
Abstract
Epithelial–mesenchymal transition (EMT) is a developmental program, which can be adopted by cancer cells to increase their migration and ability to form metastases. Transforming growth factor β (TGFβ) is a well-studied inducer of EMT. We demonstrate that TGFβ potently stimulates hyaluronan synthesis via upregulation of hyaluronan synthase 2 (HAS2) in NMuMG mammary epithelial cells. This stimulatory effect requires the kinase active type I TGFβ receptor and is dependent on Smad signaling and activation of the p38 mitogen-activated protein kinase. Knockdown of HAS2 inhibited the TGFβ-induced EMT by about 50%, as determined by the phase contrast microscopy and immunostaining using the EMT marker ZO-1. Furthermore, real-time PCR analysis of the EMT markers fibronectin, Snail1 and Zeb1 revealed decreased expressions upon HAS2 suppression, using specific small interfering RNA (siRNA) for HAS2. Removal of the extracellular hyaluronan by Streptomyces hyaluronidase or inhibiting the binding to its cell surface receptor CD44 by blocking antibodies, did not inhibit TGFβ-induced EMT. Interestingly, HAS2 suppression completely abolished the TGFβ-induced cell migration, whereas CD44 knockdown did not. These observations suggest that TGFβ-dependent HAS2 expression, but not extracellular hyaluronan, has an important regulatory role in TGFβ-induced EMT.
Similar content being viewed by others


Introduction
Epithelial–mesenchymal transition (EMT) of epithelial cells occurs during embryonic development, and is required for the formation of tissues and organs.1 EMT also has important roles during other physiological processes, such as wound healing, and during pathological conditions including cancer progression.2 During EMT, cell–cell adhesion is suppressed and epithelial cells acquire a mesenchymal phenotype exhibiting increased migratory capacity. Such an alteration in the cellular behavior is dependent on interactions between cells and the microenvironment.1
A ubiquitous component of the pericellular matrix is hyaluronan, which is enriched around proliferating and migrating cells in rapidly remodeling tissues.3 Hyaluronan has key regulatory roles in tissue homeostasis and possesses signaling properties through its interaction with cell surface receptors, such as CD44 and RHAMM.4, 5 Hyaluronan is synthesized by three related hyaluronan synthases (designated HAS1, HAS2 and HAS3) encoded by different genes.6, 7, 8, 9 The synthases are differentially expressed during mouse development and possess distinct catalytic activities.10, 11, 12 Most malignant tumors contain elevated amounts of both hyaluronan and CD44 and many reports have shown that hyaluronan-activated CD44 promotes tumor progression.13, 14, 15
The HAS2 isoform has a key role during embryogenic EMT; knockout of the mouse HAS2 gene, but not the knockout of HAS1 and HAS3 genes, leads to abnormal cardiac morphogenesis, due to failure of cushion cell endothelium to undergo mesenchymal transition, because the hyaluronan-deficient cardiac jelly fails to mediate hyaluronan–CD44–ErbB2 signaling events.16, 17 Experimental induction of the HAS2 isoform in normal epithelial cells18 and mesothelioma cells19 was found to promote EMT.
The transforming growth factor β (TGFβ) superfamily consists of cytokines with important functions during embryonal development, as well as in inflammation and homeostasis of tissues. Signaling by TGFβ is mediated via type I and type II serine/threonine kinase receptors (TβRI and TβRII, respectively) and leads to activation of receptor-regulated (R)-Smad proteins, Smad 2 and 3, which in turn form a complex with the common-mediator (Co)-Smad, Smad4. The R-Smad/Co-Smad complexes then translocate into the nucleus where they act as transcription factors regulating the transcription of certain genes involved in, for example, apoptosis, differentiation, proliferation and EMT.20, 21 TGFβ can also engage non-Smad-dependent pathways, including activation of mitogen-activated protein kinase (MAPK) pathways22, 23 and the proteolytic release and nuclear translocation of the intracellular part of TβRI.24
The levels of both TGFβ and hyaluronan are elevated in advanced cancers and fibrotic conditions.25, 26, 27 In this study, we demonstrate that TGFβ induces the HAS2 gene in mammary epithelial cells, which promotes TGFβ-induced EMT.
Results
TGFβ-induced synthesis of hyaluronan in mammary epithelial cells is due to upregulation of HAS2 and depends on Smads and the kinase activities of TβRI and p38 MAPK
Whereas there is some knowledge about the molecular mechanisms of how extracellular regulatory signals, such as platelet-derived growth factor (PDGF)-BB and TGFβ, regulate hyaluronan synthesis in cells of mesenchymal origin,26, 28, 29, 30, 31, 32 the molecular mechanisms in epithelial cells are not known. As TGFβ mediates EMT in NMuMG mammary epithelial cells33, 34 and elevated hyaluronan production via transfection with HAS2 promotes a mesenchymal and proliferative phenotype,18, 19 we investigated the effect of TGFβ on hyaluronan production by NMuMG cells. Hyaluronan was hardly detectable in unstimulated cell cultures, whereas TGFβ stimulation potently stimulated hyaluronan synthesis (Figure 1a). The TGFβ-mediated hyaluronan synthesis was nearly abrogated in cells pretreated with the TβRI kinase inhibitor GW6604 or the p38 MAPK inhibitor SB203580 (Figure 1a).

TGFβ induces hyaluronan synthesis in NMuMG cells via upregulation of HAS2 mRNA in a p38- and Smad-dependent manner. NMuMG cells were starved and stimulated for 24 h (a and b) or 6 h (c) with TGFβ, in the absence or presence of TβRI-kinase inhibitor GW6604, p38 MAPK inhibitor SB203580 or dimethyl sulfoxide control, or the hyaluronan degrading enzyme Streptomyces hyaluronidase. Cells were pretreated with the above agents or enzyme for 1 h before stimulation. (a) Conditioned medium was collected and subjected to an enzyme-linked immunosorbent-like assay for analysis of hyaluronan amount. The average of three separate experiments performed in triplicates ±s.d. is shown. (b) Before starvation and stimulation, cells were transfected with scrambled siRNA or siRNA against Smad4. Following 24 h of stimulation, the hyaluronan amount was determined. A representative experiment out of three is shown ±s.d. (c) Total RNA was prepared and reversely transcribed to complementary DNA and real-time PCR was run with primers for HAS1, 2 and 3. The mRNA copy number was related to that of 18S ribosomal RNA housekeeping gene and the average of three separate experiments ±s.d. is shown. (d) Before starvation and TGFβ stimulation, cells were transfected with scrambled siRNA or siRNA against HAS2. Following 24 h of stimulation, the amount of hyaluronan was determined. A representative experiment out of three is shown ±s.d. P-values were calculated using Student's paired t-test and *P<0.05 were considered statistically significant compared with scramble. Asterisks indicate statistically significant differences compared with cells stimulated with TGFβ alone or as indicated with lines.
To evaluate if the Smad signaling pathway is required for TGFβ-induced hyaluronan synthesis by NMuMG cells, we transfected cells with Smad4 small interfering RNA (siRNA), resulting in a marked inhibition of hyaluronan production compared with TGFβ-stimulated scrambled siRNA transfected cells (Figure 1b). Moreover, the combination of knockdown of Smad4 and inhibition of p38 MAPK by SB203580 completely blocked TGFβ-mediated hyaluronan synthesis (Figure 1b). This indicates that both Smad-dependent and Smad-independent pathways are important for the stimulatory effect of TGFβ on hyaluronan production.
In order to investigate which of the three HAS isoforms (HAS1, 2 and 3) were responsible for the dramatic induction of hyaluronan in response to TGFβ, real-time RT–PCR analysis of RNA from non-stimulated NMuMG cells or cells stimulated with TGFβ alone or in combination with inhibitors of the TβRI kinase or the p38 MAPK, was performed. In non-treated cultures, the expression levels of the three HAS isoforms were low (Figure 1c). Interestingly, upon stimulation with TGFβ the HAS2 mRNA level were induced about 10-fold, whereas those of HAS1 and HAS3 were not changed (Figure 1c), indicating that HAS2 is responsible for the TGFβ-mediated potent stimulatory effect on hyaluronan production. Cotreatment of the cells with the TβRI kinase inhibitor or the p38 MAPK inhibitor decreased the levels of HAS2 mRNA to basal levels, indicating that the TGFβ-dependent induction of HAS2 mRNA is mediated by both the TβRI kinase, most likely via the Smad pathway, and the p38 MAPK pathway. Furthermore, transfection of the cells with siRNA for HAS2 led to a significant decrease in TGFβ-mediated hyaluronan synthesis, further supporting the notion that HAS2 is the major source of the hyaluronan produced by TGFβ stimulated cells (Figure 1d).
Knockdown of HAS2, but not knockdown of CD44, inhibits TGFβ-induced EMT
Several studies have presented evidence that HAS2 expression is closely correlated to increased tumor invasion,35, 36, 37, 38 inflammation,39 and cellular migration during development.16, 40 We, therefore, examined whether HAS2 is important for TGFβ-induced EMT by knocking down HAS2 using specific siRNA. TGFβ efficiently induced EMT in NMuMG cells. Quantification of culture areas, as depicted by phase contrast microscopy, revealed that after 24 h of TGFβ stimulation about 80% of the cells had acquired a fibroblastic character, and after 48 h about 90% (Figure 2). Interestingly, depletion of HAS2 prevented the transition of the epithelial cobble-stone phenotype to spindle-like mesenchymal phenotype by about 50% after 24 h, and was sustained to similar levels 48 h after TGFβ stimulation, when the maximum TGFβ-induced EMT was observed (Figure 2).

Knockdown of HAS2 partially inhibits TGFβ-induced EMT. NMuMG cells were transfected with scrambled siRNA or siRNA against HAS2 or CD44, starved and stimulated with 1 ng/ml TGFβ for up to 48 h. Phase contrast micrographs were taken with a Zeiss Axiovision 40 phase contrast microscope. Scale bar=100 μm. The number of epithelial cells (vs mesenchymal cells) was counted in about eight photos per treatment and the average ±s.d. from three separate experiments is shown to the right. Asterisks indicate statistically significant differences comparing TGFβ-stimulated cells transfected with siRNA against HAS2 with cells transfected with scrambled siRNA.
Given that CD44 has an important role in EMT induced by the inflammatory mediator tumor necrosis factor-α,41 we investigated its importance for TGFβ-mediated EMT. Knockdown of CD44 had no effect on the TGFβ-mediated phenotypic changes of NMuMG cells (Figure 2).
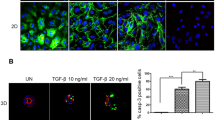
Immunofluorescence analysis demonstrated that in HAS2-expressing NMuMG cells, staining for ZO-1, a component of the intercellular epithelial cell junctions, decreased after stimulation by TGFβ and the cortical staining of F-actin was altered to stress-fibers; such changes are seen in mesenchymal cells or in epithelial cells that have undergone EMT. Interestingly, TGFβ stimulation failed to induce complete EMT in cells in which HAS2 had been knocked down as demonstrated by the presence of ZO-1 cellular junctions and the presence of cortical staining of F-actin (Figure 3a). Supperssion of the hyaluronan receptor CD44, however, did not inhibit the TGFβ-mediated switch of F-actin stress-fibers that are uniformly organized throughout each cell (Figure 3b).

Depletion of HAS2 reverts the TGFβ-mediated mesenchymal phenotype. The expression of ZO-1 (green) and F-actin (red) was analyzed by immunofluorescence in NMuMG cells transfected with scramble siRNA or siRNA against either HAS2 (a) or CD44 (b), in unstimulated or TGFβ-stimulated (1 ng/ml, 48 h) cells. Cell nucleus was visualized with DAPI (blue). Images were taken using a Zeiss Axioplan 2 immunofluorescence microscope. Scale bar=20 μm. A representative experiment out of three is shown.
In order to gain a better understanding of the role of HAS2 during EMT induced by TGFβ, we investigated the cellular distribution of HAS2 and hyaluronan, as active HASs under cellular stress can be present in intracellular compartments in addition to the cell membrane.42 HAS2 was detected all over the cell body and in particular at cell–cell contacts during the EMT process (Figure 4a). NMuMG cultures undergoing EMT in response to TGFβ stimulation all exhibited both cell-associated (Figure 4b) and intracellular (Figure 4c) hyaluronan, while untreated cells were negative. The spindle-like migratory cells had punctuated hyaluronan-positive membrane protrusions (Figure 4b). In cultures treated with siRNA for HAS2 and stimulated with TGFβ, the cells that acquired a mesenchymal phenotype were those in which HAS2 was not successfully silenced, as shown by positive staining for hyaluronan. In cells depleted of HAS2, hyaluronan staining was hardly detectable; these cells exhibited an epithelial phenotype (Figures 4b and c). Thus, exposure of NMuMG cells to TGFβ generates a mesenchymal phenotype associated with HAS2 expression and hyaluronan synthesis. We also investigated whether the intracellular localization of HAS2 and hyaluronan promoted autophagy. However, immunoblot analysis with antibodies against the autophagy marker LC3A/B, gave no evidence that TGFβ-induced HAS2 expression affected autophagy (Supplementary Figure S1).

Cellular localization of HAS2 and hyaluronan. NMuMG cells were transfected with scrambled siRNA or siRNA against HAS2 (b and c), starved, and stimulated with 1 ng/ml TGFβ for 12 h (a) or 48 h (a–c). Cells were fixed and immunostained with anti-HAS2 (green in a). Hyaluronan was visualized by biotinylated G1 probe (green in b and c), Actin filaments were visualized by TRITC-phalloidin (red) and cell nuclei with DAPI (blue). To visualize extracellular hyaluronan (b), cells were not permeabilized and to visualize intracellular hyaluronan (c), cells were first treated with 10 U/ml Streptomyces hyaluronidase to remove extracellular hyaluronan, washed extensively and then permeabilized. Arrows indicate plasma membrane protrusions stained for HAS2 or hyaluronan. A representative experiment out of two is shown.
Fibronectin and the transcription factors Snail1, Zeb1 and Zeb2 are markers of EMT. Therefore, we compared their expression levels after TGFβ stimulation in NMuMG cells expressing HAS2 or not, using real-time PCR. Stimulation with TGFβ induced a twofold upregulation of fibronectin, Zeb1 and Zeb2 mRNA, and a fivefold upregulation of Snail1 mRNA levels (Figure 5a). Interestingly, silencing of HAS2 significantly diminished the expression levels of fibronectin, Snail1 and Zeb1, but had no effect on the Zeb2 (Figure 5a). Notably, the expression levels of TGFβ-responsive genes not known to be involved in EMT, such as Smad7 and PAI1, did not change upon HAS2 depletion, indicating a certain selectivity in the effect of HAS2. Western blot analysis, after different time periods of TGFβ stimulation of cells expressing HAS2 or not, revealed a clear reduction of fibronectin expression in cells depleted of HAS2, compared with cells transfected with scrambled siRNA (Figure 5b). We also investigated whether HAS2 is required for TGFβ signaling in general using the CAGA-luciferase promoter-reporter assay (Supplementary Figure S2A) and immunoblotting for pSmad2 (Supplementary Figure S2B); no effect of knockdown of HAS2 was noticed in either of these assays. Moreover, no effect on the cleavage of the apoptotic marker caspase-3 (Supplementary Figure S2C) was seen in TGFβ-stimulated cells expressing HAS2 or not, giving no support for a particular role of HAS2 in the apoptotic response. Notably, knockdown of CD44 had neither any effect on the mRNA expression levels of fibronectin, Snail1 or Zeb1, nor on the level of fibronectin protein (Figures 5b,c).

Knockdown of HAS2, but not knockdown of CD44, inhibits TGFβ-induced upregulation of EMT markers in NMuMG cells. NMuMG cells were transfected with scrambled siRNA or siRNA against either HAS2 (a and b) or CD44 (b and c), starved and stimulated with 1 ng/ml TGFβ for up to 48 h. (a and c); total RNA was prepared after 6 h stimulation and reversely transcribed to complementary DNA. Real-time PCR was performed with primers for the EMT markers fibronectin, Snail1, Zeb1 and Zeb2, TGFβ target genes Smad7 and PAI1 and for HAS2 (a) or CD44 (c) to monitor knockdown efficiency. The mRNA copy number was related to that of 18S ribosomal RNA housekeeping gene and a representative experiment out of three experiments is shown ±s.d. Asterisks indicate statistically significant differences compared with cells transfected with scrambled siRNA. (b) Cells were lysed, and subjected to SDS–polyacrylamide gel electrophoresis and western blotting with antibodies against fibronectin and β-actin. A representative experiment out of three is shown.
Hyaluronan–CD44 interaction is not required for TGFβ-induced EMT
Next, we investigated whether the powerful production of hyaluronan by the NMuMG cells in response to TGFβ was important for the ability of TGFβ to induce EMT, using immunofluorescence detection of the tight junction protein ZO-1 and F-actin. Cotreatment of the cells with TGFβ and KM114 antibodies to block the binding of hyaluronan to CD44, or Streptomyces hyaluronidase to degrade the newly synthesized TGFβ-induced hyaluronan, did not inhibit the ability of TGFβ to switch the morphology of the cells from an epithelial cobble-stone pattern to a mesenchymal elongated shape (Figure 6a). Using flow cytometry, we verified that the binding of fluorescein isothiocyanate-conjugated hyaluronan to cells was completely abolished by KM114 antibodies (data not shown). Furthermore, high levels of hyaluronan added exogenously to NMuMG cultures, also failed to switch the morphology from cobble-stone shaped cells growing in sheets to fibroblastic-like cells (Figure 6a). Thus, exogenously added hyaluronan or blocking the binding of endogenously produced hyaluronan to CD44 did not prevent TGFβ-mediated EMT.

Extracellularly added hyaluronan or dominant negative HAS2 do not affect TGFβ-induced EMT. (a) NMuMG cells were starved and stimulated for 48 h with either hyaluronan (250 μg/ml) or TGFβ (1 ng/ml) with or without 1 h pretreatment with Streptomyces hyaluronidase or CD44-blocking antibodies KM114 (2.5 μg/ml) or control IgG (2.5 μg/ml). Cells were fixed and immunostained with anti-ZO-1 (green), TRITC-phalloidin (red) and DAPI (blue). (b) NMuMG cells were infected with ZsGreen1-fluorescent adenoviral vectors containing either wild-type HAS2 or the enzymatically inactive HAS2-K190R mutant or empty vector (mock) for 60 h;during the last 48 h cells were stimulated with 1 ng/ml TGFβ in starvation medium. Infected cells are green from the Zsgreen1 protein encoded by the adenoviral vector; infection efficiency was about 90%. Images were taken using a Zeiss Axioplan 2 immunofluorescence microscope and a representative experiment out of three is shown. Scale bar=20 μm.
Dominant negative HAS2 does not affect TGFβ-induced EMT
The finding that exogenous hyaluronan did not affect TGFβ-induced EMT, suggests that either intracellular hyaluronan is important or the HAS2 molecule itself in a manner not related to hyaluronan synthesis. To distinguish between these possibilities, we took advantage of the K190R mutant HAS2, which is devoid of hyaluronan synthesizing capacity and acts in a dominant negative manner by formation of inactive dimeric complexes with wt HAS2.43 Infections of NMuMG cells with adenovirus vectors encoding mutant HAS2, wt HAS2 or mock vector were performed (Figure 6b). Expression of the K190R HAS2 mutant did not suppress the TGFβ-induced EMT, suggesting that the role of HAS2 in TGFβ-mediated EMT is independent of its hyaluronan synthesizing activity (Figure 6b).
Knockdown of HAS2 inhibits TGFβ-induced migration of cells
Another hallmark of EMT is that cells with mesenchymal phenotype display increased motility compared with those with an epithelial phenotype. To investigate whether HAS2 suppression influences the motility of TGFβ-stimulated NMuMG cells, we measured their migration using a transwell assay. We found that cells transfected with scrambled siRNA migrated significantly faster during 48 h of TGFβ treatment than in starvation medium alone (Figure 7a). However, when HAS2 was knocked down by siRNA, TGFβ stimulation failed to increase the motility of NMuMG cells. Importantly, exogenously added hyaluronan could not significantly compensate for the lowered migratory capacity of the cells, that was induced by knockdown of HAS2. Notably, epidermal growth factor-induced cell migration was not affected by HAS2 depletion (Figure 7a), indicating that TGFβ-induced cell migration is specifically dependent on HAS2 expression.

Knockdown of HAS2 inhibits cell migration. NMuMG cells were transfected with scrambled siRNA or siRNA against HAS2 (a) or CD44 (b), starved and then stimulated with 1 ng/ml TGFβ. Cells were then maintained in Transwell assay chambers for 24 h with TGFβ, hyaluronan or epidermal growth factor as chemotactic stimuli in the bottom chamber. Before seeding the cells in the Transwell chambers, some of the cell cultures were pretreated with CD44-blocking antibody KM114 (2.5 μg/ml) or control IgG (2.5 μg/ml) for 30 min. Transmigrating cells were stained with Giemsa and counted after bright-field images were taken. Relative number of migrating cells from a representative experiment out of three ±s.d. is shown. Asterisks indicate statistically significant differences between HAS2-depleted cells compared with those expressing HAS2 and subjected to identical treatments.
We also examined whether CD44 expressed by NMuMG cells is involved in TGFβ-induced migration. As shown in Figure 7b, cells depleted of CD44 exhibited similar migration velocities as cells expressing CD44. Furthermore, use of KM114 CD44 antibodies to block the interaction between CD44 and hyaluronan had no effect on cell motility of CD44-expressing cells.
Discussion
High expression of TGFβ ligands44, 45 and elevated levels of hyaluronan15, 46 have been associated with breast cancer progression. We have demonstrated here that stimulation of immortalized mammary epithelial cells, NMuMG, with TGFβ potently induces both HAS2 expression and EMT, and that silencing of HAS2 suppresses the TGFβ-induced EMT.
Whereas our findings thus support the notion that HAS2 is important for TGFβ-induced EMT, addition of exogenous hyaluronan or removal of secreted endogenously synthesized hyaluronan by Streptomyces hyaluronidase, did not affect EMT. Moreover, blocking or downregulating the hyaluronan receptor CD44 did not either affect TGFβ-induced EMT. It is unlikely that the other hyaluronan receptor RHAMM acted on behalf of CD44 as RHAMM was found not to be expressed, or expressed at very low levels, in NMuMG cultures, whereas RHAMM was strongly expressed in mouse embryonic fibroblasts used as a positive control, as determined by immunoblotting and immunofluorescence staining (data not shown). Thus, the hyaluronan produced by TGFβ-induced HAS2 may have an intracellular function important for EMT. The presence of intracellular hyaluronan has been demonstrated.47, 48, 49, 50 Alternatively, the HAS2 molecule has another function, not related to hyaluronan production, which is important for TGFβ-induced EMT. Our finding that overexpression of a dominant negative mutant of HAS2 did not affect TGFβ-induced EMT, suggests that the involvement of HAS2 is not related to its enzymatic activity.
Recent data have implicated HAS2 in mechanisms involved in an invasive breast cancer phenotype.36, 37, 38 Estrogen/progesterone negative aggressive breast cancer cell lines, MDA-MB-231 and HS-578T, exhibit high hyaluronan synthesizing capacity whereas the non-aggressive hormone receptor positive cells, MCF-7 and Zr-75-1, synthesize minute amounts of hyaluronan.51 HAS2 expression is positively correlated with the incidence of bone metastasis of breast cancer cells and is implicated in the cooperation with stromal cells and the paracrine production of growth factors.38 In addition, HAS2 is among the 0.3% highly expressed genes that confer increased invasiveness of pancreatic cancer cells.35 Moreover, ectopic expression of HAS2 results in EMT and acquisition of a malignant migratory phenotype of non-invasive epithelial cells.18, 19 The mechanism that triggers the expression of HAS2 in certain cancers, but not HAS1 and HAS3 expression, remains to be elucidated. Our present study supports the possibility that TGFβ signaling, which is enhanced during the transition of low-grade to high-grade breast cancer, contributes to the increased HAS2 activity and increased hyaluronan levels in advanced cancers.
During TGFβ-induced EMT, a group of transcription factors is upregulated, including Snail1, Slug, Twist, ZEB1 and ZEB2, as well as the matrix molecule fibronectin. The transcription factors are expressed at much higher levels in the CD44+/CD24− breast cancer stem cell population,52 which has self-renewal properties and migratory capacity, and expresses HAS2.38 Our data clearly show that suppression of HAS2 results in lowered expression of fibronectin, Snail1 and ZEB1, but not ZEB2, during TGFβ-induced EMT (Figure 5a). Interestingly, the extracellular matrix of normal mammary tissue is soft and contains only low amount of fibronectin. However, increased synthesis of fibronectin, resulting in stiffer tissue, is seen in certain cancers.53
Our observations may have implication for disease management during breast cancer progression. As efficient TGFβ-induced EMT requires HAS2 expression, strategies to inhibit HAS2-induced hyaluronan production, or its expression, for example, with 4-methylumbelliferone, might prevent or delay invasiveness of breast cancer. The expression of HAS2 is known to be dependent on Smad and CREB-binding elements in the 5′-promoter region of HAS2.54 Our results provide evidence that TGFβ mediates its stimulatory effects on HAS2 induction both in a Smad-dependent and Smad-independent manner. Currently, experiments are in progress to elucidate the precise regulatory mechanisms through which TGFβ modulate HAS2 expression in mammary epithelial cells of different malignant phenotypes.
Materials and methods
Cell culture
NMuMG cells (normal mouse mammary epithelial cells; ATCC #CRL-1636) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, Life Technologies Europe BV, Stockholm, Sweden), supplemented with 10% fetal bovine serum (FBS; Biowest, Biotech-IgG AB, Lund, Sweden). Before stimulation, cells were starved for 24 h in Dulbecco’s modified Eagle’s medium supplemented with 2% FBS. When cells were incubated with TGFβ (TGFβ-1, Peprotech, Peprotech Nordic, Stockholm, Sweden, 1 ng/ml) and inhibitors (TβRI kinase inhibitor GW6604, American Customs Chemicals Corp, San Diego, CA, USA, 6 μM; p38 MAPK inhibitor SB203580, Calbiochem, Merck KGaA, Darmstadt, Germany, 10 μM) or CD44-blocking antibodies KM114 (BD Biosciences, BD AB, Stockholm, Sweden, 2.5 μg/ml), the inhibitors and antibodies were pre-incubated with the cells for 1 h before the addition of TGFβ. For autophagy experiments, some cells were stimulated with 50 μM chloroquine (Sigma, Sigma-Aldrich Sweden AB, Stockholm, Sweden) for 12 h before lysis.
siRNA transfection, adenoviral infection and gene reporter assay
Cells were transiently transfected with scrambled siRNA, HAS2 siRNA (20 nM, 72 h before stimulation), Smad4 siRNA (50 nM, 48 h before stimulation), or CD44 siRNA (20 nM, 72 h before stimulation), all ON-TARGETplus SMARTpool from Dharmacon (Thermo Fischer scientific, Gothenburg, Sweden), using SilentFECT transfection reagent (Biorad, Biorad laboratories AB, Sundbyberg, Sweden) according to the instructions by the manufacturer.
Green fluorescent adenoviruses expressing wild-type FLAG-mHAS2 or K190R mutant FLAG-mHAS2, which is devoid of hyaluronan synthesizing capacity,43 were made using AdenoX-Adenoviral System 3 (Clontech, Bionordica Sweden AB, Stockholm, Sweden) and transient infections of cells were performed according to the manufactureŕs protocol. For gene reporter assays, cells were infected with adenoviruses expressing LacZ and CAGA9 assays were carried out using the Firefly luciferase assay kit (Biotium, VWR International AB, Stockholm, Sweden), as previously described.55
Hyaluronan enzyme-linked immunosorbent assay-like assay
Cells (200 000/well or 30 000 cells/well for siRNA experiments), were seeded in six-well plates and grown in Dulbecco’s modified Eagle’s medium/10% FBS; after starvation for 24 h cells were stimulated with 1 ng/ml TGFβ in the absence or presence of inhibitors for another 24 h, as described above. Conditioned media were collected and the hyaluronan content was quantified by an enzyme-linked immunosorbent assay-like assay, as previously described.29 This assay is based on the specific binding of hyaluronan molecules in the samples to immobilized G1 global domain of aggrecan.
Real-time PCR
Total RNA was extracted from unstimulated or stimulated cells using the RNeasy Mini Kit (QIAGEN, Qiagen Nordic, Sollentuna, Sweden) according to the manufacturer’s instructions. One microgram of RNA was reversely transcribed to complementary DNA using iScript complementary DNA synthesis Kit (Biorad). Real-time PCR (95 °C, 2 min; 40 × (95 °C, 10 s; 55 °C, 30 s)) was carried out using iQ SYBR Green Supermix (Biorad) and primers listed in Table 1. The expression level of each target gene was normalized to that of 18S ribosomal RNA.
Microscopy
Cells seeded on coverslips in six-well plates were transfected with 20 nM of scrambled siRNA, or siRNA for HAS2 or CD44, followed by starvation and stimulation with TGFβ. Furthermore, cells were stimulated with 250 μg/ml hyaluronan alone or with 1 ng/ml TGFβ in the absence or presence of the CD44-blocking antibody KM114 (2.5 μg/ml) or control IgG (2.5 μg/ml), or 2 units/ml of Streptomyces hyaluronidase (Sigma). Cells were fixed in 3% paraformaldehyde for 15–30 min, permeabilized with 0.5% Triton X-100 for 10 min (except samples for determination of extracellular hyaluronan) and blocked in 5% FBS, 0.1 M glycine overnight at 4 °C. For visualization of intracellular hyaluronan, cells were treated with 10 U/ml Streptomyces hyaluronidase for 1 h at 37 °C and washed extensively before permeabilization. Then, cells were stained with tetramethylrhodamine isothiocyanate (TRITC)-phalloidin (0.5 μg/ml, Invitrogen) or antibodies against ZO-1 (Invitrogen; 1:200 dilution) or HAS2 (Santa cruz biotechnologies, AH Diagnostics, Stockholm, Sweden; 1:200 dilution), or with biotinylated G1 probe for hyaluronan detection, followed by Alexa Fluor 488-labeled goat anti-mouse or anti-rabbit secondary antibodies or Alexa-Fluor-488-Streptavidin-conjugate (all from Molecular Probes, Life Technologies Europe BV; 1:1000 dilution). Nuclei were counterstained with DAPI and the slides were mounted with ProLong Gold Antifade reagent (Invitrogen). Photographs were taken with a Zeiss Axioplan 2 immunofluorescence microscope (Carl Zeiss AB, Stockholm, Sweden).
Unstimulated or TGFβ-stimulated cells transfected with scrambled siRNA, or siRNA for HAS2 or CD44, as described above, were observed with a phase contrast Zeiss Axiovision 40 microscope (Carl Zeiss). Photographs were taken and the number of cells with epithelial (cobble-stone shaped and Carl Zeiss AB growing in sheets) or mesenchymal (elongated and spread) morphology per vision field was measured; ∼8 vision fields per treatment were analyzed for the fraction of epithelial cells.
Transwell migration assay
The migratory properties of cells transfected with siRNA against HAS2, CD44 or a scrambled sequence, after stimulation or not with TGFβ for 24 h, were evaluated using Transwell chambers (24-well plate, 8 μm pore size, BD Biosciences) according to the manufacturer’s instructions. As chemotactic stimuli in the bottom chamber were used 10% FBS, 1 ng/ml TGFβ, 250 μg/ml hyaluronan or 10 ng/ml epidermal growth factor. In some experiments, CD44 was blocked by pre-incubating the cells with KM114 antibodies. After 24 h, the inserts were fixed in 2.5% glutaraldehyde and stained with Giemsa. Bright-field photographs were taken using a Zeiss Axiovision 40 microscope and the number of migrating cells was counted.
Protein extraction, SDS–polyacrylamide gel electrophoresis and immunoblotting
Cells were lysed in 20 mM Tris pH 7.4, 150 mM NaCl, 10 mM EDTA, 0.5% Triton X-100, 0.5% sodium deoxycholate, supplemented with protease and phosphatase inhibitors (0.5 mg/ml Pefabloc, Roche, Roche diagnostics Scandinavia AB, Stockholm, Sweden, 10 μM Leupeptin, Sigma, Sigma-Aldrich Sweden AB, 1 μM Pepstatin Sigma, Sigma-Aldrich Sweden AB, 100 KIU/ml Aprotinin, Calbiochem, Merck KGaA and 1 mM sodium orthovanadate). After 30 min of incubation with occasional vortexing and centrifugation (10 000 g, 10 min, 4 °C), supernatants were processed for SDS-polyacrylamide gel electrophoresis (8% polyacrylamide gels) followed by immunoblotting. After protein transfer to Hybond C Extra nitrocellulose membranes (Amersham Biosciences, GE Healthcare, Uppsala, Sweden) and blocking (5% milk in tris buffered saline (TBS), 0.1% Tween 20), membranes were incubated with antibodies against fibronectin or β-actin (both from Sigma; 1:10 000 dilution), phospho-Smad2 (homemade), cleaved caspase-3 or LC3A/B (both from Cell signaling Technology, BioNordika Sweden AB, Stockholm, Sweden; 1 : 1000 dilution) at 4 °C overnight, followed by horseradish peroxide-conjugated secondary antibodies (1 : 10 000 dilution; Invitrogen); immunocomplexes were thereafter detected by chemoluminescence.
Statistical analysis
Data from three separate experiments was analyzed by paired Student’s t-test. Errors are in s.d. and a result was considered statistically significant when the P-value was below 0.05.
References
Thiery JP, Acloque H, Huang RY, Nieto MA . Epithelial-mesenchymal transitions in development and disease. Cell 2009; 139: 871–890.
Moustakas A, Heldin CH . Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci 2007; 98: 1512–1520.
Toole BP . Hyaluronan in morphogenesis. Semin Cell Dev Biol 2001; 12: 79–87.
Laurent TC, Fraser JRE . Hyaluronan. FASEB J 1992; 6: 2397–2404.
Toole BP . Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer 2004; 4: 528–539.
Watanabe K, Yamaguchi Y . Molecular identification of a putative human hyaluronan synthase. J Biol Chem 1996; 271: 22945–22948.
Itano N, Kimata K . Expression cloning and molecular characterization of HAS protein, a eukaryotic hyaluronan synthase. J Biol Chem 1996; 271: 9875–9878.
Shyjan AM, Heldin P, Butcher EC, Yoshino T, Briskin MJ . Functional cloning of the cDNA for a human hyaluronan synthase. J Biol Chem 1996; 271: 23395–23399.
Spicer AP, Olson JS, McDonald JA . Molecular cloning and characterization of a cDNA encoding the third putative mammalian hyaluronan synthase. J Biol Chem 1997; 272: 8957–8961.
Spicer AP, McDonald JA . Characterization and molecular evolution of a vertebrate hyaluronan synthase gene family. J Biol Chem 1998; 273: 1923–1932.
Itano N, Sawai T, Yoshida M, Lenas P, Yamada Y, Imagawa M et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J Biol Chem 1999; 274: 25085–25092.
Brinck J, Heldin P . Expression of recombinant hyaluronan synthase (HAS) isoforms in CHO cells reduces cell migration and cell surface CD44. Exp Cell Res 1999; 252: 342–351.
Toole BP, Slomiany MG . Hyaluronan: a constitutive regulator of chemoresistance and malignancy in cancer cells. Semin Cancer Biol 2008; 18: 244–250.
Anttila MA, Tammi RH, Tammi MI, Syrjanen KJ, Saarikoski SV, Kosma VM . High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res 2000; 60: 150–155.
Auvinen P, Tammi R, Parkkinen J, Tammi M, Agren U, Johansson R et al. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am J Pathol 2000; 156: 529–536.
Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A et al. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J Clin Invest 2000; 106: 349–360.
Camenisch TD, Schroeder JA, Bradley J, Klewer SE, McDonald JA . Heart-valve mesenchyme formation is dependent on hyaluronan-augmented activation of ErbB2-ErbB3 receptors. Nat Med 2002; 8: 850–855.
Zoltan-Jones A, Huang L, Ghatak S, Toole BP . Elevated hyaluronan production induces mesenchymal and transformed properties in epithelial cells. J Biol Chem 2003; 278: 45801–45810.
Li Y, Heldin P . Hyaluronan production increases the malignant properties of mesothelioma cells. Br J Cancer 2001; 85: 600–607.
Massague J . TGFbeta in Cancer. Cell 2008; 134: 215–230.
Moustakas A, Heldin CH . The regulation of TGFbeta signal transduction. Development 2009; 136: 3699–3714.
Sorrentino A, Thakur N, Grimsby S, Marcusson A, von Bulow V, Schuster N et al. The type I TGF-beta receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat Cell Biol 2008; 10: 1199–1207.
Yamashita M, Fatyol K, Jin C, Wang X, Liu Z, Zhang YE . TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol Cell 2008; 31: 918–924.
Mu Y, Sundar R, Thakur N, Ekman M, Gudey SK, Yakymovych M et al. TRAF6 ubiquitinates TGFbeta type I receptor to promote its cleavage and nuclear translocation in cancer. Nat Commun 2011; 2: 330.
Heldin P, Laurent TC, Heldin C-H . Effect of growth factors on hyaluronan synthesis in cultured human fibroblasts. Biochem J 1989; 258: 919–922.
Li Y, Rahmanian M, Widstrom C, Lepperdinger G, Frost GI, Heldin P . Irradiation-induced expression of hyaluronan (HA) synthase 2 and hyaluronidase 2 genes in rat lung tissue accompanies active turnover of HA and induction of types I and III collagen gene expression. Am J Respir Cell Mol Biol 2000; 23: 411–418.
Usui T, Amano S, Oshika T, Suzuki K, Miyata K, Araie M et al. Expression regulation of hyaluronan synthase in corneal endothelial cells. Invest Ophthalmol Vis Sci 2000; 41: 3261–3267.
Jacobson A, Brinck J, Briskin MJ, Spicer AP, Heldin P . Expression of human hyaluronan synthases in response to external stimuli. Biochem J 2000; 348 (Pt 1): 29–35.
Li L, Asteriou T, Bernert B, Heldin CH, Heldin P . Growth factor regulation of hyaluronan synthesis and degradation in human dermal fibroblasts: importance of hyaluronan for the mitogenic response of PDGF-BB. Biochem J 2007; 404: 327–336.
Stuhlmeier KM, Pollaschek C . Differential effect of transforming growth factor beta (TGF-beta) on the genes encoding hyaluronan synthases and utilization of the p38 MAPK pathway in TGF-beta-induced hyaluronan synthase 1 activation. J Biol Chem 2004; 279: 8753–8760.
Meran S, Thomas D, Stephens P, Martin J, Bowen T, Phillips A et al. Involvement of hyaluronan in regulation of fibroblast phenotype. J Biol Chem 2007; 282: 25687–25697.
Simpson RM, Wells A, Thomas D, Stephens P, Steadman R, Phillips A . Aging fibroblasts resist phenotypic maturation because of impaired hyaluronan-dependent CD44/epidermal growth factor receptor signaling. Am J Pathol 2010; 176: 1215–1228.
Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME et al. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol Biol Cell 2001; 12: 27–36.
Gal A, Sjoblom T, Fedorova L, Imreh S, Beug H, Moustakas A . Sustained TGF beta exposure suppresses Smad and non-Smad signalling in mammary epithelial cells, leading to EMT and inhibition of growth arrest and apoptosis. Oncogene 2008; 27: 1218–1230.
Sato N, Maehara N, Goggins M . Gene expression profiling of tumor-stromal interactions between pancreatic cancer cells and stromal fibroblasts. Cancer Res 2004; 64: 6950–6956.
Unger K, Wienberg J, Riches A, Hieber L, Walch A, Brown A et al. Novel gene rearrangements in transformed breast cells identified by high-resolution breakpoint analysis of chromosomal aberrations. Endocr Relat Cancer 2010; 17: 87–98.
Bernert B, Porsch H, Heldin P . Hyaluronan synthase 2 (HAS2) promotes breast cancer cell invasion by suppression of tissue metalloproteinase inhibitor 1 (TIMP-1). J Biol Chem 2011; 286: 42349–42359.
Okuda H, Kobayashi A, Xia B, Watabe M, Pai SK, Hirota S et al. Hyaluronan synthase HAS2 promotes tumor progression in bone by stimulating the interaction of breast cancer stem-like cells with macrophages and stromal cells. Cancer Res 2011; 72: 537–547.
Olsson M, Meadows JR, Truve K, Rosengren Pielberg G, Puppo F, Mauceli E et al. A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet 2011; 7: e1001332.
Bakkers J, Kramer C, Pothof J, Quaedvlieg NEM, Spaink HP, Hammerschmidt M . Has2 is required upstream of Rac1 to govern dorsal migration of lateral cells during zebrafish gastrulation. Development 2004; 131: 525–537.
Takahashi E, Nagano O, Ishimoto T, Yae T, Suzuki Y, Shinoda T et al. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J Biol Chem 2010; 285: 4060–4073.
Wang A, de la Motte C, Lauer M, Hascall V . Hyaluronan matrices in pathobiological processes. FEBS J 2011; 278: 1412–1418.
Karousou E, Kamiryo M, Skandalis SS, Ruusala A, Asteriou T, Passi A et al. The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J Biol Chem 2010; 285: 23647–23654.
Tang B, Vu M, Booker T, Santner SJ, Miller FR, Anver MR et al. TGF-beta switches from tumor suppressor to prometastatic factor in a model of breast cancer progression. J Clin Invest 2003; 112: 1116–1124.
Wakefield LM, Roberts AB . TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev 2002; 12: 22–29.
Boregowda RK, Appaiah HN, Siddaiah M, Kumarswamy SB, Sunila S, Thimmaiah KN et al. Expression of hyaluronan in human tumor progression. J Carcinog 2006; 5: 2.
Evanko SP, Wight T . Intracellular hyaluronan http://www.glycoforumgrjp/science/hyaluronan/HA20/HA20Ehtml 2001.
Evanko SP, Parks WT, Wight TN . Intracellular hyaluronan in arterial smooth muscle cells: association with microtubules, RHAMM, and the mitotic spindle. J Histochem Cytochem 2004; 52: 1525–1535.
Hascall VC, Majors AK, De La Motte CA, Evanko SP, Wang A, Drazba JA et al. Intracellular hyaluronan: a new frontier for inflammation? Biochim Biophys Acta 2004; 1673: 3–12.
Li Y, Li L, Brown TJ, Heldin P . Silencing of hyaluronan synthase 2 suppresses the malignant phenotype of invasive breast cancer cells. Int J Cancer 2007; 120: 2557–2567.
Heldin P, de la Torre M, Ytterberg D, Bergh J . Differential synthesis and binding of hyaluronan by human breast cancer cell lines: relationship to hormone receptor status. Oncology Rep 1996; 3: 1011–1016.
Takebe N, Warren RQ, Ivy SP . Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res 2011; 13: 211.
Williams CM, Engler AJ, Slone RD, Galante LL, Schwarzbauer JE . Fibronectin expression modulates mammary epithelial cell proliferation during acinar differentiation. Cancer Res 2008; 68: 3185–3192.
Schiller M, Dennler S, Anderegg U, Kokot A, Simon JC, Luger TA et al. Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J Biol Chem 2010; 285: 409–421.
Kowanetz M, Valcourt U, Bergstrom R, Heldin CH, Moustakas A . Id2 and Id3 define the potency of cell proliferation and differentiation responses to transforming growth factor beta and bone morphogenetic protein. Mol Cell Biol 2004; 24: 4241–4254.
Acknowledgements
This work was supported in part by grants from the Swedish Cancer Society, Agnes and Mac Rudberg Foundation, Gurli and Edward Brunnberg Foundation and Mizutani Foundation for Glycoscience. We thank Drs A Moustakas and L van der Heide (Ludwig Institute for Cancer Research, Uppsala, Sweden) for advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Porsch, H., Bernert, B., Mehić, M. et al. Efficient TGFβ-induced epithelial–mesenchymal transition depends on hyaluronan synthase HAS2. Oncogene 32, 4355–4365 (2013). https://doi.org/10.1038/onc.2012.475
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2012.475
Keywords
This article is cited by
-
Stromal hyaluronan accumulation is associated with low immune response and poor prognosis in pancreatic cancer
Scientific Reports (2021)
-
TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1
Nature (2020)
-
HAS3-induced extracellular vesicles from melanoma cells stimulate IHH mediated c-Myc upregulation via the hedgehog signaling pathway in target cells
Cellular and Molecular Life Sciences (2020)
-
Elevated expression of hyaluronan synthase 2 associates with decreased survival in diffusely infiltrating astrocytomas
BMC Cancer (2018)
-
Restriction of drug transport by the tumor environment
Histochemistry and Cell Biology (2018)
