
Key Points
-
The liver represents a key integrator of microbial responses
-
During microbial infection, the liver switches from tolerogenic towards immunogenic responses and initiates the production of acute-phase proteins
-
Overwhelming inflammatory responses contribute to development of liver injury in sepsis and to progression of acute-on-chronic liver failure in patients with liver cirrhosis
-
Sepsis-induced liver injury comprises hypoxic hepatitis, sepsis-induced cholestasis and secondary sclerosing cholangitis of critically ill patients
-
Patients with liver cirrhosis have an increased risk of microbial infections and are at high risk of death from sepsis, therefore, a fast and risk-adjusted antimicrobial therapy is important
Abstract
Sepsis and septic shock are characterized by life-threatening organ dysfunction caused by a dysregulated host response to infection. The liver has a central role during sepsis, and is essential to the regulation of immune defence during systemic infections by mechanisms such as bacterial clearance, acute-phase protein or cytokine production and metabolic adaptation to inflammation. However, the liver is also a target for sepsis-related injury, including hypoxic hepatitis due to ischaemia and shock, cholestasis due to altered bile metabolism, hepatocellular injury due to drug toxicity or overwhelming inflammation, as well as distinct pathologies such as secondary sclerosing cholangitis in critically ill patients. Hence, hepatic dysfunction substantially impairs the prognosis of sepsis and serves as a powerful independent predictor of mortality in the intensive care unit. Sepsis is particularly problematic in patients with liver cirrhosis (who experience increased bacterial translocation from the gut and impaired microbial defence) as it can trigger acute-on-chronic liver failure — a syndrome with high short-term mortality. Here, we review the importance of the liver as a guardian, modifier and target of sepsis, the factors that contribute to sepsis in patients with liver cirrhosis and new therapeutic strategies.
Similar content being viewed by others


Main
Sepsis is defined as a life-threatening organ dysfunction that results from a dysregulated host response to infection1 (Box 1). The liver has crucial roles in sepsis as it represents an important line of defence against microorganisms, as well as a frequent target of the dysregulated inflammation2,3. For the former, close interaction and communication is critical between hepatocytes and additional cell types, such as endothelial, Kupffer and hepatic stellate cells in the space of Disse3 (Fig. 1). Under physiological conditions, these cells are primed to eliminate antigens transported in the blood stream from the gut lumen in a state of homeostasis. However, during microbial infection, these cells turn into an alarm system, giving rise to signals that aim to recruit additional cells into the liver to protect the body from invading microorganisms4. This process triggers the inflammatory response of the body and contributes to efficient elimination of microorganisms. However, this protective response also results in liver injury and becomes impaired when liver cirrhosis is present. This Review describes the responses of the liver during sepsis, to highlight the mechanisms leading to liver injury during sepsis and to delineate the impaired defence against microbial infection in patients with liver cirrhosis.

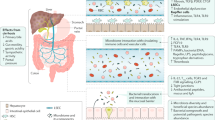
Bacteria and bacterial products such as endotoxin (lipopolysaccharide; LPS) reach the liver via hepatic arteries and the portal vein. Additionally, systemic cytokines and danger-associated molecular patterns (DAMPs) related to ischaemic hepatocyte or cholangiocyte injury accumulate in the hepatic sinusoid. The stellate cells and hepatic macrophages (Kupffer cells) respond to these danger signals (alarmins) by producing large amounts of inflammatory cytokines (TNF, IL-6, IL-1β) and chemokines (CCL2) that attract other immune cells (for example, neutrophils and monocytes) and provide the signal to hepatocytes to switch from a homeostatic to an inflammatory gene and protein expression programme. This process involves the induction of acute-phase proteins (APPs) such as serum amyloid A-1 (SAA) and other inflammatory mediators (IL-8 and CXCL1). Neutrophils (via intercellular adhesion molecule 1 (ICAM1) and lymphocyte function-associated antigen 1 (LFA1) interactions) and monocytes accumulate in the liver, contributing to antimicrobial defence (via neutrophil extracellular trap (NET) formation or phagocytosis) as well as to immune-mediated hepatocyte injury (via apoptosis induction through TNF). LSEC, liver sinusoidal endothelium cell.
Liver involvement during sepsis
The importance of the gut–liver axis. Microbial pathogens play an essential part in sepsis and frequently translocate from the gut lumen into the blood stream, where they have to pass through the liver via the portal vein2. The gut lumen harbours at least as many bacteria as the human body has eukaryotic cells, which is essential as bacteria in the gut facilitate the digestion and use of nutrients5. The digested food components enter the body via the portal blood stream, and the liver plays a crucial part in storing and processing fat, glucose and amino acids to maintain homeostasis.
The gut and the associated local immune system work together to prevent translocation of intestinal bacteria into the portal vein. After the gut intestinal epithelial barrier, the liver constitutes the second line of defence in eliminating invading bacteria and bacterial products, inhibiting a spread of bacteria into the body and, therefore, sepsis2,3. Accordingly, patients with liver cirrhosis have an increased risk of bacterial infections6, which is often due to bacterial translocation from the intestine7,8.
Liver immune surveillance mechanisms. Owing to its unique anatomical location, the liver is highly exposed to circulating antigens, endotoxins, danger signals and even microorganisms, which reach the liver either from the gastrointestinal tract via the portal vein, or from the systemic circulation via arterial blood. Thus, the liver is an important line of defence, together with the spleen, but also a major site for the regulation of immune and inflammatory responses9 (Fig. 1). The majority of mechanisms related to immune surveillance or bacterial clearance by the liver take place at the hepatic sinusoids10. Within the hepatic sinusoid, endothelial cells, hepatic stellate cells (located in the subendothelial space of Disse), resident macrophages (Kupffer cells) and manifold immune cell subsets (such as polymorphonuclear leukocytes, monocytes or natural killer T cells) encounter microorganisms, nonself-antigens, danger signals and/or other humoral factors relevant for inducing inflammatory responses9.
Infection-induced programmed cell death such as RIPK1-dependent or RIPK3-dependent necroptosis, as demonstrated for infections with intracellular bacteria, or caspase-1-dependent pyroptosis, as demonstrated for Shigella or Salmonella infections, is an important source of cytokines (IL-1β) or danger-associated molecular patterns (also known as alarmins) involved in the activation of immunity within the liver11,12,13.
Importantly, many mechanisms in the liver generally ensure suppression of immune responses to avoid accidental immune activation, hyperinflammation or autoimmunity4. For example, in a mouse model that used antigens attached to 0.5 μm latex particles, Kupffer cells rapidly cleared the particles to induce immunological tolerance via regulatory T cells14. However, in the case of concomitant, experimentally induced liver injury, the same antigens are processed in an immunogenic fashion and induce cytotoxic T-cell responses14 (Fig. 2). This finding illustrates that the liver consists of a complex network that integrates inflammatory signals to enable a rapid switch from tolerogenic towards immunogenic responses (reviewed elsewhere4).

a | A state of immune activation is characterized by activated macrophages or Kupffer cells and natural killer T (NKT) cells that produce inflammatory mediators such as interferons (IFNs), IL-1β, IL-8 and TNF. Additionally, neutrophils release reactive oxygen species and form neutrophil extracellular traps (NETs). b | To elicit immunosuppression, acute-phase proteins (APPs) recruit monocytes and neutrophils with immune-suppressive functions termed myeloid-derived suppressor cells (MDSCs), which produce anti-inflammatory cytokines such as IL-10 and transforming growth factor (TGF)-β. MDSCs also suppress T-cell responses via arginase-1, inducible nitric oxide synthase (iNOS) and via induction of regulatory T cells (Treg). LSEC, liver sinusoidal endothelial cell; TCR, T cell receptor.
During sepsis, different types of immune reactions can be distinguished, which are often categorized as type 1 or type 2 immunity15. Type 1 immunity, which is typically excessively activated in sepsis, comprises intense phagocytosis and type 1 T-helper cell activation (IFNγ, IL-2), whereas type 2 immunity is related to wound healing, type 2 T-helper cell activation (IL-4, IL-10) and resolution of inflammation15. The liver immune system participates in these processes during sepsis. Notably, the liver-induced immunogenic response represents a double-edged sword that contributes to clearance of microbial products, but can also cause organ damage due to an overwhelming systemic inflammatory response16.
Bacterial clearance and neutrophils. The liver is essential for clearing bacteria and associated toxins (such as endotoxin) from the bloodstream. In animal models of intravenous bacteria injection, >60% of the injected bacteria can be trapped in the liver within 10 min16. The liver is an important site for neutrophils, which provide immune protection against pathogens by phagocytosing bacteria or releasing antimicrobial granule proteins3,4. Furthermore, neutrophils can form a sticky web of extracellularly released nuclear DNA decorated with histones and proteases, which trap and kill bacteria17 (Fig. 2a). These neutrophil extracellular traps (NETs) are formed in the liver vasculature during sepsis18. In a mouse model of Staphylococcus-aureus-induced sepsis, NETs formed in the liver efficiently cleared bacteria and were protective against bacteraemia and sepsis17. However, the NETs also impaired blood flow in the liver and led to a mild (ischaemic) hepatic injury19, which possibly contributed to the impaired hepatic microcirculation observed in sepsis20,21. Consequently, neutrophils can also promote tissue injury in the liver22,23.
The heterogeneity of neutrophils has been recognized, with the ageing of these immune cells associated with an overly active, proinflammatory neutrophil phenotype24,25. Interestingly, the gut microbiota, as well as Toll-like receptor (TLR) signalling in myeloid cells, promoted neutrophil ageing in mouse models, and depletion of gut microbiota or myeloid TLR signalling dramatically improved endotoxin-induced septic shock25. Further studies are needed to address the heterogeneity of neutrophils and whether a switch between different subsets might represent a viable therapeutic approach.
Hepatic macrophages have the capacity to directly eliminate invading bacteria and to mount effective immune responses against pathogenic bacteria, constituting another line of antimicrobial defence26. These cells consist of functionally diverse subpopulations, such as Kupffer cells that originate from local progenitor cells and act as stationary scavengers in the sinusoid, as well as monocyte-derived macrophages that can rapidly accumulate at sites of liver injury and augment proinflammatory, but also wound-healing, responses27. Kupffer cells are equipped with danger-recognition receptors and scavenger receptors that enable them to efficiently recognize and eliminate circulating bacteria9. Interestingly, Kupffer cells intimately cooperate with platelets to limit the deleterious systemic consequences of sepsis28.
In addition to the liver, the mononuclear phagocyte system (also known as the reticuloendothelial system) in the spleen is important for clearing pathogens from the bloodstream, especially encapsulated bacteria29. The spleen also contains a specialized fraction of B cells termed innate response activator B cells, which efficiently phagocytose bacteria and protect from cytokine release and septic shock30.
Liver defence mechanisms against intracellular bacteria and viruses. The discussed mechanisms do not necessarily apply to systemic infections with intracellular bacteria or viruses. For example, in a mouse model of Listeria monocytogenes infection, Kupffer cells were not able to completely eliminate these intracellular bacteria. Instead, Listeria induced Kupffer cell-death by necroptosis, which triggered the release of hepatocytic danger signals (such as the alarmin IL-33) and monocyte recruitment that was sufficient to eliminate bacteria and restore immune homeostasis11. The accumulation of monocyte-derived macrophages in the liver also seems important for systemic viral infections. Aggregates of these cells in the liver (termed intrahepatic myeloid-cell aggregates for T-cell population expansion) were priming sites for efficient cytotoxic CD8+ T-cell responses in mouse models of different systemic viral infections (namely, recombinant adenovirus expressing a fusion protein, lymphocytic choriomeningitis virus or adenovirus vector transfer of the HBV genome)31,32.
Adaptation of hepatic biosynthesis and metabolism. Sepsis has profound implications for the function of the liver and drastically changes its gene and protein expression pattern, inducing inflammatory pathways and downregulating house-keeping functions such as metabolic, biotransformation or bile transport activities33,34,35. This adaptation considerably changes the plasma proteome and metabolome in conditions of sepsis33,36. However, although strong data on circulating biomarkers from both humans and animal models exist37,38,39, little evidence of transcriptional changes in the human liver during sepsis has been obtained40.
The hepatocellular reaction to sepsis is termed acute-phase response and results in the release of ∼30 acute-phase proteins (APPs) with various biological functions in systemic circulation41 (Table 1). Inflammatory cytokines, mainly IL-6 and IL-1-type cytokines from monocytes and macrophages, trigger the production of APPs in hepatocytes42.
IL-6 has two principle mechanisms to mediate biological effects: classic IL-6-receptor (IL-6R) signalling via a membrane-bound receptor subunit, as shown for acute-phase response induction in hepatocytes; and IL-6 trans-signalling, in which soluble IL-6R binds secreted IL-6 in a bioactive complex that has a long circulating half-life and can act on cells that lack IL-6R but express glycoprotein 130 (also known as interleukin-6 receptor subunit beta) — the common signal transducer of all IL-6 family members43 (Fig. 3).

Classic IL-6 signalling: IL-6 binds to the IL-6-receptor (IL-6R) on hepatocytes, dimerizes with gylcoprotein 130 (gp130) and activates signal transducer and activator of transcription (STAT)-3 signalling via tyrosine phosphorylation at different residues (Y2–Y6) that induces acute-phase proteins. IL-1β-dependent signals can partially modify this activation. Additional intracellular signalling pathways such as STAT1, Ras/mitogen-activated protein kinase (MAPK) are activated in parallel. In IL-6 trans-signalling, IL-6 binds to soluble IL-6R released from cell surfaces by proteolysis or alternative splicing. The agonistic IL-6–IL-6R complex can bind to gp130 to enable signalling in cells that lack the IL-6R. IL-6 trans-signalling can be antagonized by soluble gp130. NF-κB, nuclear factor κB.
Many of the APPs contribute to systemic activation of immune responses (Table 1), for instance, by acting as opsonins44, by activating neutrophils or macrophages via pattern-recognition receptors45,46 or by antimicrobial functions of the complement system47. Importantly, many of the APPs have pleiotropic functions in sepsis. For example, the C-reactive protein opsonizes bacteria, mediates complement activation and phagocytosis, but also induces reactive oxygen species and cytokine releases from macrophages48. Similarly, fibrinogen is not only involved in clot formation but also promotes leukocyte recruitment to inflammatory sites and might enhance local cytokine levels, as has been shown for fibroblast growth factor 2, which has a role in would healing49,50. Concurrently, many proteins related to homeostasis, metabolism or transporter functions (such as cytochrome P450 members or albumin) are downregulated in the liver during sepsis and are consequently termed negative acute-phase reactants51 (Table 1).
Overall, APPs are critical for controlling the systemic inflammatory response in sepsis. In mouse models with absent acute-phase response, owing either to hepatocyte-specific deletion of the glycoprotein 130 signal-transducing receptor, or to hepatocyte-specific deletion of the intracellular signalling molecule STAT3, the sepsis-induced mortality was substantially increased compared to wild-type mice, despite normal bacterial clearance52,53. The hepatic APPs serum amyloid A-1 and the chemokine CXCL1 cooperatively promoted the mobilization of myeloid-derived suppressor cells (MDSC), which prevented excessive inflammation52. These data corroborate the central role of the liver during sepsis as a modulator of systemic immune activation.
Iron mobilization and storage. Hepcidin, the liver-derived central regulator of iron homeostasis and a characteristic APP54, controls iron availability during sepsis. The IL-6-induced increase in hepcidin production leads to iron sequestration in macrophages and a decrease in circulating iron55. This process harms extracellular microorganisms that depend on the availability of exogenous iron (siderophiles) and it might also impair clearance of intracellular infections such as Salmonella or mycobacteria56. On a functional level, the downregulation of hepatic hepcidin expression in mice increased mortality during polymicrobial sepsis, organ damage and oxidative stress, and also compromised host inflammatory responses and bacterial clearance57. Furthermore, hepcidin-deficient mice were more susceptible to death from infection by the siderophilic bacterium Vibrio vulnificus compared with wild-type mice58. In line with these animal studies, increased iron availability in serum, reflected by elevated transferrin saturation, was a strong negative outcome predictor in critically ill patients59.
Liver-mediated immunosuppression. The hepatic response to sepsis involves the activation of anti-inflammatory pathways (Fig. 2b). Hepatic APPs induce the mobilization of MDSCs52 that produce the anti-inflammatory cytokines IL-10 and TGFβ, and suppress CD4+ and CD8+ T-cell responses by secreting arginase-1, inducible nitric oxide synthase (iNOS, also known as NOS2) and by inducing regulatory T cells60,61. Desensitization to lipopolysaccharide (LPS), termed endotoxin tolerance, represents another major mechanism of immunosuppression and limits overwhelming inflammatory reactions in sepsis62. For example, pretreatment of rats with daily injections of LPS reduced plasma cytokine release, alleviated multiple organ injuries and improved survival in a model of polymicrobial sepsis63. After continuous exposure to LPS, cells become refractory to TLR4 signalling and produce anti-inflammatory mediators in response to endotoxin64. Monocytes and macrophages, including the Kupffer cells of the liver, clearly display endotoxin tolerance in vitro and in vivo65. However, experimental data indicate that hepatocytes66, sinusoidal endothelial cells67 and hepatic stellate cells68 can also be desensitized to LPS.
In conclusion, the liver can trigger both defence and immunosuppressive processes and the balance between these processes during sepsis is crucial for survival.
Liver injury mechanisms during sepsis
Prognostic role of liver dysfunction. Updated consensus recommendations published in 2016 led to a partial redefinition of sepsis and septic shock to emphasize the life-threatening organ dysfunction resulting from a dysregulated host response to infection1 (Box 1). Among the different organ failures (circulatory, respiratory and kidney, among others), hepatic dysfunction has an exceptional prognostic relevance for the course of sepsis and is a powerful independent predictor of mortality69,70,71. Serum bilirubin levels, as a key marker of hepatic dysfunction, are a vital component of prognostic scores at the intensive care unit (ICU), such as the Sepsis-related Organ Failure Assessment (SOFA) score72. Recognition of sepsis-related liver dysfunction and differentiation between the clinical patterns of liver injury in sepsis is, therefore, of a great importance.
Hypoxic hepatitis. Hypoxic hepatitis (also known as 'shock liver', ischaemic hepatitis or hypoxic liver injury73) (Fig. 4), is triggered by inadequate oxygen concentration in the blood (hypoxaemic hypoxia), reduced blood flow due to decreased arterial and/or increased venous pressures (ischaemic hypoxia) or lack of oxygen carriers (anaemic hypoxia). Additionally, microthrombi can impair perfusion within the liver74. Liver sinusoidal endothelial cells also contribute to this situation as they respond to inflammatory signals such as endotoxin with an iNOS-dependent endothelial dysfunction, characterized by a decreased vasodilatatory response to acetylcholine and decreased endothelial nitric oxide synthase phosphorylation75.

Sepsis induces the development of hypoxic hepatitis, sepsis-induced cholestasis and later, the emergence of secondary sclerosing cholangitis. Hypoxic hepatitis frequently occurs within the first 48 h after admission; this condition is triggered by inadequate oxygen supply and is characterized by a massive increase in serum aspartate aminotransferase (AST), as well as alanine aminotransferase (ALT) activities. Sepsis-induced cholestasis often develops as a consequence of impaired hepatocellular bile formation or defective bile flow. The molecular mechanisms are an inflammation-mediated impairment of hepatocellular transport systems (NTCP, basolateral sodium taurocholate cotransporter; OATP, organic anion transporting protein; BSEP, canalicular bile-salt export pump; MRP2, canalicular conjugate export pump), a disruption of tight junctions and decreased intracellular biotransformation. Phosphatidylinositol 3-kinase gamma (PI3Kγ) was suggested as a key molecular switch in this process. Secondary sclerosing cholangitis is characterized by inflammation, fibrosis and destruction of the bile ducts, and is triggered by ischaemia of biliary cells. The hallmark of secondary sclerosing cholangitis is early formation of biliary casts that fill the intrahepatic ductal system. LPS, lipopolysaccharide; LSEC, liver sinusoidal endothelial cell.
Hypoxic hepatitis occurs with an incidence of ∼10% in critically ill patients and is associated with an in-hospital mortality of ∼50%76,77,78,79,80. Classic clinical criteria for the diagnosis of hypoxic hepatitis include: underlying clinical setting of cardiac, respiratory or circulatory failure; an abrupt elevation of serum aminotransferase levels (typically >20 times the upper limit of normal levels); and the exclusion of other causes of acute liver cell necrosis, such as acute viral or drug-induced hepatitis76.
Increased serum activities of aspartate aminotransferase and alanine aminotransferase can be measured a few hours after the initiation of hepatic damage and are usually accompanied by elevated lactate dehydrogenase activity. MicroRNA (miR) released from injured hepatocytes such as miR-122 might represent new sensitive and specific markers in this setting81. The same is true for circulating keratins and keratin fragments that indicate apoptotic and necrotic cell death in the liver82. Full-length (M65) and caspase-cleaved (M30) serum keratin 18 levels are indeed elevated in patients with sepsis83 and might predict patient mortality84.
Additional predictors of an unfavourable outcome are an impaired hepatic biosynthetic capacity, concomitant septic shock, increased arterial ammonia levels and sustained elevation of serum aminotransferase levels (>24 h), reflecting ongoing hepatic hypoxaemia or hypoperfusion79,85. After initial hypoxic liver damage, one-third of patients develop cholestatic hepatic dysfunction80.
Sepsis-induced cholestasis. Cholestasis is a common complication in patients with sepsis86 (Fig. 4) and results either from impaired bile formation at the hepatocellular level (hepatocellular cholestasis), or from defective bile flow at the level of small or large bile ducts (ductular cholestasis) without, necessarily, a biliary obstruction87. Experimental evidence from animal models of sepsis revealed that proinflammatory cytokines and mediators downregulate the expression of hepatocellular transport systems (such as the basolateral sodium taurocholate cotransporter, organic anion transporting protein, canalicular bile-salt export pump and canalicular conjugate export pump), thereby linking inflammation to hepatocellular cholestasis88,89,90,91,92.
Phosphatidylinositol 3-kinase gamma (PI3Kγ) was suggested to serve as an important trigger of sepsis-induced cholestasis via internalization of the proteins of the excretory apparatus and downregulating hepatocellular biotransformation. Accordingly, PI3Kγ-knockout mice were protected from sepsis-induced, experimental cholestasis71.
Additional mechanisms include altered transporter trafficking (for example, transporter retrieval from the canalicular membrane), pericanalicular reorganization of the cytoskeleton (which affects microtubules and actin microfilaments required for bile–canalicular contractions) and disrupted tight junctions between hepatocytes that affect bile secretion93,94,95. The mechanisms of ductular cholestasis in sepsis are less well understood than those of hepatocellular cholestasis. Cholangiocytes can secrete proinflammatory cytokines (TNF, IFNγ), which promote periductular inflammation (for example, neutrophil accumulation)96. Moreover, inflammatory cytokines inhibit the chloride and bicarbonate ion transport by cholangiocytes97, thereby impairing bile flow.
Cholestatic liver dysfunction (serum bilirubin levels >20 mg/l) and jaundice occur in ∼20% of critically ill patients, typically within the first 48 h after admission to the ICU, and are usually associated with severe infections such as pneumonia and Gram-negative bacterial sepsis, cardiac failure, high pressure mechanical ventilation or drugs78,98,99,100.
Clinical conditions contributing to hyperbilirubinaemia in critically ill patients with sepsis-induced cholestasis are an increased load of bilirubin (from haemolysis, trauma and/or haematoma) and extrahepatic disturbances of bile flow (choledocholithiasis and/or intraductal inflammation)101. Basic examinations in cholestatic liver dysfunction by ultrasonography, CT or MRI aim to exclude biliary pancreatitis, occlusion of bile ducts or tumorous infiltration of the liver. Endoscopic retrograde cholangiopancreatography with stent insertion and (endoscopic) ultrasonography might be useful to diagnose or treat biliary obstruction102.
Increased serum bile acid concentrations observed in cholestatic conditions interfere with physiological processes and negatively influence organ function, resulting in impaired glucose and lipid metabolism, suppression of immune response, vasodilatation and impaired renal function103,104. Notably, bile acids also act as hormones that activate at least four distinct nuclear receptors that regulate intracellular metabolic pathways in hepatocytes105. Bile-acid overload leads to increased oxidative stress, cell membrane permeability, impaired regeneration and retraction of bile transporters from the plasma membrane to the cytosol86,106. In agreement with their important biological role, plasma bile-acid levels measured at the time of diagnosis were superior to bilirubin levels in predicting short-term mortality of individuals with sepsis71.
Cholestasis and jaundice in patients with sepsis are associated with an increased risk of infections, including bacterial translocation, gastrointestinal complications and renal failure, and consequently contribute to high morbidity and at least a twofold increased mortality78,80,99. Moreover, 1-year mortality of >90% has been reported in patients with initial hypoxic hepatitis and consecutive jaundice80.
Secondary sclerosing cholangitis. Sclerosing cholangitis in critically ill patients, also known as secondary sclerosing cholangitis (SSC) or ischaemic-like cholangiopathy, is characterized by inflammation, fibrosis and destruction of the bile ducts. SSC is associated with rapid progression to liver cirrhosis (Fig. 4). As an underlying mechanism, biliary epithelial cells are exceptionally vulnerable to hypoxia, which triggers different forms of cell death such as apoptosis or necrosis107. The major risk factors for SSC in critically ill patients are severe systemic hypotension (during severe shock), trauma or acute respiratory distress syndrome37 and the systemic inflammatory response syndrome108, which suggests that ischaemia and inflammation are major triggers for this condition. In this respect, patients with SSC have a distinct bile microbial profile with a high incidence of difficult-to-treat microorganisms109. SSC in critically ill patients leads to progressive inflammatory destruction of the biliary system and patients without the option of liver transplantation have a poor prognosis110.
Current and potential future therapeutic targets. The management of hypoxic hepatitis and sepsis-induced cholestasis aims to treat the underlying disease by means described in the latest guidelines for treatment of sepsis and cardiac shock111,112.
An early appropriate haemodynamic restoration by fluid resuscitation and vasopressors that constitutes a cornerstone of sepsis treatment112, supports the improvement of hepatic perfusion and can prevent liver dysfunction. Interestingly, experimental evidence in human hepatocytes indicate that catecholamines, especially adrenaline, can induce a LPS-like inflammatory response that might exacerbate hepatic dysfunction during sepsis113. With regard to splanchnic circulation, adrenaline stimulates both the vasoconstrictory α1 and β2-adrenergic receptors. However, at high adrenaline doses in patients with sepsis, the α1-effect prevails and consequently adrenaline can impair splanchnic circulation in septic shock114. Additionally, in patients with cirrhosis who had undergone liver resection, low doses of dobutamine improved hepatic perfusion as well as oxygenation and stabilized remnant liver function115.
To date, no specific liver-targeted therapy for hypoxic hepatitis exists. In experimental studies, a protective effect of statins (inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase) has been demonstrated116,117, and one clinical study showed an association of statin medication (before ICU admission) with a decreased incidence of hypoxic hepatitis in critically ill patients118.
Animal study data suggest an immunomodulatory effect of corticosteroids on sepsis-induced cholestasis by induction of hepatobiliary transporters (through counteracting LPS-induced MRP2 downregulation) and re-establishment of bile transport119. However, on the basis of the results of the largest clinical trial on corticosteroid therapy in sepsis, corticosteroid administration is not recommended as adjuvant therapy for septic shock as it did not improve patient survival120. Remarkably, in subgroup analyses, corticosteroid-treated patients showed a faster improvement of liver failure during the first week on the ICU than the patients without corticosteroids, but this effect was not associated with decreased mortality121. Given these data, corticosteroid therapy might be beneficial in a selected subgroup of patients with liver dysfunction in an intensive care setting.
Tight glycaemic control with intensive insulin therapy has been demonstrated to reduce cholestasis and biliary sludge98. However, owing to the higher mortality reported in ICU patients treated with intensive insulin therapy compared with moderate glycaemic control, intensive insulin therapy in sepsis is not recommended122. Enteral nutrition, in comparison with parenteral nutrition, decreases the risk of cholestatic liver dysfunction, jaundice and the formation of sludge in the gall bladder123. A stabilization of the gut barrier, a reduced rate of metabolic complications in the liver and a stimulation of acid secretion promoting restoration of enterohepatic cycle might account for these beneficial effects124,125. Thus, enteral nutrition should be attempted in these patients whenever feasible. Regardless of the administration route, an excessive caloric intake should be avoided as, in addition to other serious effects, it might predispose individuals to the development of liver steatosis126.
High-volume plasma exchange with fresh frozen plasma represents an attractive option for patients with sepsis-related liver dysfunction. To this end, high-volume plasma exchange improved transplant-free survival in patients with acute liver failure and an attenuation of the systemic inflammatory response was suggested as one of the major beneficial effects of the therapy127,128.
Sepsis in liver cirrhosis
Bacterial infection in liver cirrhosis. Multiple studies demonstrated that patients with liver cirrhosis have an increased risk of developing bacterial infections6. Infections are found in 25–35% of patients with cirrhosis that have been admitted to a hospital6, and urinary infections and spontaneous bacterial peritonitis (SBP) are particularly common7,8. Other sources are subcutaneous tissue infections, respiratory infections and spontaneous bacteraemia6,7. Notably, bloodstream infections are ten times more common in patients with cirrhosis than patients without cirrhosis129. In addition, infections are more common in patients with advanced liver disease and/or renal insufficiency6,7.
Although the underlying mechanisms are incompletely understood, the involvement of a complex immune dysfunction, an altered gut microbiota and increased bacterial translocation through the intestinal wall has been suggested130,131 (Fig. 5). As predisposing events, patients with liver cirrhosis display an altered bile composition, a delayed intestinal transit time, increased intestinal permeability and a decreased production of intestinal antimicrobial peptides (Fig. 5). Notably, most of these changes correlate with the severity of liver impairment6,132. Bacterial cultures from mesenterial lymph nodes were positive in 30% of patients with Child–Pugh stage C cirrhosis, compared with only 9–15% of patients without cirrhosis133,134. Further evidence of the relevance of an intact mucosal barrier comes from patients with cirrhosis with inherited defects in microbial-defence-related genes such as NOD2 and TLR2, who have an increased risk of developing SBP and display increased mortality135,136.

Patients with liver cirrhosis display a complex immune dysfunction, an altered gut microbiota and increased bacterial translocation through the intestinal wall. The presence of portal hypertension leads to appearance of portosystemic shunts that decrease bacterial clearance in the liver. Portal hypertension might also result in overactive spleen (hypersplenism) with an increased degradation of circulating blood cells and consecutive impairment of microbial defence. An altered bile composition, a delayed intestinal transit time, decreased gastric acid production and a disturbed production of intestinal antimicrobial peptides results in small intestinal bacterial overgrowth. The systemic inflammatory response leads to an increased leakiness of tight junctions further increasing gut permeability. Bacterial translocation contributes to an additional activation of the immune response with increased levels of cytokines (TNF) and reactive oxygen species (ROS), thereby forming a vicious cycle. LPS, lipopolysaccharide.
Source and type of bacterial infection. Many of the infections (45–70%) found in patients with cirrhosis are health-care associated (diagnosed within 48 h of admission in patients with a prior hospitalization for at least 2 days in the last 6 months), whereas nosocomial and community-acquired cases are less common8,129,137. The high rate of health-care-associated infections is not surprising given the frequent hospitalization of this frail population. Similarly to nosocomial infections, the likelihood of resistant microorganisms might be increased in health-care-associated infections119. Secondary infections are detected in 20–50% of hospitalized patients and are often due to potentially preventable causes such as aspiration, catheter-associated urinary tract infection and/or Clostridium-difficile-associated colitis8. The majority of infections in cirrhosis result from Gram-negative bacteria, often of intestinal origin6,7. However, Gram-positive cocci are also important, particularly in hospitalized patients6.
Nosocomial infections are more commonly associated with the presence of nonclassic pathogens, including fungi and multidrug-resistant bacteria (pathogens resistant to three major antibiotic families including β-lactams)6,137,138. Notably, patients with multidrug-resistant bacteria are more likely to develop severe disease and also display increased mortality than patients with less resistant bacteria6,137. The infection site is also of prognostic relevance. In this respect, SBP, pneumonia or C. difficile-associated colitis are associated with high fatality rates, whereas urinary and soft-tissue infections typically display a more favourable outcome7,8,139. Development of septic shock is a particularly dreadful complication with mortality >75%140,141.
Prognosis of bacterial infections. In patients with cirrhosis, infections are more common and have a major effect on prognosis as they are associated with a fourfold increase in mortality compared with otherwise healthy individuals with an infection142. In patients with advanced liver cirrhosis and bacterial infections, the 30-day mortality is ∼30% and an additional 30% die within a year6,142. In some conditions, such as initial nonresponse to empiric antibiotic therapy or nosocomial infections, mortality often exceeds 50%143,144. Bacterial infections predispose an individual to the occurrence of gastrointestinal bleeding, development of hepatic encephalopathy, hypervolaemic hyponatraemia, kidney failure and acute-on-chronic liver failure (ACLF)6,141,145.
Importantly, bacterial infections constitute the most common precipitating event to trigger ACLF and acute kidney injury in patients with cirrhosis141,146,147. The underlying pathogenic mechanisms include haemodynamic changes with splanchnic arterial vasodilation, an inadequate cardiac output and hepatocyte injury through a strong inflammatory reaction and the resulting production of reactive oxygen or nitrogen species145,147,148. Notably, the inflammation-induced tissue damage depends on the intensity of the inflammatory response and the capacity of organs to tolerate it141,149. In this respect, patients with ACLF have been suggested to possess a decreased tolerance of different target organs to the inflammatory response141.
Early diagnosis of infection in patients with cirrhosis is of crucial importance but is highly challenging. Advanced liver disease is often characterized by a persistent immune activation and a high degree of systemic inflammation that can result in elevated levels of inflammatory markers such as C-reactive protein or procalcitonin, even in absence of a bacterial infection6. Similarly, signs of systemic inflammatory response might be present even without an infection owing to hyperdynamic circulation, hepatic encephalopathy and fluid overload150. By contrast, patients with advanced sepsis can present with normal heart rate owing to the use of β-blockers and the lack of other systemic inflammatory response criteria because of immune dysfunction and hypersplenism6. Additionaly, the synthesis of C-reactive protein produced primarily by hepatocytes might not be appropriately elevated in individuals with advanced liver disease6,51. Novel methods might improve early detection of bacterial infection in cirrhosis, including real-time PCR-based multipathogen assays and mass-spectroscopy-based methods detecting antibiotic-resistant pathogens in patient samples such as blood or ascites fluid6. However, not all of these proposed new methods (such as PCR for bacterial DNA from blood or ascites) could predict mortality in patients with cirrhosis in prospective multicentre trials132, highlighting the need for additional tools and studies.
Preventing and treating bacterial infections. Given the detrimental role of bacterial infections in patients with cirrhosis, antibiotic treatment is of major relevance. Patients with severely decompensated cirrhosis were shown to benefit from primary antibiotic prophylaxis with norfloxacin151,152, and norfloxacin can also be used as a secondary prophylaxis for patients after SBP until liver function becomes compensated153. In patients with cirrhosis and gastrointestinal bleeding, immediate antibiotic therapy with broad coverage against intestinal bacteria (such as ceftriaxone intravenously for at least 5 days) is essential to improve survival154,155,156. Conversely, long-term antibiotic prophylaxis is associated with emergence of multidrug-resistant pathogens and might contribute to development of C. difficile-associated diarrhoea6,157. To minimize this outcome, the principles of correct antibiotic stewardship should be followed whenever possible157. In addition, a prophylactic treatment with rifaximin might become an interesting future option as its effect is largely restricted to the intestinal lumen and does not lead to major alterations in intestinal microbiota6,157.
In patients with cirrhosis and with a bacterial infection, a fast and appropriate antibiotic therapy is vital as both a delay in drug administration and an inappropriate therapy result in increased mortality140,158. The empiric therapy should be chosen on the basis of the risk profile of the patient, the local occurrence of resistant bacteria, the site of infection and previous exposure to antibiotics. Third-generation cephalosporins are often suitable for many community-acquired infections, whereas nosocomial and health-care-associated infections often require the use of broader-spectrum antibiotics6,143,159. In patients with SBP, antibiotic treatment should be accompanied by intravenous albumin supplementation that reduces the incidence of renal failure and decreases mortality160. Furthermore, a response to treatment of SBP should be assessed by repeated paracentesis 48 h after initial diagnosis161. In contrast to SBP, albumin supplementation did not provide an obvious benefit in other infection types, although an association with a temporary improvement in renal and/or circulatory function was observed162,163. Thus, further studies are needed to delineate the effects of albumin in this setting.
The role of nonselective β-blockers as a prophylaxis for oesophageal varices in patients with decompensated liver cirrhosis is heavily debated as studies have demonstrated controversial results ranging from improved survival in refractory ascites164 to increased mortality in SBP165. Interestingly, nonselective β-blockers decreased intestinal permeability and bacterial translocation in patients with cirrhosis166 and reduced portal hypertension167. In patients with ACLF, the use of nonselective β-blockers was associated with lowered ACLF grades, reduced severity of systemic inflammation and even improved 28-day mortality; however, no effect on 6-month and 1-year mortality was noted168. Although further studies are needed to better understand the risks and benefits of β-blockers in patients with advanced liver cirrhosis, the authors discourage their use in patients with sepsis.
Other useful approaches include a combined treatment with the vasoactive drug terlipressin and intravenous albumin that have been studied in patients with acute kidney insufficiency and cirrhosis associated with sepsis169. This therapy improved arterial pressure and patients responding to this treatment displayed an improved 3-month survival170. Interestingly, combined treatment of granulocyte colony-stimulating factor and erythropoietin improved 1-year survival in patients with decompensated cirrhosis171. Although further data are needed to confirm this single-centre randomized trial, one could speculate that the supplementation with granulocyte colony-stimulating factor corrects the impaired neutrophil function that occurs in patients with cirrhosis. In fact, this effect might explain the reduced occurrence of septic shock seen in the treatment group171.
Conclusions
As highlighted in this Review, the liver represents a crucial conductor of antimicrobial response that contributes both to the clearance of pathogens and the triggers of immunosuppression that provide protection from an overwhelming immune response. A dysregulation of this complex interface leads to sepsis-induced liver injury and increased sepsis-related mortality. The importance of the liver as an immune organ becomes particularly apparent in patients with liver cirrhosis who have an increased risk of acquiring microbial infections, but also have an increased mortality from sepsis. Thus, further research elucidating the hepatic response to microbial infection as well as alterations in this response in liver cirrhosis are crucial to increase the survival of patients with sepsis and liver cirrhosis.
References
Singer, M. et al. The third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315, 801–810 (2016).
Schnabl, B. & Brenner, D. A. Interactions between the intestinal microbiome and liver diseases. Gastroenterology 146, 1513–1524 (2014).
Protzer, U., Maini, M. K. & Knolle, P. A. Living in the liver: hepatic infections. Nat. Rev. Immunol. 12, 201–213 (2012).
Heymann, F. & Tacke, F. Immunology in the liver — from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 13, 88–110 (2016).
Marchesi, J. R. et al. The gut microbiota and host health: a new clinical frontier. Gut 65, 330–339 (2016).
Jalan, R. et al. Bacterial infections in cirrhosis: a position statement based on the EASL Special Conference 2013. J. Hepatol. 60, 1310–1324 (2014).
Singal, A. K., Salameh, H. & Kamath, P. S. Prevalence and in-hospital mortality trends of infections among patients with cirrhosis: a nationwide study of hospitalised patients in the United States. Aliment. Pharmacol. Ther. 40, 105–112 (2014).
Bajaj, J. S. et al. Second infections independently increase mortality in hospitalized patients with cirrhosis: the North American consortium for the study of end-stage liver disease (NACSELD) experience. Hepatology 56, 2328–2335 (2012).
Jenne, C. N. & Kubes, P. Immune surveillance by the liver. Nat. Immunol. 14, 996–1006 (2013).
Brunt, E. M. et al. Pathology of the liver sinusoids. Histopathology 64, 907–920 (2014).
Bleriot, C. et al. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 42, 145–158 (2015).
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Bleriot, C. & Lecuit, M. The interplay between regulated necrosis and bacterial infection. Cell. Mol. Life Sci. 73, 2369–2378 (2016).
Heymann, F. et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 62, 279–291 (2015).
Spellberg, B. & Edwards, J. E. Jr. Type 1/type 2 immunity in infectious diseases. Clin. Infect. Dis. 32, 76–102 (2001).
Yan, J., Li, S. & Li, S. The role of the liver in sepsis. Int. Rev. Immunol. 33, 498–510 (2014).
McDonald, B., Urrutia, R., Yipp, B. G., Jenne, C. N. & Kubes, P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12, 324–333 (2012).
Kolaczkowska, E. et al. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 6, 6673 (2015).
Jenne, C. N. et al. Neutrophils recruited to sites of infection protect from virus challenge by releasing neutrophil extracellular traps. Cell Host Microbe 13, 169–180 (2013).
Tanaka, K. et al. In vivo characterization of neutrophil extracellular traps in various organs of a murine sepsis model. PLoS ONE 9, e111888 (2014).
Wu, Y. et al. Laser speckle contrast imaging for measurement of hepatic microcirculation during the sepsis: a novel tool for early detection of microcirculation dysfunction. Microvasc. Res. 97, 137–146 (2015).
Moles, A. et al. A TLR2/S100A9/CXCL-2 signaling network is necessary for neutrophil recruitment in acute and chronic liver injury in the mouse. J. Hepatol. 60, 782–791 (2014).
Huebener, P. et al. The HMGB1/RAGE axis triggers neutrophil-mediated injury amplification following necrosis. J. Clin. Invest. 125, 539–550 (2015).
Nauseef, W. M. & Borregaard, N. Neutrophils at work. Nat. Immunol. 15, 602–611 (2014).
Zhang, D. et al. Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532 (2015).
Abdullah, Z. & Knolle, P. A. Scaling of immune responses against intracellular bacterial infection. EMBO J. 33, 2283–2294 (2014).
Tacke, F. & Zimmermann, H. W. Macrophage heterogeneity in liver injury and fibrosis. J. Hepatol. 60, 1090–1096 (2014).
Wong, C. H., Jenne, C. N., Petri, B., Chrobok, N. L. & Kubes, P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat. Immunol. 14, 785–792 (2013).
Tarantino, G., Scalera, A. & Finelli, C. Liver–spleen axis: intersection between immunity, infections and metabolism. World J. Gastroenterol. 19, 3534–3542 (2013).
Rauch, P. J. et al. Innate response activator B cells protect against microbial sepsis. Science 335, 597–601 (2012).
Huang, L. R. et al. Intrahepatic myeloid-cell aggregates enable local proliferation of CD8+ T cells and successful immunotherapy against chronic viral liver infection. Nat. Immunol. 14, 574–583 (2013).
Knolle, P. A., Bottcher, J. & Huang, L. R. The role of hepatic immune regulation in systemic immunity to viral infection. Med. Microbiol. Immunol. 204, 21–27 (2015).
Bauer, M., Press, A. T. & Trauner, M. The liver in sepsis: patterns of response and injury. Curr. Opin. Crit. Care 19, 123–127 (2013).
Cobb, J. P. et al. Sepsis gene expression profiling: murine splenic compared with hepatic responses determined by using complementary DNA microarrays. Crit. Care Med. 30, 2711–2721 (2002).
Yang, Q. et al. Dynamics of hepatic gene expression profile in a rat cecal ligation and puncture model. J. Surg. Res. 176, 583–600 (2012).
Langley, R. J. et al. Integrative “omic” analysis of experimental bacteremia identifies a metabolic signature that distinguishes human sepsis from systemic inflammatory response syndromes. Am. J. Respir. Crit. Care Med. 190, 445–455 (2014).
Tacke, F. et al. Levels of circulating miR-133a are elevated in sepsis and predict mortality in critically ill patients. Crit. Care Med. 42, 1096–1104 (2014).
Benz, F., Roy, S., Trautwein, C., Roderburg, C. & Luedde, T. Circulating microRNAs as biomarkers for sepsis. Int. J. Mol. Sci. 17, E78 (2016).
Lambeck, S., Weber, M., Gonnert, F. A., Mrowka, R. & Bauer, M. Comparison of sepsis-induced transcriptomic changes in a murine model to clinical blood samples identifies common response patterns. Front. Microbiol. 3, 284 (2012).
Marshall, J. C. New translational research provides insights into liver dysfunction in sepsis. PLoS Med. 9, e1001341 (2012).
Bode, J. G., Albrecht, U., Haussinger, D., Heinrich, P. C. & Schaper, F. Hepatic acute phase proteins — regulation by IL-6- and IL-1-type cytokines involving STAT3 and its crosstalk with NF-κB-dependent signaling. Eur. J. Cell Biol. 91, 496–505 (2012).
Tacke, F., Luedde, T. & Trautwein, C. Inflammatory pathways in liver homeostasis and liver injury. Clin. Rev. Allergy Immunol. 36, 4–12 (2009).
Hunter, C. A. & Jones, S. A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457 (2015).
Shah, C., Hari-Dass, R. & Raynes, J. G. Serum amyloid A is an innate immune opsonin for gram-negative bacteria. Blood 108, 1751–1757 (2006).
Medzhitov, R. Recognition of microorganisms and activation of the immune response. Nature 449, 819–826 (2007).
Cheng, N., He, R., Tian, J., Ye, P. P. & Ye, R. D. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J. Immunol. 181, 22–26 (2008).
Ward, P. A. & Gao, H. Sepsis, complement and the dysregulated inflammatory response. J. Cell. Mol. Med. 13, 4154–4160 (2009).
Lelubre, C., Anselin, S., Zouaoui Boudjeltia, K., Biston, P. & Piagnerelli, M. Interpretation of C-reactive protein concentrations in critically ill patients. Biomed. Res. Int. 2013, 124021 (2013).
Ugarova, T. P. & Yakubenko, V. P. Recognition of fibrinogen by leukocyte integrins. Ann. N. Y. Acad. Sci. 936, 368–385 (2001).
Farrell, D. H. Pathophysiologic roles of the fibrinogen gamma chain. Curr. Opin. Hematol. 11, 151–155 (2004).
Gabay, C. & Kushner, I. Acute-phase proteins and other systemic responses to inflammation. N. Engl. J. Med. 340, 448–454 (1999).
Sander, L. E. et al. Hepatic acute-phase proteins control innate immune responses during infection by promoting myeloid-derived suppressor cell function. J. Exp. Med. 207, 1453–1464 (2010).
Sakamori, R. et al. Signal transducer and activator of transcription 3 signaling within hepatocytes attenuates systemic inflammatory response and lethality in septic mice. Hepatology 46, 1564–1573 (2007).
Pietrangelo, A. et al. STAT3 is required for IL-6-gp130–dependent activation of hepcidin in vivo. Gastroenterology 132, 294–300 (2007).
Nicolas, G. et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J. Clin. Invest. 110, 1037–1044 (2002).
Drakesmith, H. & Prentice, A. M. Hepcidin and the iron-infection axis. Science 338, 768–772 (2012).
Zeng, C. et al. Hepatic hepcidin protects against polymicrobial sepsis in mice by regulating host iron status. Anesthesiology 122, 374–386 (2015).
Arezes, J. et al. Hepcidin-induced hypoferremia is a critical host defense mechanism against the siderophilic bacterium Vibrio vulnificus. Cell Host Microbe 17, 47–57 (2015).
Tacke, F. et al. Iron parameters determine the prognosis of critically ill patients. Crit. Care Med. 44, 1049–1058 (2016).
Ren, D. et al. Myeloid-derived suppressor cells accumulate in the liver site after sepsis to induce immunosuppression. Cell. Immunol. 279, 12–20 (2012).
Hammerich, L. & Tacke, F. Emerging roles of myeloid derived suppressor cells in hepatic inflammation and fibrosis. World J. Gastrointest. Pathophysiol. 6, 43–50 (2015).
Lopez-Collazo, E. & del Fresno, C. Pathophysiology of endotoxin tolerance: mechanisms and clinical consequences. Crit. Care 17, 242 (2013).
Shi, D. W. et al. LPS pretreatment ameliorates multiple organ injuries and improves survival in a murine model of polymicrobial sepsis. Inflamm. Res. 60, 841–849 (2011).
Broad, A., Jones, D. E. & Kirby, J. A. Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr. Med. Chem. 13, 2487–2502 (2006).
Collins, P. E. & Carmody, R. J. The regulation of endotoxin tolerance and its impact on macrophage activation. Crit. Rev. Immunol. 35, 293–323 (2015).
Scott, M. J., Liu, S., Shapiro, R. A., Vodovotz, Y. & Billiar, T. R. Endotoxin uptake in mouse liver is blocked by endotoxin pretreatment through a suppressor of cytokine signaling-1-dependent mechanism. Hepatology 49, 1695–1708 (2009).
Uhrig, A. et al. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J. Leukoc. Biol. 77, 626–633 (2005).
Dangi, A. et al. Selective expansion of allogeneic regulatory T cells by hepatic stellate cells: role of endotoxin and implications for allograft tolerance. J. Immunol. 188, 3667–3677 (2012).
Dizier, S. et al. Early hepatic dysfunction is associated with a worse outcome in patients presenting with acute respiratory distress syndrome: a post-hoc analysis of the ACURASYS and PROSEVA studies. PLoS ONE 10, e0144278 (2015).
Koch, A. et al. Increased liver stiffness denotes hepatic dysfunction and mortality risk in critically ill non-cirrhotic patients at a medical ICU. Crit. Care 15, R266 (2011).
Recknagel, P. et al. Liver dysfunction and phosphatidylinositol–3-kinase signalling in early sepsis: experimental studies in rodent models of peritonitis. PLoS Med. 9, e1001338 (2012).
Vincent, J. L. et al. The SOFA (Sepsis-related Organ Failure Assessment) score to describe organ dysfunction/failure. On behalf of the Working Group on Sepsis-Related Problems of the European Society of Intensive Care Medicine. Intensive Care Med. 22, 707–710 (1996).
Lescot, T., Karvellas, C., Beaussier, M. & Magder, S. Acquired liver injury in the intensive care unit. Anesthesiology 117, 898–904 (2012).
Spapen, H. Liver perfusion in sepsis, septic shock, and multiorgan failure. Anat. Rec. (Hoboken) 291, 714–720 (2008).
La Mura, V. et al. Liver sinusoidal endothelial dysfunction after LPS administration: a role for inducible-nitric oxide synthase. J. Hepatol. 61, 1321–1327 (2014).
Henrion, J. et al. Hypoxic hepatitis: clinical and hemodynamic study in 142 consecutive cases. Medicine (Baltimore) 82, 392–406 (2003).
Birrer, R., Takuda, Y. & Takara, T. Hypoxic hepatopathy: pathophysiology and prognosis. Intern. Med. 46, 1063–1070 (2007).
Kramer, L. et al. Incidence and prognosis of early hepatic dysfunction in critically ill patients — a prospective multicenter study. Crit. Care Med. 35, 1099–1104 (2007).
Fuhrmann, V. et al. Hypoxic hepatitis: underlying conditions and risk factors for mortality in critically ill patients. Intensive Care Med. 35, 1397–1405 (2009).
Jager, B. et al. Jaundice increases the rate of complications and one-year mortality in patients with hypoxic hepatitis. Hepatology 56, 2297–2304 (2012).
Roderburg, C. et al. Elevated miR-122 serum levels are an independent marker of liver injury in inflammatory diseases. Liver Int. 35, 1172–1184 (2015).
Ku, N. O., Strnad, P., Bantel, H. & Bishr Omary, M. Keratins: biomarkers and modulators of apoptotic and necrotic cell death in the liver. Hepatology 64, 966–976 (2016).
Moore, D. J. et al. A pilot study assessing the prognostic value of CK18 and nDNA biomarkers in severe sepsis patients. Clin. Drug Investig. 32, 179–187 (2012).
Lorente, L. et al. Serum levels of caspase-cleaved cytokeratin-18 and mortality are associated in severe septic patients: pilot study. PLoS ONE 9, e109618 (2014).
Drolz, A. et al. Clinical impact of arterial ammonia levels in ICU patients with different liver diseases. Intensive Care Med. 39, 1227–1237 (2013).
Bhogal, H. K. & Sanyal, A. J. The molecular pathogenesis of cholestasis in sepsis. Front. Biosci. (Elite Ed.) 5, 87–96 (2013).
Geier, A., Fickert, P. & Trauner, M. Mechanisms of disease: mechanisms and clinical implications of cholestasis in sepsis. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 574–585 (2006).
Geier, A. et al. Cytokine-dependent regulation of hepatic organic anion transporter gene transactivators in mouse liver. Am. J. Physiol. Gastrointest. Liver Physiol. 289, G831–G841 (2005).
Trauner, M., Arrese, M., Lee, H., Boyer, J. L. & Karpen, S. J. Endotoxin downregulates rat hepatic ntcp gene expression via decreased activity of critical transcription factors. J. Clin. Invest. 101, 2092–2100 (1998).
Kim, P. K., Chen, J., Andrejko, K. M. & Deutschman, C. S. Intraabdominal sepsis down-regulates transcription of sodium taurocholate cotransporter and multidrug resistance-associated protein in rats. Shock 14, 176–181 (2000).
Kosters, A. & Karpen, S. J. The role of inflammation in cholestasis: clinical and basic aspects. Semin. Liver Dis. 30, 186–194 (2010).
Elferink, M. G. et al. LPS-induced downregulation of MRP2 and BSEP in human liver is due to a posttranscriptional process. Am. J. Physiol. Gastrointest. Liver Physiol. 287, G1008–G1016 (2004).
Haussinger, D., Schmitt, M., Weiergraber, O. & Kubitz, R. Short-term regulation of canalicular transport. Semin. Liver Dis. 20, 307–321 (2000).
De Maio, A., Gingalewski, C., Theodorakis, N. G. & Clemens, M. G. Interruption of hepatic gap junctional communication in the rat during inflammation induced by bacterial lipopolysaccharide. Shock 14, 53–59 (2000).
Han, X., Fink, M. P., Uchiyama, T., Yang, R. & Delude, R. L. Increased iNOS activity is essential for hepatic epithelial tight junction dysfunction in endotoxemic mice. Am. J. Physiol. Gastrointest. Liver Physiol. 286, G126–G136 (2004).
Strazzabosco, M., Fabris, L. & Spirli, C. Pathophysiology of cholangiopathies. J. Clin. Gastroenterol. 39, S90–S102 (2005).
Spirli, C. et al. Proinflammatory cytokines inhibit secretion in rat bile duct epithelium. Gastroenterology 121, 156–169 (2001).
Mesotten, D. et al. The effect of strict blood glucose control on biliary sludge and cholestasis in critically ill patients. J. Clin. Endocrinol. Metab. 94, 2345–2352 (2009).
Brienza, N. et al. Jaundice in critical illness: promoting factors of a concealed reality. Intensive Care Med. 32, 267–274 (2006).
Whitehead, M. W., Hainsworth, I. & Kingham, J. G. The causes of obvious jaundice in south west Wales: perceptions versus reality. Gut 48, 409–413 (2001).
Chand, N. & Sanyal, A. J. Sepsis-induced cholestasis. Hepatology 45, 230–241 (2007).
Berzosa, M. et al. Diagnostic bedside EUS in the intensive care unit: a single-center experience. Gastrointest. Endosc. 77, 200–208 (2013).
Horvatits, T., Trauner, M. & Fuhrmann, V. Hypoxic liver injury and cholestasis in critically ill patients. Curr. Opin. Crit. Care 19, 128–132 (2013).
Trauner, M. et al. Targeting nuclear bile acid receptors for liver disease. Dig. Dis. 29, 98–102 (2011).
de Aguiar Vallim, T. Q., Tarling, E. J. & Edwards, P. A. Pleiotropic roles of bile acids in metabolism. Cell Metab. 17, 657–669 (2013).
Liu, H. X., Keane, R., Sheng, L. & Wan, Y. J. Implications of microbiota and bile acid in liver injury and regeneration. J. Hepatol. 63, 1502–1510 (2015).
Luedde, T., Kaplowitz, N. & Schwabe, R. F. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 147, 765–783.e4 (2014).
Leonhardt, S. et al. Trigger mechanisms of secondary sclerosing cholangitis in critically ill patients. Crit. Care 19, 131 (2015).
Voigtlander, T. et al. Microbiological analysis of bile and its impact in critically ill patients with secondary sclerosing cholangitis. J. Infect. 70, 483–490 (2015).
Kulaksiz, H., Heuberger, D., Engler, S. & Stiehl, A. Poor outcome in progressive sclerosing cholangitis after septic shock. Endoscopy 40, 214–218 (2008).
Dellinger, R. P. et al. Surviving sepsis campaign: international guidelines for management of severe sepsis and septic shock: 2012. Crit. Care Med. 41, 580–637 (2013).
O'Gara, P. T. et al. 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction: executive summary: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 127, 529–555 (2013).
Aninat, C. et al. Catecholamines induce an inflammatory response in human hepatocytes. Crit. Care Med. 36, 848–854 (2008).
De Backer, D., Creteur, J., Silva, E. & Vincent, J. L. Effects of dopamine, norepinephrine, and epinephrine on the splanchnic circulation in septic shock: which is best? Crit. Care Med. 31, 1659–1667 (2003).
Taura, P. et al. The use of β-adrenergic drugs improves hepatic oxygen metabolism in cirrhotic patients undergoing liver resection. J. Hepatol. 52, 340–347 (2010).
Russo, L. et al. Addition of simvastatin to cold storage solution prevents endothelial dysfunction in explanted rat livers. Hepatology 55, 921–930 (2012).
Llacuna, L. et al. Targeting cholesterol at different levels in the mevalonate pathway protects fatty liver against ischemia-reperfusion injury. J. Hepatol. 54, 1002–1010 (2011).
Drolz, A. et al. Statin therapy is associated with reduced incidence of hypoxic hepatitis in critically ill patients. J. Hepatol. 60, 1187–1193 (2014).
Kubitz, R., Wettstein, M., Warskulat, U. & Haussinger, D. Regulation of the multidrug resistance protein 2 in the rat liver by lipopolysaccharide and dexamethasone. Gastroenterology 116, 401–410 (1999).
Sprung, C. L. et al. Hydrocortisone therapy for patients with septic shock. N. Engl. J. Med. 358, 111–124 (2008).
Moreno, R. et al. Time course of organ failure in patients with septic shock treated with hydrocortisone: results of the Corticus study. Intensive Care Med. 37, 1765–1772 (2011).
Finfer, S., Heritier, S. & NICE Study Managament Committee and SUGAR Study Executive Committee. The NICE-SUGAR (Normoglycaemia in Intensive Care Evaluation and Survival Using Glucose Algorithm Regulation) Study: statistical analysis plan. Crit. Care Resusc. 11, 46–57 (2009).
Kreymann, K. G. et al. ESPEN guidelines on enteral nutrition: intensive care. Clin. Nutr. 25, 210–223 (2006).
Braga, M. et al. Early postoperative enteral nutrition improves gut oxygenation and reduces costs compared with total parenteral nutrition. Crit. Care Med. 29, 242–248 (2001).
Hu, Q. G. & Zheng, Q. C. The influence of enteral nutrition in postoperative patients with poor liver function. World J. Gastroenterol. 9, 843–846 (2003).
Paquot, N. & Delwaide, J. Fatty liver in the intensive care unit. Curr. Opin. Clin. Nutr. Metab. Care 8, 183–187 (2005).
Larsen, F. S. et al. High-volume plasma exchange in patients with acute liver failure: an open randomised controlled trial. J. Hepatol. 64, 69–78 (2016).
Karvellas, C. J. & Stravitz, R. T. High volume plasma exchange in acute liver failure: dampening the inflammatory cascade? J. Hepatol. 64, 10–12 (2016).
Bartoletti, M. et al. Epidemiology and outcomes of bloodstream infection in patients with cirrhosis. J. Hepatol. 61, 51–58 (2014).
Wiest, R., Lawson, M. & Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 60, 197–209 (2014).
Macnaughtan, J. & Jalan, R. Clinical and pathophysiological consequences of alterations in the microbiome in cirrhosis. Am. J. Gastroenterol. 110, 1399–1410 (2015).
Bruns, T., Zimmermann, H. W. & Stallmach, A. Risk factors and outcome of bacterial infections in cirrhosis. World J. Gastroenterol. 20, 2542–2554 (2014).
O'Boyle, C. J. et al. Microbiology of bacterial translocation in humans. Gut 42, 29–35 (1998).
Cirera, I. et al. Bacterial translocation of enteric organisms in patients with cirrhosis. J. Hepatol. 34, 32–37 (2001).
Appenrodt, B. et al. Nucleotide-binding oligomerization domain containing 2 (NOD2) variants are genetic risk factors for death and spontaneous bacterial peritonitis in liver cirrhosis. Hepatology 51, 1327–1333 (2010).
Nischalke, H. D. et al. Toll-like receptor (TLR) 2 promoter and intron 2 polymorphisms are associated with increased risk for spontaneous bacterial peritonitis in liver cirrhosis. J. Hepatol. 55, 1010–1016 (2011).
Sargenti, K., Prytz, H., Strand, A., Nilsson, E. & Kalaitzakis, E. Healthcare-associated and nosocomial bacterial infections in cirrhosis: predictors and impact on outcome. Liver Int. 35, 391–400 (2015).
Fernandez, J. et al. Bacterial infections in cirrhosis: epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology 35, 140–148 (2002).
Pereira, G. et al. Renal failure and hyponatremia in patients with cirrhosis and skin and soft tissue infection. A retrospective study. J. Hepatol. 56, 1040–1046 (2012).
Arabi, Y. M. et al. Antimicrobial therapeutic determinants of outcomes from septic shock among patients with cirrhosis. Hepatology 56, 2305–2315 (2012).
Moreau, R. et al. Acute-on-chronic liver failure is a distinct syndrome that develops in patients with acute decompensation of cirrhosis. Gastroenterology 144, 1426–1437.e9 (2013).
Arvaniti, V. et al. Infections in patients with cirrhosis increase mortality four-fold and should be used in determining prognosis. Gastroenterology 139, 1246–1256.e5 (2010).
Merli, M. et al. An empirical broad spectrum antibiotic therapy in healthcare-associated infections improves survival in cirrhotics: a randomized trial. Hepatology 63, 1632–1639 (2016).
Piano, S. et al. The empirical antibiotic treatment of nosocomial spontaneous bacterial peritonitis: results of a randomized, controlled clinical trial. Hepatology 63, 1299–1309 (2016).
Bernardi, M., Moreau, R., Angeli, P., Schnabl, B. & Arroyo, V. Mechanisms of decompensation and organ failure in cirrhosis: from peripheral arterial vasodilation to systemic inflammation hypothesis. J. Hepatol. 63, 1272–1284 (2015).
Barreto, R. et al. Type-1 hepatorenal syndrome associated with infections in cirrhosis: natural history, outcome of kidney function, and survival. Hepatology 59, 1505–1513 (2014).
Angeli, P., Tonon, M., Pilutti, C., Morando, F. & Piano, S. Sepsis-induced acute kidney injury in patients with cirrhosis. Hepatol. Int. 10, 115–123 (2016).
Nesseler, N. et al. Clinical review: the liver in sepsis. Crit. Care 16, 235 (2012).
Medzhitov, R., Schneider, D. S. & Soares, M. P. Disease tolerance as a defense strategy. Science 335, 936–941 (2012).
Thabut, D. et al. Model for end-stage liver disease score and systemic inflammatory response are major prognostic factors in patients with cirrhosis and acute functional renal failure. Hepatology 46, 1872–1882 (2007).
Fernandez, J. et al. Primary prophylaxis of spontaneous bacterial peritonitis delays hepatorenal syndrome and improves survival in cirrhosis. Gastroenterology 133, 818–824 (2007).
Saab, S., Hernandez, J. C., Chi, A. C. & Tong, M. J. Oral antibiotic prophylaxis reduces spontaneous bacterial peritonitis occurrence and improves short-term survival in cirrhosis: a meta-analysis. Am. J. Gastroenterol. 104, 993–1001 (2009).
Gines, P. et al. Norfloxacin prevents spontaneous bacterial peritonitis recurrence in cirrhosis: results of a double-blind, placebo-controlled trial. Hepatology 12, 716–724 (1990).
de Franchis, R. Expanding consensus in portal hypertension: report of the Baveno VI Consensus Workshop: stratifying risk and individualizing care for portal hypertension. J. Hepatol. 63, 743–752 (2015).
Tripathi, D. et al. U. K. guidelines on the management of variceal haemorrhage in cirrhotic patients. Gut 64, 1680–1704 (2015).
Fernandez, J. et al. Norfloxacin versus ceftriaxone in the prophylaxis of infections in patients with advanced cirrhosis and hemorrhage. Gastroenterology 131, 1049–1056 (2006).
Fernandez, J., Tandon, P., Mensa, J. & Garcia-Tsao, G. Antibiotic prophylaxis in cirrhosis: good and bad. Hepatology 63, 2019–2031 (2016).
Karvellas, C. J., Abraldes, J. G., Arabi, Y. M. & Kumar, A. Appropriate and timely antimicrobial therapy in cirrhotic patients with spontaneous bacterial peritonitis-associated septic shock: a retrospective cohort study. Aliment. Pharmacol. Ther. 41, 747–757 (2015).
Fernandez, J. & Acevedo, J. New antibiotic strategies in patients with cirrhosis and bacterial infection. Expert Rev. Gastroenterol. Hepatol. 9, 1495–1500 (2015).
Sort, P. et al. Effect of intravenous albumin on renal impairment and mortality in patients with cirrhosis and spontaneous bacterial peritonitis. N. Engl. J. Med. 341, 403–409 (1999).
Rimola, A. et al. Diagnosis, treatment and prophylaxis of spontaneous bacterial peritonitis: a consensus document. J. Hepatol. 32, 142–153 (2000).
Guevara, M. et al. Albumin for bacterial infections other than spontaneous bacterial peritonitis in cirrhosis. A randomized, controlled study. J. Hepatol. 57, 759–765 (2012).
Thevenot, T. et al. Effect of albumin in cirrhotic patients with infection other than spontaneous bacterial peritonitis. A randomized trial. J. Hepatol. 62, 822–830 (2015).
Leithead, J. A. et al. Non-selective β-blockers are associated with improved survival in patients with ascites listed for liver transplantation. Gut 64, 1111–1119 (2015).
Mandorfer, M. et al. Nonselective β blockers increase risk for hepatorenal syndrome and death in patients with cirrhosis and spontaneous bacterial peritonitis. Gastroenterology 146, 1680–1690.e1 (2014).
Reiberger, T. et al. Non-selective betablocker therapy decreases intestinal permeability and serum levels of LBP and IL-6 in patients with cirrhosis. J. Hepatol. 58, 911–921 (2013).
Reiberger, T. et al. Carvedilol for primary prophylaxis of variceal bleeding in cirrhotic patients with haemodynamic non-response to propranolol. Gut 62, 1634–1641 (2013).
Mookerjee, R. P. et al. Treatment with non-selective beta blockers is associated with reduced severity of systemic inflammation and improved survival of patients with acute-on-chronic liver failure. J. Hepatol. 64, 574–582 (2016).
Boyer, T. D. et al. Terlipressin plus albumin is more effective than albumin alone in improving renal function in patients with cirrhosis and hepatorenal syndrome type 1. Gastroenterology 150, 1579–1589.e2 (2016).
Rodriguez, E. et al. Terlipressin and albumin for type-1 hepatorenal syndrome associated with sepsis. J. Hepatol. 60, 955–961 (2014).
Kedarisetty, C. K. et al. Combination of granulocyte colony-stimulating factor and erythropoietin improves outcomes of patients with decompensated cirrhosis. Gastroenterology 148, 1362–1370.e7 (2015).
Acknowledgements
Our research was supported by the Interdisciplinary Center for Clinical Research (IZKF) within the faculty of Medicine at the RWTH Aachen University (P.S.) and by the Deutsche Forschungsgemeinschaft (DFG) SFB/TRR57 (P.S., F.T. and C.T.), DFG TR 285/10-1 (C.T.) and SPP 1782 (P.S.).
Author information
Authors and Affiliations
Contributions
The authors contributed equally to the review.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Strnad, P., Tacke, F., Koch, A. et al. Liver — guardian, modifier and target of sepsis. Nat Rev Gastroenterol Hepatol 14, 55–66 (2017). https://doi.org/10.1038/nrgastro.2016.168
Published:
Issue Date:
DOI: https://doi.org/10.1038/nrgastro.2016.168
This article is cited by
-
Implications of neonatal absence of innate immune mediated NFκB/AP1 signaling in the murine liver
Pediatric Research (2024)
-
OTUD5 promotes the inflammatory immune response by enhancing MyD88 oligomerization and Myddosome formation
Cell Death & Differentiation (2024)
-
Bovine Colostrum in Increased Intestinal Permeability in Healthy Athletes and Patients: A Meta-Analysis of Randomized Clinical Trials
Digestive Diseases and Sciences (2024)
-
ACE2 Expressed on Myeloid Cells Alleviates Sepsis-Induced Acute Liver Injury via the Ang-(1–7)–Mas Receptor Axis
Inflammation (2024)
-
The P2Y2 receptor as a sensor of nucleotides and cell recruitment during inflammatory processes of the liver
Purinergic Signalling (2024)
