
Abstract
Degeneration of basal forebrain cholinergic circuitry represents an early event in the development of Alzheimer’s disease (AD). These alterations in central cholinergic function are associated with disruptions in arousal, sleep/wake architecture, and cognition. Changes in sleep/wake architecture are also present in normal aging and may represent a significant risk factor for AD. M1 muscarinic acetylcholine receptor (mAChR) positive allosteric modulators (PAMs) have been reported to enhance cognition across preclinical species and may also provide beneficial effects for age- and/or neurodegenerative disease-related changes in arousal and sleep. In the present study, electroencephalography was conducted in young animals (mice, rats and nonhuman primates [NHPs]) and in aged mice to examine the effects of the selective M1 PAM VU0453595 in comparison with the acetylcholinesterase inhibitor donepezil, M1/M4 agonist xanomeline (in NHPs), and M1 PAM BQCA (in rats) on sleep/wake architecture and arousal. In young wildtype mice, rats, and NHPs, but not in M1 mAChR KO mice, VU0453595 produced dose-related increases in high frequency gamma power, a correlate of arousal and cognition enhancement, without altering duration of time across all sleep/wake stages. Effects of VU0453595 in NHPs were observed within a dose range that did not induce cholinergic-mediated adverse effects. In contrast, donepezil and xanomeline increased time awake in rodents and engendered dose-limiting adverse effects in NHPs. Finally, VU0453595 attenuated age-related decreases in REM sleep duration in aged wildtype mice. Development of M1 PAMs represents a viable strategy for attenuating age-related and dementia-related pathological disturbances of sleep and arousal.
Similar content being viewed by others

Introduction
Declining integrity of the central cholinergic system is associated with disruptions in sleep/wake architecture, arousal, and cognition in normal aging and neurodegenerative disease [1, 2]. Alterations in multiple synaptic markers of basal forebrain cholinergic integrity and function have been reported in aging and Alzheimer’s disease (AD) patient populations [3,4,5,6]. In addition, degeneration of the basal forebrain cholinergic projection system is a robust and reliable predictor of entorhinal and neocortical neurodegeneration and constitutes an early event in the development of AD [7]. Specifically, decreases in the cortical expression of cholinergic markers have been correlated with age- and/or neurodegenerative disease-related impairments in attention, memory, and executive functions [5, 8,9,10,11]. Similar deteriorations in cortical cholinergic integrity and cognitive performance have been well documented across aged rodent and nonhuman primate (NHP) species [12, 13]. The impact of deteriorating central cholinergic circuitry has also been linked with abnormalities in sleep/wake architecture and arousal, which are thought to directly contribute to and exacerbate the cognitive impairments observed in individuals with advanced age and dementia [1, 2]. Accumulating evidence indicates that disruptions in sleep represent a significant risk factor for AD, with older dementia patients exhibiting shorter sleep duration and fragmented sleep, elevated rates of sleep disordered breathing and altered circadian rest/activity patterns [14]. Collectively, these findings have led to therapeutic approaches for the enhancement of central cholinergic signaling to ameliorate symptoms associated with pathologic changes in aging and neurodegenerative diseases such as AD [14].
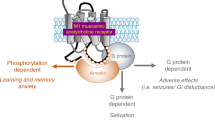
To date, acetylcholinesterase inhibitors (AChEIs) represent the only FDA-approved treatment for the cognitive impairments associated with AD that specifically target the cholinergic system. AChEIs block the degradation of acetylcholine (ACh) resulting in increased synaptic levels of ACh [5]. While AChEIs produce modest therapeutic effects on cognitive impairments during the early stages of AD, these drugs are associated with dose-limiting adverse effects due to nonselective activation of central and peripheral muscarinic acetylcholine receptors (mAChRs) [15]. Of the five different mAChR subtypes activated by ACh (M1–M5), M1 is highly expressed postsynaptically in brain regions that regulate arousal, sleep, and cognition, including the cortex, striatum, and hippocampus [16,17,18,19,20]. Thus, activation of M1 mAChRs is thought to be a promising strategy for the symptomatic treatment of AD-related cognitive deficits. Early clinical studies with xanomeline, an M1/M4-mAChR subtype-preferring orthosteric agonist, showed significant efficacy in ameliorating behavioral disturbances in AD patients with a trend toward improvements in reaction time and verbal memory deficits [21]. Xanomeline, as well as M1-preferring orthosteric agonists, also produced pro-cognitive effects in rodents and NHPs [22], yet have failed in clinical development due to off-target activation of peripheral mAChRs similar to those observed with AChEIs [21].
Using an alternative strategy for the development of subtype-selective activators of M1, our group and others have focused on identification of ligands that target less highly conserved regions of the receptor, termed allosteric sites, which are distinct from the highly conserved ACh binding site. This approach has resulted in the discovery of multiple M1 positive allosteric modulators (PAMs), including VU0453595, with greater than tenfold selectivity for M1 over the other mAChR subtypes and suitable pharmacokinetic properties for dosing in rodent and NHP species [22,23,24]. VU0453595 does not directly activate M1, but potentiates the effects of the endogenous neurotransmitter ACh, thereby maintaining the spatial and temporal pattern of endogenous cholinergic signaling [25]. Previous studies have shown that VU0453595, like other M1 mAChR PAMs, enhances cognitive performance without dose-limiting adverse effects in rodents [26,27,28,29,30,31,32,33]. More recently, our group reported that the investigational drug candidate VU319, a highly optimized M1 PAM, was well tolerated in a Phase I single ascending dose clinical study with dose-related changes indicative of target engagement, including improved reaction times and increased amplitudes of event-related potentials in an incidental memory task [34, 35].
While accumulating evidence supports the further development of M1 PAMs for cognitive decline associated with AD, there is limited information on the impact of selective activation of M1 on sleep/wake architecture and arousal during normal aging. The central cholinergic system plays a critical role in the regulation of normal sleep–wake patterns across species. ACh levels are highest in the morning during peak wakefulness, then decrease throughout the day to lowest levels during the early stages of non-rapid eye movement (NREM) sleep, followed by subsequent increases with the transition from NREM to the later stages of rapid eye movement (REM) sleep [36]. Increases in ACh stimulate higher frequency electroencephalography (EEG) activity consistent with wakefulness and REM sleep [36,37,38]. Previous studies in young adult humans or NHPs have reported that M1 PAMs alter arousal by decreasing power in low frequency ranges, i.e., shifting power from low to high frequency ranges [39], or by attenuating the increase in lower frequency power elicited by the nonselective mAChR antagonist scopolamine in NHPs [40].
The present study is the first systematic evaluation of the effects of the M1 mAChR PAM VU0453595 on sleep/wake architecture and arousal using EEG in young rats, mice, and NHPs and in normally aged mice. In addition, these studies examined the therapeutic index between doses that modulate sleep/wake architecture and/or arousal relative to dose-limiting adverse effects in comparison with the AChEI donepezil and xanomeline. These data provide a critical foundation for future studies of M1 PAMs in preclinical dementia models and AD patients.
Methods and materials
Subjects
Young adult male Sprague-Dawley rats (n = 12, 250–275 g; Envigo, Indianapolis, IN), young (4–6 month; n = 13), or aged (22–26 month; n = 10) adult male wildtype mice or young adult male M1 KO mice (4–6 months; n = 6) with the same genetic background (C57BL/6NTac; Taconic) and drug-naïve (n = 8) young adult (4–8 year old at start of study) male cynomolgus macaques (Macaca fascicularis) served as subjects. All animals were socially housed prior to surgery.
Animals lived in a temperature and humidity controlled environment under a 12/12 h light-dark cycle with water available ad libitum. Rodents had ad libitum access to food in their home cages. NHPs were weighed weekly and fed enough food daily (Purina LabDiet 5045, St. Louis, MO, USA and fresh fruit and vegetables) to maintain healthy body weights and appearance as determined by daily inspection and periodic veterinary examinations. All animals were individually housed following implantation of EEG devices. All experiments were approved by the Vanderbilt University (mice/rats) or Wake Forest School of Medicine (NHPs) Animal Care and Use Committees, and experimental procedures conformed to guidelines established by the National Research Council Guide for the Care and Use of Laboratory Animals. Environmental enrichment was provided as outlined in the Animal Care and Use Committee of Wake Forest University Non-Human Primate Environmental Enrichment Plan.
Compounds
VU0453595, BQCA, and xanomeline L-tartrate were synthesized in-house [30, 41]; donepezil was obtained from Sigma Aldrich (St. Louis, MO). VU0453595 and BQCA were formulated as a microsuspension in 5% and 20% beta-cyclodextrin, respectively, in sterile water. Donepezil and xanomeline were formulated in sterile saline and water, respectively, as aqueous solutions. All compounds’ formulations were adjusted to pH 6–7. Compounds were administered at 10 mL/kg (mice) intraperitoneally (i.p.) and 2 mL/kg (rats) i.p. except for BQCA (administered subcutaneously [s.c.]). For NHPs, VU0453595 was administered orally (i.g.) via a nasogastric tube at 5 mg/mL (3.0 and 10 mg/kg) or 10 mg/mL (30 mg/kg). Xanomeline (1.0, 3.0 mg/kg) was administered s.c. at 0.1 mL/kg. Donepezil (3.0, 10 mg/kg) was administered orally as a powder mixed in a palatable treat and hand-fed to each NHP (EEG) or via nasogastric tube in saline (pharmacokinetic studies). The dose ranges tested have previously been shown to increase cognitive performance in rodents [26, 27, 29] or reverse pharmacological disruptions in rodent or NHPs [30, 42]. Administration of each compound followed a within-subject, counter-balanced design such that each animal received all doses with a minimum of 5 days (washout) between doses; separate vehicle determinations were conducted for each compound.
Electroencephalography
Surgery
All animals used in telemetry studies were surgically implanted under isoflurane anesthesia with a telemetric transmitter (Data Sciences International [DSI], Minneapolis, MN) for the wireless recording of EEG, electromyographic (EMG), and motor activity as previously described [43,44,45,46] (for detailed procedures see Supplementary methods). Following surgery, animals were individually housed.
Examining sleep/wake architecture and qEEG
For all studies, EEG and EMG were recorded from the home cage of each animal continuously for 24 h beginning at the onset of the light cycle on the day of each study. This study design allowed us to assess wake-promoting and sleep altering effects in rodents during the time period they predominately sleep, and to assess effects on arousal in NHPs (since NHPs rarely sleep during the light period, effects on sleep were not directly examined).
Sleep staging and analysis
Trained observers blinded to condition (age, genotype, or pharmacological challenge) scored each 10-s epoch (rats, NHPs) or 5-s epochs (mice) using Neuroscore 3.0 software (DSI) to determine sleep/wake stages, including wake, NREM, or REM sleep based on accepted characteristic oscillatory patterns as previously published by our group [43,44,45]. The amount of time in each stage (wake, NREM, REM) in 1-h (rat, NHPs) or 2-h (mice) bins across a 24-h period served as primary dependent measures to determine effects of age, genotype, or acute pharmacological challenge.
qEEG spectral power analysis
Following sleep staging, quantitative EEG (qEEG) relative power spectra were computed in 1 Hz bins from 0.5 to 100 Hz (rat) or 50 Hz (mouse, NHP) using a Fast Fourier transform with a Hamming window and overlap ratio of 0.5. Relative power within each 1-Hz increment was subsequently binned by stage (wake, NREM, or REM), then averaged across a select time period to yield the state-dependent relative power spectrum for each animal and condition. Differences in spectral power between genotypes or dose-effect determinations were examined in 1-h bins (mouse, rat) or 4-h bins (NHPs) in a state-dependent (Wake, NREM, REM) manner (see Supplementary methods for further description). For NHP studies, the spectral power was averaged for the three vehicle-treatment conditions, and doses of each compound and their respective vehicle treatment were normalized to this 3-day mean.
Assessing effects of M1 mAChR PAM VU0453595, BQCA, and donepezil on sleep/wake duration and qEEG in young rats
To examine selective versus nonselective effects of enhancing cholinergic function, VU0453595 (3.0–30 mg/kg, i.p.), BQCA (3.0–30 mg/kg, s.c.), and donepezil (1.0–10 mg/kg, i.p.) or their respective vehicle were administered 2 h after light onset (quiescent period) in young rats. EEG, EMG, and activity were monitored continuously for 24 h.
Assessing effects of VU0453595, donepezil, and xanomeline on sleep/wake and qEEG in young cynomolgus NHPs
As a proof-of concept study to examine translatability of EEG as a biomarker of CNS function in higher order, gyrencephalic species, VU0453595 (3.0–30 mg/kg, i.g.), donepezil (3.0–10 mg/kg, p.o.), and xanomeline (1.0–3.0 mg/kg, s.c.) were tested in five young adult cynomolgus macaques. Test compounds were administered 30 min after light onset (when arousal levels were presumably low).
Assessing unconditioned behavioral effects and plasma concentrations of VU0453595, xanomeline, and donepezil in young cynomolgus NHPs
To establish dose-effect relationships and a relative therapeutic index, we determined plasma concentrations of each compound. To test the hypothesis that M1 mAChR PAMs elicit less severe adverse effects than nonselective agonists or AChEIs, we implemented a qualitative rating scale to compare the effects of VU0453595, donepezil, and xanomeline in cynomolgus macaques on cholinergic-mediated changes in somatomotor and autonomic function (see Supplementary Table S1).
Vehicle, VU0453595 (3.0–30 mg/kg, i.g.), donepezil (3.0–10 mg/kg, i.g.), and xanomeline (1.0, 3.0 mg/kg, s.c.) were administered 30 min after light onset. Blood samples were collected from 4 NHPs at 0.25, 0.5, 1, 2, 4, 8, 12, and 24 h post dosing to determine plasma concentrations (see Supplementary methods for blood collection and analysis methods). Prior to compound administration and just prior to blood collection at each time point, a brief (<5 min) assessment of general health and autonomic/somatomotor function was conducted to assess potential cholinergic-mediated adverse effects. This assessment incorporated aspects of prior batteries examining adverse or off-target effects across species [30, 42, 43, 47,48,49]. Briefly, trained observers examined each NHP for changes in 18 measures of autonomic function (including functions known to be sensitive to cholinergic stimulation such as salivation, lacrimation, urination, and defecation, body temperature), as well as 13 measures of somatomotor function. Ratings were assigned on a scale of 0, 1, or 2; where 0 = normal or no change from baseline, 1 = a slight effect, and 2 = a marked effect. Scores at each time point were averaged across all NHPs that received each dose of each compound.
Assessing effects of M1 mAChR PAM VU0453595 on sleep/wake duration and qEEG in young and aged mice
VU0453595 (3.0–30 mg/kg, i.p.) or vehicle were administered 2 h after light onset (quiescent period, when rodents predominantely sleep) to young (4–6 months) or aged (22–26 months) wildtype mice or to young (4–6 months) M1 KO mice (30 mg/kg VU0453595 and vehicle only) to confirm M1 PAM selectivity.
Statistics
Sleep/wake architecture and qEEG data are presented as means ± S.E.M. and plasma concentrations are shown as means ± S.D. When possible, a repeated measures two-way analysis of variance (matching both factors) was applied. When group sizes were uneven, a repeated measures, mixed effects model (REML) was applied (see Supplementary methods and Supplementary statistics Table S2 for complete details of tests, factors and results). In all cases, main effects were followed by Dunnett’s or Bonferroni’s multiple comparison test (see Table S2). GraphPad Prism version 8.0 was used for all graphing and statistical applications.
Results
M1 mAChR PAM VU0453595, BQCA, and donepezil produced differential effects on sleep/wake architecture in young rats when dosed 2 h into the inactive period
The M1 mAChR PAM VU0453595 did not alter sleep/wake architecture in young adult rats. There was a main effect of time for all three stages (Wake, NREM, REM; all p < 0.001); and a main effect of VU0453595 dose on REM sleep only (p < 0.05), but no interaction for any stage (Fig. 1a–c; see Supplementary statistics Table S2 for details). There was a main effect of time and time × dose interaction for BQCA on awake duration and NREM sleep (Fig. 1d, e; all p < 0.001). There was a main effect of time (p < 0.001) on REM sleep duration (Fig. 1f). BQCA increased time awake and decreased NREM and REM sleep.

Shown is the duration of time awake (a, d, g), in non-REM (NREM) sleep (b, e, h), or in REM sleep (c, f, i). Following compound administration 2 h into the light (inactive) phase (see arrowhead), VU0453595 did not change time awake (a), in non-REM (NREM) sleep (b), or in REM sleep (c). 3 mg/kg BQCA decreased time awake at ZT 13; 10 mg/kg BQCA increased time awake at ZT 4 and 6, and 30 mg/kg BQCA increased time awake at ZT 4 and 5, and decreased time awake at 13, 14, and 22 (d). 10 mg/kg BQCA decreased NREM sleep at ZT 4 and 6; 30 mg/kg BQCA decreased NREM sleep at ZT 4–6, and increased NREM sleep duration at ZT 13, 14, and 22 (e). 30 mg/kg BQCA decreased REM sleep time at ZT 4, and increased REM sleep at 13–14 (f). 1.0 mg/kg donepezil increased duration of time awake at the ZT 3, 4, and decreased time awake at ZT 24. 3.0 mg/kg donepezil increased time awake at ZT 3–7, and decreased time awake at ZT 16, 20, 23, 24. 10 mg/kg donepezil increased time awake at ZT 3–11, and decreased time awake ZT 16, 17, 20, 23, and 24 (g). 1.0 mg/kg donepezil decreased duration of NREM sleep at ZT 3 and 4 time points with an increase at ZT 24; 3.0 mg/kg donepezil decreased duration of NREM sleep at ZT 3–7 and increased NREM sleep duration at ZT 16, 20, and 23.10 mg/kg donepezil decreased NREM duration at ZT 3–10 with an increase at ZT 16, 17, 20 23, and 24 (h). 3.0 mg/kg donepezil decreased duration of REM sleep at ZT 4–6 and increased REM sleep duration at ZT 9 and 20, while 10 mg/kg donepezil decreased REM sleep duration at ZT 4–11, and increased REM sleep duration at ZT 16, 17, 20, 21, 23 and 24 (i). Gray shading represents 12-h dark period. Data are means ± S.E.M of 1-h bins; n = 8–12/group; open symbols, p < 0.05 compared to vehicle (Dunnett’s test).
In contrast, there was a main effect of donepezil dose, time, and dose × time interaction on duration of time awake (Fig. 1g; all p < 0.001), NREM sleep (Fig. 1h; p < 0.001), and REM sleep time (effect of dose (p < 0.05), time, and dose × time interaction (both p < 0.001)) (Fig. 1l). Donepezil increased time awake and decreased NREM and REM sleep.
M1 mAChR PAM VU0453595, BQCA, and donepezil produced differential effects on relative spectral power in awake epochs 1–2 h post dosing in young rats
There was a main effect of frequency and dose × frequency interaction (both p < 0.001) of VU0453595 on spectral power. In total, 3.0 mg/kg VU0453595 decreased power in the delta band (red horizontal line), while 30 mg/kg VU0453595 (green horizontal line) decreased power in alpha band and increased power in the high gamma band in the frontal cortex (Fig. 2a). There was a main effect of frequency and dose × frequency interaction (both p < 0.001) of BQCA on spectral power. BQCA decreased power in alpha and low-beta ranges and increased power in the gamma band in the frontal cortex (Fig. 2b). There was a main effect of dose (p = 0.001), frequency and dose × frequency interaction (both p < 0.001) of donepezil on spectral power (Fig. 2c). Specifically, 3.0 and 10 mg/kg donepezil (blue and green lines, respectively) increased low frequency delta power, and decreased power in theta, alpha, and low-beta ranges. 3.0 mg/kg donepezil increased, whereas 10 mg/kg donepezil decreased power in the gamma band range.

Shown are changes in relative spectral power in the frontal cortex, during waking epochs only, in the 1- to 2-h period following administration of VU0453595 (a), BQCA (b), and donepezil (c). 3 mg /kg VU0453595 decreased frequencies 0.5 and 1. 10 mg/kg VU0453595 increased frequency 9. 30 mg/kg VU0453595 decreased frequencies 11–13 and increased frequencies 60 and 62–99 (a). 3 mg/kg BQCA decreased frequencies 9–11, 13, 14, 17 and increased frequencies 60–62 and 67–99. 10 mg/kg BQCA decreased frequencies 12 and 13 and increased frequencies 60 and 61. 30 mg/kg BQCA decreased frequencies 9–17 and increased frequencies 57–99 (b). 3 mg/kg donepezil decreased frequencies 0.5, 5–17 and increased frequencies 31–79, 83–85, and 90. 10 mg/kg donepezil decreased frequencies 0.5–2, 4–20, 83–99 and increased frequencies 28, 29, 44 (c). Gray/tan shading represents frequency bands (∆, delta 0.5–4 Hz; θ theta 4–8 Hz; α alpha, 8–13 Hz; β beta, 13–30 Hz; γ gamma 30–100 Hz). Data are means ± S.E.M.; n = 7–12/group; corresponding colored horizontal dots/lines represent frequencies at which each dose group was statistically different from vehicle-treated rats, p < 0.05, Dunnett’s post hoc test.
In NREM sleep epochs, 1–2 h following dosing, donepezil decreased delta power (p < 0.01), a measure of sleep quality, whereas BQCA and VU0453595 did not significantly affect delta power (p > 0.05; see Supplementary Fig. S1).
M1 mAChR PAM VU0453595 increased high frequency beta and gamma power in young adult male NHPs
Neither VU0453595, xanomeline, nor donepezil altered the duration of time awake, in NREM, or in REM sleep (since dosing occurred during early active period, long lasting effects on sleep were not seen nor expected; hence, data not shown). For VU0453595, qEEG analysis revealed a dose × frequency interaction (p < 0.01; Fig. 3a). In total, 10 and 30 mg/kg increased power in the gamma frequency and 30 mg/kg increased power in the beta frequency range (see Supplementary Fig. S2 for individual data). Xanomeline treatment caused a significant main effect of frequency band (p < 0.01) and dose × frequency interaction (p < 0.05; Fig. 3b), with 3.0 mg/kg xanomeline decreasing power in the delta, theta, and alpha bands (see Supplementary Fig. S3 for individual data). In donepezil-treated NHPs, there were no significant main effects of dose, frequency band, nor a dose × frequency interaction (all p > 0.05), despite qualitative increases in beta (3.0 mg/kg) and gamma bands (3.0 and 10 mg/kg) (Fig. 3c; see Supplementary Fig. S4 for individual data).

Shown are changes in relative spectral power collapsed into spectral bands (to minimize variability) following administration of VU0453595 (a), xanomeline (b), and donepezil (c) immediately after light onset; power bands are defined as delta (0.5–4 Hz), theta (4–8 Hz), alpha (8–13 Hz), sigma (13–18 Hz), beta (18–30 Hz), gamma (30–50 Hz). All 10-s epochs during the first 4 h post dosing were combined and expressed as a percent change from spectral power within the same frequency band and time period from a mean of three vehicle-treated conditions. Data are presented as mean ± S.E.M.; n = 5 (VU0453595), n = 4 (xanomeline, donepezil); *p < 0.05 compared to vehicle (Dunnett’s test).
M1 mAChR PAM VU0453595 displayed a reduced adverse side effect profile compared to xanomeline and donepezil in young adult male NHPs
All three compounds demonstrated dose-proportional increases in plasma concentrations; VU0453595 demonstrated a relatively long rate of elimination following oral administration (Supplementary Fig. S5). VU0454595 (3.0–30 mg/kg) appeared to cause a slight increase in urination, and a qualitatively assessed decrease in respiration rate at the 30 mg/kg dose (Supplementary Table S3). Occasional changes in posture, motor coordination, and leg weakness were noted in one NHP following 30 mg/kg VU0453595 (Supplementary Table S3). To confirm sensitivity of this scale, donepezil and xanomeline were also examined. Xanomeline (1.0 and 3.0 mg/kg) induced miosis, vasoconstriction, increased arousal, irritability, and salivation. In addition, in some NHPs 3.0 mg/kg xanomeline induced oral dyskinesias (Supplementary Table S4). Donepezil (10 mg/kg) induced urination, defecation, emesis, ptosis, vasoconstriction, irritability, and in some cases tremors (Supplementary Table S5).
M1 mAChR PAM VU0453595 attenuated reductions in REM sleep in aged wildtype mice when dosed 2 h into the inactive period
In aged wildtype mice treated with VU0453595, there was a significant effect of time (p < 0.001; Fig. 4a) and dose × time interaction (p < 0.01), on duration of time awake in 2-h bins across the 24-h period. There was a significant effect of time (p < 0.001; Fig. 4b) and dose × time interaction (p < 0.05), on duration of NREM sleep time. There was a significant effect of time and dose × time interaction (both p < 0.001; Fig. 4c), on duration of REM sleep. VU0453595 transiently increased wake and decreased NREM sleep followed by sustained increases in REM sleep duration (Fig. 4a–c) in aged wildtype mice.

Shown is the duration of time awake (a, d, g), in non-REM (NREM) sleep (b, e, h), or in REM sleep (c, f, i) in aged (22–26 month old (a–c)) and young adult (4–6 month old) mice (d–f). Following compound administration 2 h into the light (quiescent) phase (see arrowhead), VU453595 increased REM sleep duration in aged (22–26 month old) mice (c) without affecting REM sleep in young (4–6 month old) mice (f). Specifically, 30 mg/kg VU0453595 produced a significant increase in duration of time awake at ZT 4 (a) and a decrease in NREM sleep from ZT 4 and 6 in aged mice (b). 3 mg/kg VU0453595 increased REM sleep at ZT 6, 8 and decreased REM sleep at ZT 20, 10 mg/kg VU0453595 increased REM sleep at ZT 6 and 8 and 30 mg/kg VU0453595 increased REM sleep duration from ZT 8 and 10 in aged wildtype mice (c). In young wildtype mice, 30 mg/kg VU0453595 produced significant increases in duration of time awake at ZT 4 (d). 10 mg/kg VU0453595 produced a significant decrease in duration of NREM sleep at ZT 6 and increase at ZT 24 and 30 mg/kg VU0453595 produced a significant decrease in NREM sleep from ZT 4 (e); but no significant effects on REM sleep duration (f). For comparison, vehicle-treated groups were replotted in (g–i) to better illustrate the age-related decreases in REM sleep duration. Aged mice showed significantly higher durations of time awake at ZT 8 and 22 (g) and significantly lower durations of REM sleep at the ZT 6–10 and 22 compared to young mice (i). Grey shading represents 12-h dark period. Data are means ± S.E.M. of 2-h bins; n = 10–13/group; Open symbols, p < 0.05 compared to vehicle (Dunnett’s test (a–f); Bonferroni (g–i)).
In young wildtype mice treated with VU0453595, there was a significant effect of time (p < 0.001; Fig. 4d) and dose (p < 0.05) on duration of time awake in 2-h bins across the 24-h period. There was a significant effect of time (p < 0.001; Fig. 4e) and dose × time interaction (p < 0.05) on duration of NREM sleep time. There was a significant effect of time (p < 0.001; Fig. 4f) on duration of REM sleep time. In young wildtype mice, similar to aged mice, transient effects of VU0453595 were present on wake and NREM but there were no effects on REM sleep (Fig. 4d–f).
When comparing vehicle-treated aged wildtype mice to vehicle-treated young wildtype mice there was a significant effect of time (Fig. 4g) and time × age interaction (both p < 0.001) on duration of time awake in 2-h bins across the 24-h period. There was a significant effect of time (p < 0.001; Fig. 4h) and time × age interaction (p < 0.01) on duration of NREM sleep in 2-h bins across the 24-h period. There was a significant effect of age (Fig. 4i), time and time × age interaction (all p < 0.001) on duration of REM sleep in 2-h bins across the 24-h period. Aged wildtype mice showed greater time awake and less time in REM sleep compared to young mice (Fig. 4g, i).
M1 mAChR PAM VU0453595 increased high frequency gamma power in awake epochs 1–2 h post dosing in young and aged mice
In young wildtype mice treated with VU453595, there was a main effect of dose, frequency, and dose × frequency interaction (all p < 0.001) on spectral power in the frontal cortex (Fig. 5a). In aged wildtype mice (Fig. 5b), there was a significant effect of frequency and dose × frequency interaction (both p < 0.001). In both young and aged mice, VU0453595 increased high frequency gamma power and 30 mg/kg VU0453595 decreased power in the low frequency range. The effects of 30 mg/kg VU0453595 on sleep–wake architecture and qEEG were absent in young M1 knockout mice (Supplementary Fig. S6).

Shown are changes in relative spectral power in the frontal cortex, during waking epochs only, in the 1- to 2-h period following administration of VU0453595 in young adult (4–6 month old (a)) and aged (22–26 month old) wildtype mice (b). In young wildtype mice, 30 mg/kg VU0453595 decreased power distribution in 0.5–2 range and increased power at 30–50 Hz (a). In aged wildtype mice, 10 mg/kg VU0453595 decreased power distribution at 3 Hz, and increased power at 40 and 43–50 Hz; 30 mg/kg VU0453595 decreased power distribution at 0.5–2 Hz and increased power at 5–6, 44, and 46–50 Hz (b). Comparison of spectral power in vehicle-treated young and aged mice shown as percent difference from young mice (c). Aged mice showed a decrease at 0.5 Hz and an increase at 3 Hz compared to young wildtype mice. Gray/tan shading represents frequency bands (∆, delta 0.5–4 Hz; θ theta 4–8 Hz; α alpha, 8–13 Hz; β beta, 13–30 Hz; γ gamma 30–100 Hz). Data are means ± S.E.M.; in (c), error bars on the young mice group represent SEM following calculations of individual percent differences from the group mean. n = 11–12/group; corresponding colored horizontal dots/lines represent frequencies at each dose that were statistically different from vehicle-treated mice. p < 0.05 (Dunnett’s post hoc test).
Comparison of vehicle-treated aged and young mice revealed a significant effect of frequency (p < 0.0001) and frequency × age interaction (p < 0.0001; Fig. 5c).
Discussion
Disruptions in normal sleep/wake architecture and arousal are commonly observed symptoms in normal aging and neurodegenerative disorders and may be a significant prodromal risk factor for the development of AD [14, 50]. Accumulating evidence suggests selective activation of M1 mAChRs may represent an alternative therapeutic strategy for the normalization of cognitive deficits and potentially the sleep/wake abnormalities associated with neurodegenerative diseases. In the present study, the M1 PAM VU0453595 produced dose-related increases in high frequency gamma power in young mice, rats, and NHPs, a well-characterized correlate of arousal and cognitive enhancement [51], without changing the duration of time spent in the different sleep/wake stages. These effects were absent in young M1 mAChR KO mice, confirming an M1 mAChR-selective effect. Importantly, doses of VU0453595 that increased gamma power in monkeys did not induce cholinergic-mediated adverse effects that were present following administration of donepezil and xanomeline. This qEEG signature of selective M1 mAChR engagement was recapitulated by the M1 ago-PAM BQCA. In contrast, the AChEI donepezil produced a distinct qEEG signature, dose-dependently increased time awake, nonselectively decreased sleep duration in young rats, and decreased delta power in NREM sleep indicative of decreased sleep quality [52,53,54,55]. In aged mice, VU0453595 produced a robust attenuation of age-related changes in sleep, specifically enhancement of REM duration, which is shown to be significantly decreased with normal aging. In combination, these findings in young and aged animals produce an important baseline for the future evaluation of M1 PAM effects on sleep wake/architecture and EEG in preclinical species and clinical populations.
Interestingly, in aged mice, the magnitude of VU0453595-dependent increases in high frequency gamma power was lower in comparison with effects observed in young mice. Since an M1 PAM enhances effects of endogenous ACh, these data suggest possible differences in endogenous ACh signaling, which can vary with circadian rhythm, age, and disease state. Under normal conditions, stimulation of cholinergic projections from midpontine cholinergic nuclei increases the transition from NREM selectively to REM sleep [56]. In contrast, stimulation of the cholinergic basal forebrain neurons during NREM promotes the transition to either REM sleep or wakefulness [57] and is directly responsible for the increased gamma and theta oscillations during waking states [58]. The basal forebrain cholinergic system degenerates in AD [59], whereas the midpontine projection neurons are spared [60]. As similar, though less severe, cholinergic changes are present in normal aging, M1 mAChR PAMs may enhance ACh-mediated functions through intact midpontine projections resulting in increased REM sleep. However, once age-related basal forebrain cholinergic degeneration becomes severe, insufficient endogenous ACh signaling may preclude an M1 PAM from having the same magnitude of effect on arousal during wakefulness.
In this study, animals were dosed during the inactive period (when cholinergic tone is presumed to be low) to enable maximal possible dynamic range to observe increases in arousal. However, in aged animals with declining integrity of the central cholinergic system, dosing during the active period may be the optimal time to observe enhancements in arousal. In young animals, enhancing arousal during the inactive period is possible, yet increasing REM sleep above optimal levels may be difficult. Ongoing studies are focused on understanding circadian rhythm fluctuations and age-related decline in cholinergic synaptic integrity in preclinical species, to determine whether there is sufficient endogenous ACh to observe efficacy with VU0453595 alone, or if cholinergic tone needs to be boosted in combination with a subthreshold dose of an AChEI. This information may be critical in determining the design of future clinical trials with regard to patient demographics, time of drug administration, and whether effects of an M1 PAM may be sufficient as a stand-alone treatment or required to be administered as an adjunct treatment to maximize clinical efficacy.
Several promising M1 mAChR allosteric modulators have progressed into clinical trials as potential treatments for cognitive impairments associated with AD or neuropsychiatric disorders, including a proof-of-concept study examining efficacy of MK-7622 as an adjunct treatment to AChEIs in AD patients [61]. Unfortunately, these programs were halted due to a lack of true subtype-selectivity and off-target adverse side effect liability [39, 62, 63] (Merck, ClinicalTrial ID: NCT01852110). Interestingly, while VU0453595 increased power in high frequency ranges in young NHPs similar to young mice and rats, the M1 PAM MK-7622 dose-dependently decreased power in delta to sigma power bands in young NHPs [39], similar to our present finding with xanomeline. Differences between MK-7622 effects and VU0453595 in NHPs may be attributed to methodology (e.g., electrode placement, data collection, analytical techniques). An alternative interpretation is that an increase in high frequency gamma power was achieved by dose escalation that is precluded by dose-limiting adverse effects of less selective M1 PAMs, direct agonists, or indirect agonists (e.g., MK-7622, xanomeline, donepezil). Indeed, MK-7622 displays robust agonist activity at the M1 mAChR in cell-based assays, seizure activity [27] and cholinergic-mediated adverse effects within dose ranges that improve cognition in rodents [64]; all effects not seen with the pure M1 PAM VU0453595 [27]. While quantitatively different from the actions of VU0453595, qualitatively, both compounds shifted power distribution from lower to higher frequency ranges, which likely corresponds with modest increases in arousal and behavioral effects. Importantly, prior studies with MK-7622 were promising in that dose ranges predicted from preclinical studies produced reliable changes on qEEG in healthy humans, notably increased power in sigma and beta power bands [39]. Specific to VU0453595, it remains to be seen whether this increase in gamma power will correlate with greater efficacy for cognitive enhancement. Recently, the Warren Center for Neuroscience Drug Discovery, in collaboration with the Vanderbilt Center for Cognitive Medicine, completed a Phase I study in healthy volunteers with the M1 PAM VU319 and appeared to show dose-related changes in both cognitive and EEG measures of central M1-mediated target engagement [34, 35]; future studies will assess whether this translates to efficacy in clinical populations.
In summary, the present findings suggest selective M1 PAMs may be beneficial in enhancing not only cognition and/or arousal, but also in normalizing REM sleep deficits observed in pathologic aging and neurodegenerative diseases with minimal adverse effects and support the utility of EEG as a highly translational marker of central M1 target engagement for future clinical M1 PAM development.
Funding and disclosures
CWL, TMB, ALB, and PJC hold patents that protect different classes of muscarinic acetylcholine allosteric modulators including M1 PAMs recently licensed to Acadia Pharmaceuticals for therapeutic development. PJC and CWL receive research support from Acadia Pharmaceuticals and Boehringer Ingleheim. The remaining authors have nothing to disclose.
This work was supported by the National Institute of Health Grants AG054622 (CKJ), MH073676 (PJC), MH093366 (PJC), MH087965 (PJC), the William K. Warren Foundation and a PhRMA Foundation postdoctoral fellowship grant in Pharmacology and Toxicology (RWG).
References
Prinz PN, Peskind ER, Vitaliano PP, Raskind MA, Eisdorfer C, Zemcuznikov HN, et al. Changes in the sleep and waking EEGs of nondemented and demented elderly subjects. J Am Geriatr Soc. 1982;30:86–92.
Lloret MA, Cervera-Ferri A, Nepomuceno M, Monllor P, Esteve D, Lloret A. Is sleep disruption a cause or consequence of Alzheimer’s disease? Reviewing its possible role as a biomarker. Int J Mol Sci. 2020;21:1168–86.
Bartus RT, Dean IIIRL, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–17.
Terry AV, Buccafusco JJ. The cholinergic hypothesis of age and Alzheimer’s disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther. 2003;306:821–7.
Dumas JA, Newhouse PA. The cholinergic hypothesis of cognitive aging revisited again: cholinergic functional compensation. Pharmacol Biochem Behav. 2011;99:254–61.
Aghourian M, Legault-Denis C, Soucy J-P, Rosa-Neto P, Gauthier S, Kostikov A, et al. Quantification of brain cholinergic denervation in Alzheimer’s disease using PET imaging with [18F]-FEOBV. Mol Psychiatry. 2017;22:1531–8.
Fernández-Cabello S, Kronbichler M, Van Dijk KRA, Goodman JA, Spreng RN, Schmitz TW. Basal forebrain volume reliably predicts the cortical spread of Alzheimer’s degeneration. Brain. 2020;143:993–1009.
Drachman DA, Leavitt J. Human memory and the cholinergic system. Arch Neurol. 1974;30:113.
Mesulam M. The cholinergic lesion of Alzheimer’s disease: pivotal factor or side show? Learn Mem. 2004;11:43–9.
Richter N, Allendorf I, Onur OA, Kracht L, Dietlein M, Tittgemeyer M, et al. The integrity of the cholinergic system determines memory performance in healthy elderly. Neuroimage. 2014;100:481–8.
Schliebs R, Arendt T. The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. J Neural Transm. 2006;113:1625–44.
Wu CF, Bertorelli R, Sacconi M, Pepeu G, Consolo S. Decrease of brain acetylcholine release in aging freely-moving rats detected by microdialysis. Neurobiol Aging. 1988;9:357–61.
Voytko ML, Mach RH, Gage HD, Ehrenkaufer RL, Efange SM, Tobin JR. Cholinergic activity of aged rhesus monkeys revealed by positron emission tomography. Synapse. 2001;39:95–100.
Bubu OM, Brannick M, Mortimer J, Umasabor-Bubu O, Sebastião YV, Wen Y, et al. Sleep, cognitive impairment, and Alzheimer’s disease: a systematic review and meta-analysis. Sleep. 2017;40:zsw032.
Galimberti D, Scarpini E. Old and new acetylcholinesterase inhibitors for Alzheimer’s disease. Expert Opin Investig Drugs. 2016;25:1181–7.
Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–26.
Levey AI, Edmunds SM, Koliatsos V, Wiley RG, Heilman CJ. Expression of m1-m4 muscarinic acetylcholine receptor proteins in rat hippocampus and regulation by cholinergic innervation. J Neurosci. 1995;15:4077–92.
Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined m1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci USA. 1998;95:11465–70.
Rouse ST, Gilmor ML, Levey AI. Differential presynaptic and postsynaptic expression of m1-m4 muscarinic acetylcholine receptors at the perforant pathway/granule cell synapse. Neuroscience. 1998;86:221–32.
Rouse ST, Marino MJ, Potter LT, Conn PJ, Levey AI. Muscarinic receptor subtypes involved in hippocampal circuits. Life Sci. 1999;64:501–9.
Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–73.
Jones CK, Byun N, Bubser M. Muscarinic and nicotinic acetylcholine receptor agonists and allosteric modulators for the treatment of schizophrenia. Neuropsychopharmacology. 2012;37:16–42.
Conn PJ, Lindsley CW, Jones CK. Activation of metabotropic glutamate receptors as a novel approach for the treatment of schizophrenia. Trends Pharmacol Sci. 2009;30:25–31.
Bubser M, Byun N, Wood MR, Jones CK. Muscarinic receptor pharmacology and circuitry for the modulation of cognition. 2012;208:121–66.
Conn PJ, Jones CK, Lindsley CW. Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci. 2009;30:148–55.
Lv X, Dickerson JW, Rook JM, Lindsley CW, Conn PJ, Xiang Z. M1muscarinic activation induces long-lasting increase in intrinsic excitability of striatal projection neurons. Neuropharmacology. 2017;118:209–22.
Moran SP, Dickerson JW, Cho HP, Xiang Z, Maksymetz J, Remke DH, et al. M1-positive allosteric modulators lacking agonist activity provide the optimal profile for enhancing cognition. Neuropsychopharmacology. 2018;43:1763–71.
Grannan MD, Mielnik CA, Moran SP, Gould RW, Ball J, Lu Z, et al. Prefrontal cortex-mediated impairments in a genetic model of NMDA receptor hypofunction are reversed by the novel M1 PAM VU6004256. ACS Chem Neurosci. 2016;7:1706–16.
Gould RW, Dencker D, Grannan M, Bubser M, Zhan X, Wess J, et al. Role for the M1 muscarinic acetylcholine receptor in top-down cognitive processing using a touchscreen visual discrimination task in mice. ACS Chem Neurosci. 2015;6:1683–95.
Ghoshal A, Rook JM, Dickerson JW, Roop GN, Morrison RD, Jalan-Sakrikar N, et al. Potentiation of M1 muscarinic receptor reverses plasticity deficits and negative and cognitive symptoms in a schizophrenia mouse model. Neuropsychopharmacology. 2016;41:598–610.
Rook JM, Bertron JL, Cho HP, Garcia-Barrantes PM, Moran SP, Maksymetz JT, et al. A novel M1 PAM VU0486846 exerts efficacy in cognition models without displaying agonist activity or cholinergic toxicity. ACS Chem Neurosci. 2018;19:2274–85.
Uslaner JM, Eddins D, Puri V, Cannon CE, Sutcliffe J, Chew CS, et al. The muscarinic M1 receptor positive allosteric modulator PQCA improves cognitive measures in rat, cynomolgus macaque, and rhesus macaque. Psychopharmacology. 2013;225:21–30.
Ma L, Seager MA, Wittmann M, Jacobson M, Bickel D, Burno M, et al. Selective activation of the M1 muscarinic acetylcholine receptor achieved by allosteric potentiation. Proc Natl Acad Sci USA. 2009;106:15950–5.
Newhouse P, Conley A, Key A, Blackford J, Lindsley C, Conn P, et al. Development of the muscarinic cholinergic PAM VU319 for cognitive enhancement: phase 1 tests of safety and target engagement. Alzheimer’s Dement J Alzheimer’s Assoc. 2019;15:P574.
Conley A, Key A, Blackford J, Conn P, Lindsley C, Jones C, et al. Cognitive and electrophysiological measures of a phase 1 single dose study of the muscarinic positive allosteric modulator VU319. Alzheimer’s Dement J Alzheimer’s Assoc. 2019;15:P253.
Brown RE, Basheer R, McKenna JT, Strecker RE, McCarley RW. Control of sleep and wakefulness. Physiol Rev. 2012;92:1087–187.
Graef S, Schönknecht P, Sabri O, Hegerl U. Cholinergic receptor subtypes and their role in cognition, emotion, and vigilance control: an overview of preclinical and clinical findings. Psychopharmacology. 2011;215:205–29.
Platt B, Riedel G. The cholinergic system, EEG and sleep. Behav Brain Res. 2011;221:499–504.
Uslaner JM, Kuduk SD, Wittmann M, Lange HS, Fox SV, Min C, et al. Preclinical to human translational pharmacology of the novel M1 positive allosteric modulator MK-7622. J Pharmacol Exp Ther. 2018;365:556–66.
Kurimoto E, Nakashima M, Kimura H, Suzuki M. TAK-071, a muscarinic M1 receptor positive allosteric modulator, attenuates scopolamine-induced quantitative electroencephalogram power spectral changes in cynomolgus monkeys. PLoS One. 2019;14:e0207969.
Shirey JK, Brady AE, Jones PJ, Davis AA, Bridges TM, Kennedy JP, et al. A selective allosteric potentiator of the M1 muscarinic acetylcholine receptor increases activity of medial prefrontal cortical neurons and restores impairments in reversal learning. J Neurosci. 2009;29:14271–86.
Vardigan JD, Cannon CE, Puri V, Dancho M, Koser A, Wittmann M, et al. Improved cognition without adverse effects: novel M1 muscarinic potentiator compares favorably to donepezil and xanomeline in rhesus monkey. Psychopharmacology. 2015;232:1859–66.
Gould RW, Nedelcovych MT, Gong X, Tsai E, Bubser M, Bridges TM, et al. State-dependent alterations in sleep/wake architecture elicited by the M4 PAM VU0467154—relation to antipsychotic-like drug effects. Neuropharmacology. 2016;102:244–53.
Rook JM, Xiang Z, Lv X, Ghoshal A, Dickerson JW, Bridges TM, et al. Biased mGlu5-positive allosteric modulators provide invivo efficacy without potentiating mGlu5 modulation of NMDAR currents. Neuron. 2015;86:1029–40.
Nedelcovych MT, Gould RW, Zhan X, Bubser M, Gong X, Grannan M, et al. A rodent model of traumatic stress induces lasting sleep and quantitative electroencephalographic disturbances. ACS Chem Neurosci. 2015;6:485–93.
Fisher NM, Gould RW, Gogliotti RG, McDonald AJ, Badivuku H, Chennareddy S, et al. Phenotypic profiling of mGlu7 knockout mice reveals new implications for neurodevelopmental disorders. Genes Brain Behav. 2020. https://doi.org/10.1111/gbb.12654.
Bubser M, Bridges TM, Dencker D, Gould RW, Grannan M, Noetzel MJ, et al. Selective activation of M4 muscarinic acetylcholine receptors reverses MK-801-induced behavioral impairments and enhances associative learning in rodents. ACS Chem Neurosci. 2014;5:920–42.
Andersen MB, Fink-Jensen A, Peacock L, Gerlach J, Bymaster F, Lundbæk JA, et al. The muscarinic M1/M4receptor agonist xanomeline exhibits antipsychotic-like activity in cebus apella monkeys. Neuropsychopharmacology. 2003;28:1168–75.
Patel S, Freedman S, Chapman KL, Emms F, Fletcher AE, Knowles M, et al. Biological profile of L-745,870, a selective antagonist with high affinity for the dopamine D4 receptor. J Pharmacol Exp Ther. 1997;283:636–47.
Frohnhofen H, Schlitzer J, Netzer N. Sleep in older adults and in subjects with dementia. Z Gerontol Geriatr. 2017;50:603–8.
Buzsáki G, Silva da FL. High frequency oscillations in the intact brain. Prog Neurobiol. 2012;98:241–9.
Nissen C, Nofzinger EA, Feige B, Waldheim B, Radosa MP, Riemann D, et al. Differential effects of the muscarinic M1 receptor agonist RS-86 and the acetylcholine-esterase inhibitor donepezil on REM sleep regulation in healthy volunteers. Neuropsychopharmacology. 2006;31:1294–300.
Riemann D, Gann H, Dressing H, Müller WE, Aldenhoff JB. Influence of the cholinesterase inhibitor galanthamine hydrobromide on normal sleep. Psychiatry Res. 1994;51:253–67.
Iwata N, Kozuka M, Hara M, Kaneko T, Tonohiro T, Sugimoto M, et al. Activation of cerebral function by CS-932, a functionally selective M1 partial agonist: neurochemical characterization and pharmacological studies. Jpn J Pharmacol. 2000;84:266–80.
Jung JY, Roh M, Ko KK, Jang HS, Lee SR, Ha JH, et al. Effects of single treatment of anti-dementia drugs on sleep-wake patterns in rats. Korean J Physiol Pharmacol. 2012;16:231–6.
Van Dort CJ, Zachs DP, Kenny JD, Zheng S, Goldblum RR, Gelwan NA, et al. Optogenetic activation of cholinergic neurons in the PPT or LDT induces REM sleep. Proc Natl Acad Sci USA. 2015;112:584–9.
Han Y, Shi Y, Xi W, Zhou R, Tan Z, Wang H, et al. Selective activation of cholinergic basal forebrain neurons induces immediate sleep-wake transitions. Curr Biol. 2014;24:693–8.
Cape EG, Manns ID, Alonso A, Beaudet A, Jones BE. Neurotensin-induced bursting of cholinergic basal forebrain neurons promotes gamma and theta cortical activity together with waking and paradoxical sleep. J Neurosci. 2000;20:8452–61.
Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer’s disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–9.
Woolf NJ, Jacobs RW, Butcher LL. The pontomesencephalotegmental cholinergic system does not degenerate in Alzheimer’s disease. Neurosci Lett. 1989;96:277–82.
Voss T, Li J, Cummings J, Farlow M, Assaid C, Froman S, et al. Randomized, controlled, proof-of-concept trial of MK-7622 in Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv. 2018;4:173–81.
Bradley SJ, Molloy C, Bundgaard C, Mogg AJ, Karen J, Dwomoh L, et al. Bitopic binding mode of an M1 muscarinic acetylcholine receptor agonist associated with adverse clinical trial outcomes. Mol Pharmacol. 2018;93:645–56.
Nathan PJ, Watson J, Lund J, Davies CH, Peters G, Dodds CM, et al. The potent M1 receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int J Neuropsychopharmacol. 2013;16:721–31.
Mandai T, Sako Y, Kurimoto E, Shimizu Y, Nakamura M, Fushimi M, et al. T-495, a novel low cooperative M1 receptor positive allosteric modulator, improves memory deficits associated with cholinergic dysfunction and is characterized by low gastrointestinal side effect risk. Pharmacol Res Perspect. 2020;8:e00560.
Acknowledgements
We would like to thank Erica Williams and Xuewen Gong for their efforts with data processing, Weimin Peng and Dina McGinness for technical support, Ken Szeliga and Susan Nader for assistance with data collection, and David Devilbiss for thoughtful advice on data analysis and presentation. Studies were performed in part through the use of the Murine Neurobehavioral Core laboratory at the Vanderbilt University Medical Center.
Author information
Authors and Affiliations
Contributions
RWG: conceptualization, methodology, investigation, visualization, formal analysis, writing-original draft, and editing. JKR: formal analysis, writing–review, and editing. MTN: methodology, investigation. MB: supervision, visualization, writing–review and editing. ALB: formal analysis. TMB: formal analysis. PAN: writing–review and editing. PJC: resources. CWL: resources. MAN: resources, writing–review and editing. CKJ: conceptualization, methodology, resources, writing–review and editing, funding acquisition
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Gould, R.W., Russell, J.K., Nedelcovych, M.T. et al. Modulation of arousal and sleep/wake architecture by M1 PAM VU0453595 across young and aged rodents and nonhuman primates. Neuropsychopharmacol. 45, 2219–2228 (2020). https://doi.org/10.1038/s41386-020-00812-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-020-00812-7
This article is cited by
-
Automatic wavelet-based assessment of behavioral sleep using multichannel electrocorticography in rats
Sleep and Breathing (2021)
