based on the down- or upregulation of a cellular co-factor involved in viral replication. In addition, shorter time periods between compound addition (15 or 30 min) can be used instead of the 1-h interval.
References
Pannecouque, C., Daelemans, D. & De Clercq, E. Tetrazolium-based colorimetric assay for the detection of HIV replication inhibitors: revisited 20 years later. Nat. Protoc. 3, 427–434 (2008).
Pauwels, R. et al. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J. Virol. Methods 20, 309–321 (1988).
De Clercq, E. Suramin: a potent inhibitor of the reverse transcriptase of RNA tumor viruses. Cancer Lett. 8, 9–22 (1979).
Furman, P.A. et al. Phosphorylation of 3′-azido-3′-deoxythymidine and selective interaction of the 5′-triphosphate with human immunodeficiency virus reverse transcriptase. Proc. Natl Acad. Sci. USA 83, 8333–8337 (1986).
Chandra, P., Vogel, A. & Gerber, T. Inhibitors of retroviral DNA polymerase: their implication in the treatment of AIDS. Cancer Res. 45, 4677s–4684s (1985).
Balzarini, J. et al. 9-[(2RS)-3-fluoro-2-phosphonylmethoxypropyl] derivatives of purines: a class of highly selective antiretroviral agents in vitro and in vivo. Proc. Natl Acad. Sci. USA 88, 4961–4965 (1991).
Parniak, M.A., Min, K.L., Budihas, S.R., Le Grice, S.F. & Beutler, J.A. A fluorescence-based high-throughput screening assay for inhibitors of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H activity. Anal. Biochem. 322, 33–39 (2003).
Engelman, A. & Craigie, R. Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro. J. Virol. 66, 6361–6369 (1992).
Debyser, Z., Cherepanov, P., Pluymers, W. & De Clercq, E. Assays for the evaluation of HIV-1 integrase inhibitors. Methods Mol. Biol. 160, 139–155 (2001).
Dreyer, G.B. et al. Inhibition of human immunodeficiency virus 1 protease in vitro: rational design of substrate analogue inhibitors. Proc. Natl Acad. Sci. USA 86, 9752–9756 (1989).
Vercruysse, T., Pardon, E., Vanstreels, E., Steyaert, J. & Daelemans, D. An intrabody based on a llama single-domain antibody targeting the N-terminal alpha-helical multimerization domain of HIV-1 rev prevents viral production. J. Biol. Chem. 285, 21768–21780 (2010).
Vercruysse, T. et al. Measuring cooperative Rev protein-protein interactions on Rev-responsive RNA by fluorescence resonance energy transfer. RNA Biol. 8, 316–324 (1984).
Mitsuya, H. et al. Suramin protection of T cells in vitro against infectivity and cytopathic effect of HTLV-III. Science 226, 172–174 (1984).
Broder, S. et al. Effects of suramin on HTLV-III/LAV infection presenting as Kaposi's sarcoma or AIDS-related complex: clinical pharmacology and suppression of virus replication in vivo. Lancet 2, 627–630 (1985).
De Clercq, E. Suramin in the treatment of AIDS: mechanism of action. Antiviral Res. 7, 1–10 (1987).
Yahi, N. et al. Suramin inhibits binding of the V3 region of HIV-1 envelope glycoprotein gp120 to galactosylceramide, the receptor for HIV-1 gp120 on human colon epithelial cells. J. Biol. Chem. 269, 24349–24353 (1994).
Hamy, F. et al. An inhibitor of the Tat/TAR RNA interaction that effectively suppresses HIV-1 replication. Proc. Natl Acad. Sci. USA 94, 3548–3553 (1997).
Daelemans, D. et al. A second target for the peptoid Tat/transactivation response element inhibitor CGP64222: inhibition of human immunodeficiency virus replication by blocking CXC-chemokine receptor 4-mediated virus entry. Mol. Pharmacol. 57, 116–124 (2000).
Pluymers, W. et al. Viral entry as the primary target for the anti-HIV activity of chicoric acid and its tetra-acetyl esters. Mol. Pharmacol. 58, 641–648 (2000).
Robinson, W.E. Jr. L-chicoric acid, an inhibitor of human immunodeficiency virus type 1 (HIV-1) integrase, improves on the in vitro anti-HIV-1 effect of Zidovudine plus a protease inhibitor (AG1350). Antiviral Res. 39, 101–111 (1998).
Daelemans, D., Lu, R., De Clercq, E. & Engelman, A. Characterization of a replication-competent, integrase-defective human immunodeficiency virus (HIV)/simian virus 40 chimera as a powerful tool for the discovery and validation of HIV integrase inhibitors. J. Virol. 81, 4381–4385 (2007).
Getchell, J.P. et al. Continuous production of a cytopathic human T-lymphotropic virus in a permissive neoplastic T-cell line. J. Clin. Microbiol. 23, 737–742 (1986).
Tabarrini, O. et al. Studies of anti-HIV transcription inhibitor quinolones: identification of potent N1-vinyl derivatives. ChemMedChem 5, 1880–1892 (2010).
Van Neck, T. et al. Inhibition of the CRM1-mediated nucleocytoplasmic transport by N-azolylacrylates: structure-activity relationship and mechanism of action. Bioorg. Med. Chem. 16, 9487–9497 (2008).
Daelemans, D., Pannecouque, C., Pavlakis, G.N., Tabarrini, O. & De Clercq, E. A novel and efficient approach to discriminate between pre- and post-transcription HIV inhibitors. Mol. Pharmacol. 67, 1574–1580 (2005).
Mitsuya, H. et al. Dextran sulfate suppression of viruses in the HIV family: inhibition of virion binding to CD4+ cells. Science 240, 646–649 (1988).
Daelemans, D., De Clercq, E. & Vandamme, A.M. A quantitative GFP-based bioassay for the detection of HIV-1 Tat transactivation inhibitors. J. Virol. Methods 96, 183–188 (2001).
Morikawa, Y. et al. Complete inhibition of human immunodeficiency virus Gag myristoylation is necessary for inhibition of particle budding. J. Biol. Chem. 271, 2868–2873 (1996).
Pauwels, R. et al. Potent and selective inhibition of HIV-1 replication in vitro by a novel series of TIBO derivatives. Nature 343, 470–474 (1990).
Frankel, A.D. & Young, J.A. HIV-1: fifteen proteins and an RNA. Annu. Rev. Biochem. 67, 1–25 (1998).
Keyaerts, E., Vijgen, L., Maes, P., Neyts, J. & Van Ranst, M. In vitro inhibition of severe acute respiratory syndrome coronavirus by chloroquine. Biochem. Biophys. Res. Commun. 323, 264–268 (2004).
Keyaerts, E. et al. Plant lectins are potent inhibitors of coronaviruses by interfering with two targets in the viral replication cycle. Antiviral Res. 75, 179–187 (2007).
Pasternak, A.O. et al. Highly sensitive methods based on seminested real-time reverse transcription-PCR for quantitation of human immunodeficiency virus type 1 unspliced and multiply spliced RNA and proviral DNA. J. Clin. Microbiol. 46, 2206–2211 (2008).
Witvrouw, M. et al. Polyanionic (i.e., polysulfonate) dendrimers can inhibit the replication of human immunodeficiency virus by interfering with both virus adsorption and later steps (reverse transcriptase/integrase) in the virus replicative cycle. Mol. Pharmacol. 58, 1100–1108 (2000).
Ito, M. et al. Inhibitory effect of dextran sulfate and heparin on the replication of human immunodeficiency virus (HIV) in vitro. Antiviral Res. 7, 361–367 (1987).
De Clercq, E. et al. Highly potent and selective inhibition of human immunodeficiency virus by the bicyclam derivative JM3100. Antimicrob. Agents Chemother. 38, 668–674 (1994).
Pannecouque, C. et al. Inhibition of HIV-1 replication by a bis-thiadiazolbenzene-1,2-diamine that chelates zinc ions from retroviral nucleocapsid zinc fingers. Antimicrob. Agents Chemother. 54, 1461–1468 (2010).
Nakashima, H. et al. Inhibition of replication and cytopathic effect of human T cell lymphotropic virus type III/lymphadenopathy-associated virus by 3′-azido-3′-deoxythymidine in vitro. Antimicrob. Agents Chemother. 30, 933–937 (1986).
Merluzzi, V.J. et al. Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science 250, 1411–1413 (1990).
Hazuda, D.J. et al. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc. Natl Acad. Sci. USA 101, 11233–11238 (2004).
Tabarrini, O. et al. Structure modifications of 6-aminoquinolones with potent anti-HIV activity. J. Med. Chem. 47, 5567–5578 (2004).
Kempf, D.J. et al. ABT-538 is a potent inhibitor of human immunodeficiency virus protease and has high oral bioavailability in humans. Proc. Natl Acad. Sci. USA 92, 2484–2488 (1995).
Reed, L.J. & Muench, H. A simple method for estimating fifty percent endpoints. Am. J. Hyg. 27, 493–497 (1938).
Acknowledgements
We thank K. Erven, C. Heens and L. Bral for fine technical and editorial assistance.
Author information
Authors and Affiliations
Contributions
R.P. and E.D.C. conceived the original protocol and D.D. and C.P. adjusted it to its current form and co-wrote the manuscript with E.D.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Daelemans, D., Pauwels, R., De Clercq, E. et al. A time-of–drug addition approach to target identification of antiviral compounds. Nat Protoc 6, 925–933 (2011). https://doi.org/10.1038/nprot.2011.330
Published:
Issue Date:
DOI: https://doi.org/10.1038/nprot.2011.330
This article is cited by
-
Withania somnifera extracts induced attenuation of HIV-1: a mechanistic approach to restrict viral infection
Virology Journal (2023)
-
Antiviral and ROS scavenging potential of Carica papaya Linn and Psidium guajava leaves extract against HIV-1 infection
BMC Complementary Medicine and Therapies (2023)
-
Statin-mediated disruption of Rho GTPase prenylation and activity inhibits respiratory syncytial virus infection
Communications Biology (2021)
-
Potent sialic acid inhibitors that target influenza A virus hemagglutinin
Scientific Reports (2021)
-
Effect of fetal calf serum on propagation of duck hepatitis A virus genotype 3 in duck embryo fibroblast cells
BMC Veterinary Research (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
