
Abstract
Positive, prosocial interactions are essential for survival, development, and well-being. These intricate and complex behaviors are mediated by an amalgamation of neural circuit mechanisms working in concert. Impairments in prosocial behaviors, which occur in a large number of neuropsychiatric disorders, result from disruption of the coordinated activity of these neural circuits. In this review, we focus our discussion on recent findings that utilize modern approaches in rodents to map, monitor, and manipulate neural circuits implicated in a variety of prosocial behaviors. We highlight how modulation by oxytocin, serotonin, and dopamine of excitatory and inhibitory synaptic transmission in specific brain regions is critical for regulation of adaptive prosocial interactions. We then describe how recent findings have helped elucidate pathophysiological mechanisms underlying the social deficits that accompany neuropsychiatric disorders. We conclude by discussing approaches for the development of more efficacious and targeted therapeutic interventions to ameliorate aberrant prosocial behaviors.
Similar content being viewed by others

Introduction
Social behaviors, broadly defined, comprise any interaction between members of a species and thus consist of a complex set of behaviors including those required for survival, reproduction, nurturing offspring, and overall well-being [1, 2]. It is likely that many of the neural mechanisms mediating adaptive social behaviors that are necessary for reproduction and survival have been evolutionarily conserved. Furthermore, impairments in adaptive, prosocial behaviors that promote well-being commonly occur in a broad range of neuropsychiatric disorders including schizophrenia (SZ), substance use disorder, mood disorders and, most notably, in autism spectrum disorder (ASD) [3].
In this brief review, we limit our discussion to recent findings on the neural circuit mechanisms regulating non-sexual, non-aggressive, prosocial behaviors in rodents, which include behaviors that assess sociability, social reward, social memory, as well as behavioral antecedents related to empathy (e.g., emotional contagion). We will not cover sexual, parental, or aggressive behaviors and refer the reader to several recent reviews on these topics [4,5,6,7,8,9]. Here, we operationally define prosocial behaviors based on assays that measure direct social interactions, social preference, social novelty, and social “reward”. We also discuss some of the neural circuit mechanisms thought to underlie deficits in prosocial behaviors that occur in prominent neuropsychiatric disorders and conclude by discussing potential novel therapeutic interventions to treat these deficits.
A major advance in the current understanding of mammalian prosocial behaviors came from seminal studies that investigated the role of oxytocin (OXT) in voles, a neuropeptide that is well established to play critical roles in social and affiliative behaviors [9, 10]. Monogamous prairie voles, who maintain a pair bond for life, were found to have a significantly higher density of OXT receptors in the nucleus accumbens (NAc), a critical node in the brain’s reward circuitry [11, 12], when compared to non-monogamous montane voles [13]. The critical importance of these receptors was subsequently established by demonstrating that direct infusion of an OXT receptor antagonist into the NAc prevented pair bond formation in prairie voles [10]. These findings provided an important initial clue to some of the critical neural circuit mechanisms governing one specific form of adaptive social behavior. In addition to OXT being a critical regulator of affiliative social behavior, it became apparent that the prominent neuromodulators, serotonin (5-HT) and dopamine (DA), which are implicated in a variety of cognitive and emotional processes [3, 12, 14,15,16,17], also play pivotal roles in regulating prosocial behaviors such as social approach [3, 18]. Together, a body of work supported the idea that these neuromodulators regulate social behavior through alterations in neuronal activity at key nodes of the mesolimbic reward system [19, 20]. However, until the advent of now standard technologies that provide genetic access to discrete neuronal populations to monitor and manipulate activity patterns in defined cell types, progress to more comprehensively delineate the neural circuits regulating prosocial behaviors was limited [21, 22]. Here, we will focus on more recent studies that leverage the methodological advances that have occurred over the last two decades and which most commonly are applied to mice.
Standard behavioral assays of prosocial behavior
Mice are social creatures and provide an excellent species in which to investigate the neural circuit mechanisms underlying normal prosocial interactions as well as what goes awry in disease states because of their genetic accessibility, which has led to the generation of abundant knockout and transgenic lines. Their popularity as an experimentally accessible species has led to the establishment of numerous behavioral assays, which have been validated to assess a variety of closely related prosocial behaviors [23].
Juvenile interaction assay
The juvenile interaction assay assesses direct, non-aggressive social interaction between an adult test mouse (>7 weeks of age) and novel conspecific juvenile mouse (commonly 3–5 weeks of age) of the same sex [24]. The use of a juvenile mouse as the object of the adult mouse’s attention greatly reduces the likelihood of aggression. It is routinely performed in the home cage of the test animal, allowing for free interaction and a more naturalistic measure of sociability. Social interactions are times during which the test mouse actively explores the juvenile mouse as defined by active pursuit, sniffing of any region (including the snout, body, and anogenital area) as well as grooming. While this assay can provide useful information, there are potential confounds. The behavior of test mice can be highly variable ranging from total lack of interest in the juvenile to the rare instance of overt aggression. Thus, it is critical to perform analyses blindly when making comparisons between any types of experimental manipulation. Nevertheless, the ease with which this assay can be performed makes it a worthwhile tool when assessing this simple form of sociability.
Recently there has been a surge in the development of machine learning based tools for both pose-estimation tracking of animals, as well as behavioral classifiers [25,26,27]. Currently, three main categories of programs exist: (1) pure pose-estimation tracking programs, such as Social LEAP Estimates Animal Poses, DeepPoseKit, and Multi-Animal DeepLabCut [28,29,30], which employ user-specified points to guide tracking; (2) behavioral classifiers that build upon pose-estimation programs, for example Simple Behavioral Analysis [31]; and (3) one-stop programs that allow for both pose-estimation and quantification of behavior, such as AlphaTracker, Motion Sequencing, and the Mouse Action Recognition System [32,33,34]. One advantage of such programs is that they allow for the parameters of behavioral classification to be quantitatively defined and more easily compared among research groups. Each of these programs has unique advantages, such as speed, ease of use or user-specified versus unsupervised analytic approaches. However, they also have limitations, such as specific equipment requirements or problems with identity swapping of individuals. These limitations should be considered when choosing to employ these technologies in lieu of the gold standard of manual scoring by expert behaviorists [35, 36].
Three-chamber sociability assay
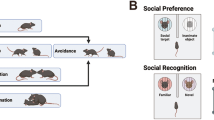
Unlike the direct contact that occurs in the juvenile interaction assay, the three-chamber sociability task assesses the preference a test mouse has for a “social chamber,” housing a novel conspecific (either age-matched or juvenile) under a wired cup versus an “empty chamber” with an empty wired cup [37]. Test mice freely explore the three-chamber apparatus for a fixed amount of time (e.g., 20 min), and a simple, quantitative readout of social approach and preference can be generated by calculating the amount of time the test mice spend in the social versus the empty chambers. This assay is simple to implement and provides a compliment to the juvenile interaction assay by addressing some of the issues that can arise with direct interactions, such as fighting or aggression, while still allowing for sensory interactions. However, this assay may be difficult to interpret given that the social stimulus is confined, making the motivation for social approach challenging to assess. For example, the test mouse might approach the social stimulus to engage in aggressive or sexual behavior rather than some form of affiliative behavior. In addition, there are many variables that may influence social approach behavior, such as prior stress, illness, or previous aggressive behavior within a home cage [38].
Social conditioned place preference (CPP)
CPP assays have traditionally been used to investigate the rewarding properties of stimuli, such as drugs of abuse and natural rewards [39]. More recently, modifications in the CPP assay have been made to expand its use to investigate the rewarding properties of prosocial interactions [40, 41]. In one version of social CPP, chambers are distinguished by two novels sets of bedding. After baseline preference is established on day 1 (pre-conditioning trial), mice spend 24 h on one type of bedding with cage mates (termed social conditioning), followed by 24 h on the other type of bedding without cage mates (termed isolation conditioning). Immediately following the isolation conditioning, a 30 min post-conditioning trial is performed to determine preference for the two conditioned bedding cues. Because this assay assesses the ability to learn and remember the “rewarding” component of non-aggressive social interactions, when changes in social CPP are elicited, it is important to perform appropriate control assays, such as drug or food induced CPP, to test whether any observed deficits are due to impairments in memory mechanisms. It has also been reported that there is a critical developmental window during adolescence when mice can optimally learn social rewarding cues and thereby exhibit social CPP [42].
Three-chamber social memory assay
While social CPP depends on a memory for the context in which social interactions occurred, memory for individual interactions between rodents, often termed social memory, is commonly measured as the preference for a novel over a familiar conspecific using a three-chamber behavioral assay [43]. This assay consists of two sessions separated by varying intervals to determine the duration of the social memory. The first session is a standard sociability assay where one chamber has an empty wired cup and the other a wired cup housing a novel conspecific. For the second trial, a novel conspecific is placed in the previously empty chamber with the now familiar conspecific from the first session housed in the same chamber. The test subjects will routinely spend more time in the chamber containing the novel conspecific indicating that they remembered their interaction with the now familiar conspecific.
Neural mechanisms of prosocial behavior and social reward
The seminal studies investigating the role of OXT release in the NAc on pair bonding in prairie voles provided motivation for further work on OXT’s mechanism of action in the NAc. In mice, both pharmacologic blockade and molecular ablation of presynaptic OXT receptors in the NAc impaired social reward processing, as assayed by social CPP [41]. Additional experimental manipulations suggested that OXT induces 5-HT release from dorsal raphe (DR) inputs in the NAc, which acts on presynaptic 5-HT1b receptors that are also required for social reward [41]. For decades, 5-HT release in the central nervous system has been implicated in regulating a broad range of behaviors, including sociability [3, 18, 44,45,46]. However, whether 5-HT release specifically in the NAc directly regulated sociability remained unknown. Work utilizing a circuit-based approach to examine the role of 5-HT release from DR inputs in the NAc revealed that optogenetic activation of DR 5-HT inputs increased sociability while optogenetic inhibition of these inputs decreased sociability, as measured by the juvenile interaction and three-chamber sociability assays [47]. Thus, modulations of DR 5-HT input activity in the NAc bidirectionally influenced sociability. Furthermore, pharmacological blockade of 5-HT1b receptors in the NAc via local infusion of a selective antagonist prevented the enhanced sociability due to activation of DR 5-HT neurons [47].
Further support for the notion that 5-HT release in the NAc promotes sociability came from the study of the mechanism of action of (±) 3,4-methylenedioxymetham-phetamine (MDMA), the Schedule 1 recreational drug, which has powerful prosocial effects in humans and is a potent releaser of monoamines, which preferentially increases 5-HT release [48,49,50]. In mice, systemic administration of a prosocial dose of MDMA enhanced sociability in the three-chamber assay, an effect that was mimicked by direct infusion of MDMA into the NAc, prevented by pharmacological inhibition of the 5-HT transporter (SERT) or its genetic deletion, and blocked by direct infusion into the NAc of a 5-HT1b receptor antagonist [51]. In contrast, the actions of MDMA on the DA transporter (DAT) were not required for this prosocial effect, as prior administration of a DAT inhibitor did not influence MDMA’s effects in the three-chamber assay. Furthermore, administration of methamphetamine, a potent DA releaser, did not enhance sociability [51]. This is a surprising result, which deserves further exploration, given that optogenetic activation of NAc-projecting DA neurons can influence prosocial behaviors [52]. Importantly, unlike the release of DA in the NAc caused by optogenetic stimulation or administration of drugs of abuse [11, 21, 53, 54], 5-HT release in the NAc is not acutely reinforcing [11, 21, 47] but see ref. [55]. Together, these findings suggest that 5-HT release, but not DA release, in the NAc is required for the prosocial effects of MDMA. Given the different behavioral effects of DA and 5-HT release in the NAc, an important topic for future research will be to elucidate how these two neuromodulators differ in their effects on NAc ensemble neuronal activity.
Recently, to interrogate motivation for social interactions, social operant tasks have been employed [56, 57]. Consistent with an involvement of DA in the reinforcing properties of a social interaction, VTA DA activity increased during lever pressing in a social instrumental operant task and optogenetic inhibition of VTA DA neurons impaired social reinforcement learning [57]. This increase in VTA DA activity appears in part to be driven by the activity of GABAergic neurons in the medial amygdala, via their projections to the medial preoptic nucleus that in turn directly connects to the VTA [58]. Furthermore, several lines of evidence suggest that OXT contributes to promoting sociability and social reward not only by enhancing 5-HT release in the NAc, but also by increasing excitatory drive on VTA DA neurons that project to NAc [59,60,61]. An additional provocative proposal is that administration of MDMA extends a developmental critical period for social reward learning via activation of NAc OXT receptors, which in turn enhance 5-HT release [42]. Given the critical importance of sociability and social reward, it is perhaps not surprising that multiple different neuromodulators influence these behaviors via multiple actions in key regions of mesolimbic reward circuitry (Fig. 1).

This simplified schematic highlights the key circuits, neuromodulators and receptors discussed in this review with a focus on inputs to the nucleus accumbens (NAc). Oxytocin release from paraventricular nucleus (PVN) inputs to the NAc promotes serotonin (5-HT) release via binding to oxytocin receptors (OXTR) on 5-HT inputs from the dorsal raphe (DR). Oxytocin also stimulates NAc-projecting dopamine (DA) neurons in the ventral tegmental area (VTA). 5-HT release in the NAc paradoxically can regulate oxytocin release via 5-HT4 receptors (5-HT4R) on PVN inputs. 5-HT also filters incoming information to the NAc by depressing glutamate release from excitatory inputs coming from the paraventricular nucleus of the thalamus (PVT), ventral hippocampus (vHip), and basolateral amygdala (BLA) via 5-HT1b receptors but does not affect medial prefrontal (mPFC) inputs. In contrast, DA depresses glutamate release only from PVT inputs, potentially via actions on presynaptic D2 receptors (D2R). Furthermore, DA acting on D1 receptors (D1R) presumably on NAc D1 MSNs is necessary for normal social interactions. (Arrow heads represent release and binding of neuromodulators at specific receptors. Minus signs (−) represent depressed glutamate release).
While the electrophysiological actions of DA in the dorsal and ventral striatum have been the subject of investigation for decades [62, 63], relatively little is known about the actions of 5-HT in these structures. Over a decade ago, application of 5-HT to slices of either the dorsal or ventral striatum was found to cause a long-term depression (LTD) of excitatory synaptic transmission via activation of presynaptic 5-HT1b receptors [64]. This simple form of 5-HT1b receptor-triggered synaptic plasticity was subsequently found to be generated by bath application of OXT or MDMA [41, 51], results consistent with the 5-HT releasing properties of these agents.
A limitation of all of these previous in vitro brain slice studies was that unknown excitatory inputs in the NAc were stimulated to generate excitatory postsynaptic responses. To determine whether 5-HT may have input-specific modulatory effects, activating opsins were expressed in vivo in the four major brain regions that send excitatory projections to the NAc medial shell: the ventral hippocampus (vHip), basolateral amygdala (BLA), medial prefrontal cortex (mPFC) and paraventricular thalamus (PVT), allowing for optogenetic activation of these inputs in acute NAc slices [65]. Bath application of 5-HT or MDMA (to release endogenous 5-HT) depressed excitatory postsynaptic responses in D1 medium spiny neurons generated by vHip, BLA and PVT inputs but surprisingly spared those generated by mPFC inputs. In marked contrast, bath application of DA or methamphetamine (to release endogenous DA) depressed responses generated by PVT inputs while having minimal effects on those generated by the three other brain regions.
Does this input-specific filtering of excitatory inputs to the NAc by 5-HT and DA have relevance to prosocial behaviors? A review of relevant findings does not provide a clear answer. Optogenetic inhibition of BLA-to-NAc inputs was found to enhance sociability in wild-type mice and reverse social deficits in Shank3−/− mice while activation of these inputs reduced sociability in wild-type mice [66]. Consistent with these findings, we found that inhibition of BLA-to-NAc inputs enhanced sociability but surprisingly observed a decrease in sociability when vHip-to-NAc inputs were optogenetically inhibited [65]. In contrast, inhibition of either PVT-to-NAc or mPFC-to-NAc inputs did not alter sociability although the latter manipulation prevented the enhanced sociability elicited by systemic MDMA administration [65]. Additional confusion is provided by the observation that activation of inputs from the prelimbic cortex to the NAc core reduced sociability [67]. Clearly, much more work will be necessary to generate heuristically useful hypotheses of the complex mechanisms by which DA, 5-HT and OXT modulate activity in the NAc and other critical brain regions to regulate sociability and social reward. An additional important topic, which deserves appropriate attention, is the potential sex-specific effects of these neuromodulators and sex differences generally in social behaviors [1, 2, 6, 69,70,71]. For example, sex-specific differences in the effects of activating OXT receptor positive mPFC interneurons have been reported with activation inducing anxiety in males, but promoting social behavior in females [68, 69].
Neural mechanisms of social memory
The ability to recognize and remember individuals is critical to the formation and maintenance of social relationships. Utilizing the three-chamber social memory assay in rodents, it was found that OXT activity in the septum and amygdala is required for social memory [43, 70]. This common molecular mediator of both social memory and social reward supports the notion that the ability to consistently recognize individuals is a fundamental component of social motivation.
Perhaps not surprisingly, the hippocampus also plays a role in this form of memory [71] but only recently have specific hippocampal subregions been implicated [72,73,74,75,76,77,78,79,80,81,82,83]. Using the novel Amigo2-Cre transgenic driver line that targets pyramidal neurons specifically in the CA2 region, it was demonstrated that inactivation of this subregion selectively impaired social memory while leaving sociability and other forms of memory intact [72]. Excitotoxic lesions of CA2 also blocked the formation of a social memory but not an olfactory memory [84]. Furthermore, CA2 pyramidal cell activity remapped during social interactions, but unlike CA1 pyramidal cell activity, did not remap to changes in spatial cues [74]. Although dorsal CA1 activity does not appear to be critical for social memory, inhibition of ventral CA1 [81] and its projections to both the NAc and mPFC impair social memory [81, 83].
The cellular mechanisms governing CA2 regulation of social memory appear complex. Consistent with the high levels of receptors for OXT and vasopressin in CA2, genetic and pharmacological manipulations of OXT receptors and vasopressin 1b receptors (Avpr1bRs) suggest that OXT and vasopressin-mediated enhancement of CA2 pyramidal cell activity is required for social memory [77,78,79,80]. Specifically, optogenetic activation of PVN Avp inputs to CA2 enhances social memory in an Avpr1bR-dependent manner [80]. Similarly, OXT receptor activation induces burst firing of CA2 pyramidal neurons and conditional deletion of these receptors abolishes social memory [77,78,79]. Modulation of CA2 parvalbumin-positive (PV) interneurons is also critical for social memory in that they exhibit a heterosynaptic form of input-timing-dependent inhibitory LTD, the blockade of which impairs social memory [75, 76].
Recent work has expanded our understanding of the circuit basis of social memory by demonstrating a critical role of inputs to CA2 pyramidal neurons from the medial septum (MS) [85]. Chemogenetic inhibition of MS neurons projecting to CA2 impaired social memory while having no effects on basal sociability or inanimate object memory. Ex vivo slice recordings revealed that a novel social interaction, but not interaction with a novel object, increased the strength of excitatory synapses between MS glutamatergic neurons and dorsal CA2 pyramidal neurons. Importantly, reversing this synaptic strength increase following a social interaction by in vivo optogenetic application of LTD inducing stimuli prevented the expression of social memory. MS neurons express both OXT receptors and several different subtypes of 5-HT receptors. However, local infusion of specific receptor antagonists into the MS revealed that OXT receptors were not required for social memory, which was prevented by a selective 5-HT1b receptor antagonist. Several additional findings strongly support a critical role for 5-HT induced modulation of MS activity in social memory. First, fiber photometry recordings of GCaMP6f fluorescence revealed a 5-HT1b receptor-dependent increase in the activity of MS cells projecting to CA2 during a novel social interaction but not during interactions with a familiar mouse or a novel object. Second, optogenetic manipulation of 5-HT inputs in the MS from the median raphe bidirectionally influenced social memory. Third, local MS infusion of a 5-HT1b receptor agonist prolonged the duration of social memory. MS slice electrophysiology revealed that 5-HT increases the activity of MS neurons projecting to CA2 at least in part via disinhibition.
This body of work begins to elucidate components of a complex neural network regulating social memory involving 5-HT regulation of a MS-to-CA2 circuit with further direct modulation of dorsal CA2 by OXT and Avp. This processing of social information by dorsal CA2, possibly in the form of sharp-wave ripples [86], is then relayed to ventral CA1 that in turn influences downstream limbic structures including the NAc and mPFC.
Neural mechanisms of “empathy” related behaviors
Empathy is often defined as the ability to detect and respond to the emotional or affective state of others and thus can play a critical role in regulating appropriate social interactions [87]. While once thought to be a purely human capability, rodents are able to perceive the affective state of conspecifics as evidenced by, for example, the phenomenon of emotional contagion, during which perceiving a behavioral change in one individual reflexively elicits a similar state in the observer [88]. Such observational learning or social transfer has been observed for pain, fear, stress, food preference, consolation and helping behavior [89,90,91,92,93,94]. For example, exposure to a conspecific being shocked depresses operant responding in the test subject [95] or the joint experience of a painful noxious stimuli influences the degree of hyperalgesia in both subjects [96]. These types of observations set the stage for investigating the evolutionarily conserved neural basis of empathy-related behaviors.
Social transfer of pain
Foundational studies established that pain-related behaviors induced by a noxious stimulus (e.g., paw injection of formalin) were increased if experienced with a conspecific also in pain [96]. Even the mere experience of being housed with a conspecific in an aversive or painful state can induce allodynia and hyperalgesia in mice [90, 97, 98]. This type of emotional contagion is often referred to as the “social transfer of pain.” Multiple sensory modalities are implicated in mediating the social transfer of pain, with both the assay and species under investigation critically determining the dominant modality [88, 89].
Human brain imaging studies implicate the insula and anterior cingulate cortex (ACC) as important regulators of empathic behaviors [99, 100]. In mice, cFos mapping studies suggest that enhanced activity in the mPFC, insula, ACC, NAc and amygdala occurs in “bystander” mice following an interaction with a cage-mate in pain [98, 101]. At the circuit level, optogenetic modulation of ACC-to-NAc core projections during the social interaction bidirectionally regulated the social transfer of pain [98]. Inhibition of this circuit element also suppressed the social transfer of analgesia, which occurred when bystander mice in pain were exposed to a mouse in pain that was experiencing morphine-induced analgesia [98]. Surprisingly this same manipulation did not influence the social transfer of fear, which was reduced by inhibition of ACC-to-BLA projections, as previously demonstrated [102].
Social transfer of fear
Social transfer of fear, routinely assayed as increased freezing by an observer, is a particularly robust form of emotional contagion and can occur via direct observation of a demonstrator being shocked or indirectly via interaction with a conditioned subject responding to a cue [103,104,105]. Similar to the social transfer of pain, the details of the experimental procedure as well as the species being studied may influence the neural mechanisms mediating the phenomenon. For example, prior experience of the unconditioned stimuli by the observer differentially impacts the effectiveness of the social transfer of fear in mice versus rats [106].
Projections from the ACC and insula to the BLA have been implicated in this form of emotional contagion. Specifically, there is an increase in ACC activity in voles exposed to a stressed partner [93] and inhibition of ACC projections to the BLA prevents the acquisition of fear, as assessed by freezing, in observer mice [98, 102, 103]. On the other hand, inhibition of insula-to-BLA circuits alter social approach to a distressed conspecific, although the effects of this manipulation on social transfer of fear were not investigated [107]. Recently hippocampal-to-BLA circuits were found to be necessary for enhanced social fear learning in mice that previously experienced a footshock [108]. In rats, an additional intra-amygdala circuit from the lateral nucleus to the medial nucleus is also necessary for the social transfer of fear [109].
OXT modulation of ACC- and insula-to-BLA circuits also regulates social transfer of fear. Antagonism of insula OXT receptors recapitulates the effects of optogenetic inhibition of this circuit [107] and inhibition of OXT receptors in the ACC similarly alters interaction with a stressed partner [93]. Furthermore, inhibition of central amygdala projecting PVN OXT neurons, but not projections to the NAc, mPFC, or CA2, impairs the discrimination of fear in a novel conspecific [110]. However, species-specific differences remain an important factor in assessing the translational relevance of these findings as OXT receptor expressing neurons are found in the ACC of prairie voles but not mice [111]. Together, these findings highlight recent advances in mapping the circuit mechanisms in this specific form of empathy, which has recently been more extensively reviewed [112].
Social transfer of stress
Stress is another emotional contagion that occurs with either direct observation or interaction with a previously stressed individual. Many studies employ a witness or vicarious social defeat paradigm where an observer that is forced to witness a conspecific undergoing the physical component of social defeat stress develops similar depressive- and anxiety-like behaviors [91, 113]. To date there is no direct investigations of the circuit mechanism underlying this phenotype; preliminary evidence suggests some overlap with similarly adaptations occurring in the VTA, dorsal hippocampus and striatum following either stressor [113,114,115].
Overall, current evidence supports the hypothesis that cortico-limbic circuits regulate empathy-related behaviors [116]. It remains unclear how these circuits are impacted by neuromodulators such as OXT and 5-HT, which regulate other aspects of social behavior. Additional important questions are whether the differences in circuit regulation for specific types of emotional contagions are due primarily to differences in the sensory modality involved and whether other variables known to impact social transfer, such as sex, age and familiarity are governed by separate or overlapping neural systems.
Prosocial behavioral deficits in neuropsychiatric disorders
Impairments in adaptive prosocial behaviors are present in many brain disorders including ASD, SZ, substance use disorder and depression [3, 117]. Here, we will briefly review recent findings on some of the neural mechanisms underlying the social impairments present in mouse models of these disorders.
Autism spectrum disorder
A cardinal feature of ASD is impairments in social interactions [117] and thus there is a large literature on this topic, only some of which we will review here. Because ASD etiology has a strong genetic component, mouse models for ASDs based on genetic variants, such as single nucleotide variations and copy number variations (CNVs), provide an invaluable tool for investigating pathophysiology satisfying both construct and face validity [118]. While there are innumerable mouse models for ASD that exhibit wide variations in behaviors, a common phenotype is deficits in various social behaviors [119].
One of the most common etiologies of ASD is a CNV on chromosome 16p11.2. In a 16p11.2 deletion mouse model, selective deletion of the syntenic chromosomal region specifically in DR 5-HT neurons induced deficits in sociability, which was associated with decreases in DR 5-HT neuron activity during social encounters and reduction in their intrinsic excitability [47]. Optogenetic activation of DR 5-HT inputs in the NAc rescued the social deficits present in these mice, and this rescue was dependent upon activation of presynaptic 5-HT1b receptors [47]. Consistent with a critical role of deficits in 5-HT signaling in ASD, mice expressing a gain-of-function SERT variant that decreases 5-HT levels exhibit impaired sociability as well as altered basal firing of DR 5-HT neurons [120, 121]. Another CNV mouse model that exhibits social behavior deficits, chromosomal 15q11.3 duplication, exhibits reduced excitatory transmission in DR 5-HT neurons and low glucose metabolism in the DR, suggesting reduced DR 5-HT activity [122]. Furthermore, several studies have reported that increasing 5-HTergic tone rescues social deficits present in various mouse models for ASD [47, 122,123,124]. Together, these findings support the hypothesis that dysfunction in 5-HT-induced modulation of specific target structures is a critical feature underlying social deficits in ASD.
Another mechanism that has been implicated in various mouse models for ASD is imbalance of excitatory and inhibitory synaptic transmission, or E/I imbalance, in certain brain regions [125]. Specifically, social deficits present in mice lacking the Cntnap2 gene were rescued by correcting E/I imbalance via optogenetic activation of inhibitory PV neurons or inhibition of excitatory pyramidal neurons in the mPFC [126]. Additionally, these mice exhibit a reduction in both excitatory and inhibitory synaptic inputs onto L2/3 pyramidal neurons and perturbed oscillatory activity in the mPFC [127]. Consistent with a critical role for mPFC dysfunction in mouse models of ASD, restoration of Shank3 expression in the mPFC rescued sociability deficits in Shank3 deletion mice [128]. Surprisingly, however, a different group found that E/I imbalance in the ACC, but not the mPFC, contribute to social deficits in Shank3 deletion mice [129].
An alternative simple strategy to influence E/I imbalance is to directly modulate GABA receptors. Indeed, systemic administration of R-Baclofen, a GABAB receptor agonist, reversed the social deficits present in two different 16p11.2 deletion mouse models [130]. However, in four etiologically distinct ASD mouse models, altered E/I balance was observed in the somatosensory cortex, without any corresponding changes in overall circuit excitability, suggesting altered E/I balance may be homeostatic compensation and not a primary mechanism mediating social impairments [131].
Schizophrenia
SZ is a severe neurodevelopmental disorder characterized by both positive symptoms, such as hallucinations and delusions, as well as negative symptoms, such as social withdrawal, anhedonia and impaired cognition. CNVs, both deletions and duplications, have been implicated in SZ [132,133,134] but only a small number of studies have explored the neural mechanisms underlying social deficits in mouse models of these CNVs. In the Df(16)A+/− mouse model of the human 22q11.2 microdeletion, impaired social memory has been suggested to be a consequence of reduced density of PV interneurons in area CA2 of the hippocampus and the consequent impaired feedforward inhibition of CA2 pyramidal neurons [73]. Additionally, these mice exhibited decrease firing of CA2 pyramidal neurons during the three-chamber sociability assay, perhaps due to increased current through a K+ channel (TREK-1) [73]. Consistent with this suggestion, systemic administration of a TREK-1 antagonist rescued deficits in social memory and CA2 firing properties [135].
Carriers of the 16p11.2 duplication also have a high probability of developing SZ but unlike individuals with a 22q11.2 deletion, only a small percentage of these subjects also present with intellectual disabilities [136, 137]. Mice with 16p11.2 duplication exhibit impaired social behavior, as well as compromised hippocampal-orbitofrontal cortex-amygdala functional connectivity, which was assessed using network analysis of 14C-2-deoxyglucose imaging data [138]. This neural network has been implicated in the cognitive processes related to social memory [139], suggesting that social deficits present in SZ may involve impaired recognition as well as reduced motivation.
Substance use disorder
Withdrawal from drugs of abuse often results in social isolation and general impairments in sociability [140]. However, little research to date has identified the neural mechanisms underlying the reduced social motivation accompanying substance use disorder. One study reported that 4 weeks of withdrawal from chronic morphine administration in mice induced social deficits, which were accompanied by impaired DR 5-HTnergic activity paralleling the deficits observed in ASD models [141]. Morphine withdrawal was also found to enhance cytokine signaling in the lateral habenula, ultimately decreasing the activity of neurons projecting to the DR [142]. Furthermore, chemogenetic inhibition of this lateral habenula-to-DR circuit mimicked the withdrawal-induced social deficits [142]. In prairie voles, repeated amphetamine exposure inhibited pair bonding, decreased OXT levels in the mPFC and increased NAc DA levels [143]. Direct infusion of OXT into the mPFC restored pair bonding and normalized NAc DA levels [143]. These limited results are consistent with a critical role for OXT and 5-HT-triggered modulation in mediating adaptive prosocial behaviors. However, given the devastation caused by the high prevalence of substance abuse disorder, clearly much more work needs to be done on this important topic.
The neural mechanisms that mediate the influence of social interactions on substance abuse-related behaviors also deserve attention. For example, the presence of a drug-naïve peer reduces drug seeking while the presence of an individual associated with previous drug use promotes relapse [144]. Furthermore, the opportunity to socialize reduces drug seeking for both methamphetamine and heroin [145, 146]. These observations demonstrate the high appetitive valence of prosocial interactions and the powerful effects they can exert on decreasing pathological behaviors associated with substance use disorder.
Social deficits in stress-related disorders
Stress-related disorders, in particular depression, are frequently accompanied by deficits in adaptive prosocial behaviors. In mice, impaired sociability is commonly caused by exposing subjects to a form of severe chronic stress such as chronic social defeat stress (CSDS) [147]. A number of studies have found that chronic stressors cause maladaptive adaptations in the mesolimbic DA system [148], the details of which seem to be influenced by the nature and the degree of the stressor. With a severe stressor such as CSDS, increased release of BDNF from VTA DA inputs to the NAc contributes to social avoidance [149], whereas chronic mild stress-induced social behavioral deficits are associated with decreased DA release in limbic target regions [150].
Social behavior impairments as a consequence of early life stress also seem to stem from aberrations of the DA system, including long-lasting transcriptional changes in VTA DA neurons [151] and impaired DA signaling in the lateral septum [152]. While alterations in VTA DA neuron function contribute to stress-induced social avoidance, they do not occur in isolation. CSDS also increases inhibitory transmission from the ventral pallidum-to-VTA, the importance of which was demonstrated by the finding that inhibiting this circuit restored normal social interactions [153]. Surprisingly, acute social isolation stress was found to induce synaptic adaptations on DA neurons in the DR, but not in the VTA [154]. Furthermore, inhibition of DR DA neurons promoted social avoidance only following an experience of social isolation [154]. However, 1 week of social isolation during adolescence resulted in increased excitability of mPFC-projecting VTA DA neurons and increased social interaction but decreased social novelty preference [155]. These neural and behavioral adaptations were reversed via chemogenetic inhibition of OXT neurons in the PVN [155].
Alterations in VTA DA activity will affect activity in multiple downstream targets, including the NAc and mPFC, which appear to be particularly important for mediating social avoidance [149, 150, 156,157,158,159]. Specifically, CSDS induced enhancement of synaptic transmission at thalamic inputs, but not cortical inputs to the NAc, promoted social avoidance [160]. In contrast, weakening of a mPFC-to- dorsal periaqueductal gray projection also appears to play an important role in CSDS induced social avoidance [161]. Additionally, infusion of OXT into the mPFC following CSDS had antidepressant actions, which were associated with increased DA levels, as well as enhanced excitatory synaptic transmission, in the mPFC [162]. Thus, acute stress may promote social interactions at least in part via upregulation of DA input activity in the mPFC [155], whereas chronic stressors appear to lead to a hypodopaminergic state [162]. Yet, in both cases upregulation of mPFC excitatory transmission promoted sociability. CSDS-induced changes in the activity of NAc cholinergic interneurons also appears to contribute to the social behavior deficits [163], perhaps via regulation of local DA release [164] or excitatory synaptic transmission [165].
Of course, stress-induced modifications of brain network activity likely occur in circuits other than the canonical mesolimbic reward circuitry. For example, mPFC-to-PVT activity is impaired during social interactions following 2 weeks of adolescent social isolation, and activation of this circuit rescued isolation induced social deficits [166]. On the other hand, inhibition of BLA-to-mPFC or BLA-to-vHip inputs decreased anxiety while also enhancing social interactions [167,168,169]. One approach that may help investigators understand and integrate these individual findings involves brain-wide electrophysiological monitoring of network activity [157]. In mice, this method revealed a stress vulnerability network that included many of the brain regions mentioned thus far including mPFC, amygdala, NAc, vHip and VTA [157]. Surprisingly, this vulnerability network was distinct from the network activity underlying the expression of aberrant behaviors elicited by CSDS [157].
Novel therapeutic directions
Current pharmacological therapies for neuropsychiatric disorders do not routinely treat discrete behavioral impairments. Thus, there are no specific treatments for the social impairments that accompany any of the disorders we have discussed. Given the often highly damaging consequences of social isolation and withdrawal, this is a glaring deficiency in the therapeutic armamentarium that clinicians have at their disposal. It is likely that when effective in treating depression, standard treatments such as selective 5-HT reuptake inhibitors (SSRIs) or 5-HT and norepinephrine reuptake inhibitors (SNRIs) ameliorate sociability deficits but these are all too commonly ineffective in treating large proportions of subjects with mood disorders [3, 170, 171]. For ASD, there are no approved medications for any of the core symptoms including sociability deficits. Similarly, the anti-psychotics used to treat SZ have minimal effects on negative symptoms, a prominent one of which is social isolation and withdrawal. Finally, there is also clearly a need for intervention that effectively reduces the social isolation that commonly accompanies substance abuse and often leads to relapse following withdrawal and abstinence.
Despite the paucity of treatments for social deficits, there is reason to be hopeful. Consistent with the important action of OXT and vasopressin in social reward and social memory, intranasal OXT holds some promise [172, 173], although it may only be beneficial in subsets of patients with low OXT levels [174]. Peripheral levels of vasopressin appear to be an accurate predictor of ASD and was effective at ameliorating social deficits when administered intranasally in a small trial [175, 176]. Of particular interest are drugs such as MDMA, which are termed entactogens or even empathogens [177]. MDMA has well documented prosocial effects in human subjects and a pilot study found that MDMA-assisted psychotherapy reduced social anxiety in ASD adults [178]. Indeed, MDMA ameliorated sociability deficits in four different mouse models for ASD [124]. Because of its abuse potential and potential toxicity [51, 177], it is unlikely that MDMA itself can ever be used as a daily treatment for sociability deficits. However, analogs that preferentially target the 5-HT transporter and not the DAT may be worth testing [51, 179].
Another valuable strategy may be to target the critical receptors that mediate the social effects of 5-HT. We have presented evidence that activation of 5-HT1b receptors in the NAc is critical for promoting sociability while these same receptors in the MS play a critical role in social memory. Consistent with the therapeutic potential of targeting 5-HT1b receptors, administration of a specific 5-HT1b receptor agonist rescued sociability deficits in six different mouse models for ASD [124]. The potential role of relative increases in E/I balance in ASD suggests that targeting GABA receptors may also be worthwhile. Indeed, R-baclofen, a GABAB receptor agonist ameliorated behavioral deficits in mouse models for Fragile X syndrome [180] as well as in two lines of 16p11.2 deletion mice [130]. Although, clinical trials have not found it effective in improving global social withdrawal symptoms [181], there remains hope that in appropriate subpopulations of ASD individuals, it might yet prove efficacious [182].
Neuromodulation approaches offer a non-pharmacological strategy for the treatment of social deficits, with deep brain stimulation (DBS), repetitive transcranial magnetic stimulation (rTMS), and transcranial direct current stimulation (tDCS) being the primary techniques currently employed. While non-invasive strategies (i.e., rTMS and tDCS) are clearly more desirable than the invasive option of DBS, these methods currently only allow manipulation of relatively shallow cortical regions [183]. This limitation is particularly problematic for targeting the deeper subcortical structures, which have been implicated in mediating prosocial behaviors and social memory. However, engineering advances in TMS may enable stimulation of deeper structures and ultrasound-mediated stimulation, which can more readily target deep structures, is vigorously being pursued. It may also be possible to use current non-invasive techniques to stimulate cortical regions, such as the mPFC or orbital frontal cortex, in a manner that therapeutically modifies pathological brain networks mediating social deficits. In fact, a few small studies have found rTMS effective in improving social interactions in ASD patients, but these clearly need to be replicated in larger randomized, double-blind clinical trials [184]. Although DBS is used routinely to treat Parkinson’s disease and is actively being investigated for its efficacy in depression and obsessive compulsive disorder, understandably it has not been vigorously pursued as a treatment for ASD. Nevertheless, we were able to find one case study reporting DBS of the BLA in a young self-injurious male with ASD to be effective in blocking self-injury and improving social behavior [185].
Future research directions
Given the complexity of social interactions and their importance for survival, it is not surprising that many neuromodulators and distinct brain regions and circuits have been implicated in the variety of adaptive prosocial behaviors that have been examined in rodents. Continued application of the armamentarium of modern techniques in genetically tractable species will be important to further elucidate the complex neural circuit mechanisms that mediate adaptive prosocial behaviors and how these work in concert to coordinate appropriate social interactions. Of particular interest will be comparing the actions of individual neuromodulators on ensemble activity in critical target regions such as the NAc and mPFC. However, neuromodulators do not work in isolation and investigators will also need to elucidate how neuromodulators work in concert to influence social as well as other behaviors (Fig. 1).
Of course, a critical question that applies to all behavioral animal research is whether mechanisms elucidated in non-human species have in fact been evolutionarily conserved to the degree that they provide insights into the brain activity mediating comparable human behaviors. There remain many challenges to breaking down the barriers between pre-clinical animal researchers and clinical investigators studying human subjects [8, 186]. Nevertheless, several experimental approaches may provide valuable bridges such as applying various forms of neuromodulation and testing the effects of novel pharmacological manipulations. Monitoring and manipulating the same or similar circuits in both animal and human subjects will be of particular value as can be done, for example, with DBS [187, 188] or administering drugs such as MDMA [124, 178, 179]. Furthermore, it will be important to examine rodent behaviors in more ethologically relevant natural conditions. For example, wireless stimulation of paraventricular hypothalamic nucleus (PVN) OXT neurons in mice was found to enhance both prosocial and agnostic behavior in a context-dependent manner [189].
A more sophisticated and nuanced understanding of the neural mechanisms underlying adaptive and pathological social behaviors should provide foundational knowledge that will be essential for developing more effective treatments specifically targeting the deficient prosocial behaviors that are a major source of morbidity in prevalent brain disorders. Furthermore, given the current state of the world, we cannot imagine a more important topic for neuroscience researchers than to advance understanding of the neural mechanisms that promote empathic and compassionate prosocial interactions.
References
de Waal FBM. Mama’s last hug: animal emotions and what they tell us about ourselves. 1st ed. New York: W.W. Norton & Company; 2019.
Anderson DJ. The nature of the beast: how emotions guide us. 1st ed. New York: Basic Books; 2022.
Charney DS, Sklar PB, Buxbaum JD, Nestler EJ. Charney & Nestler’s neurobiology of mental illness. 5th ed. New York: Oxford University Press; 2018.
Wei D, Talwar V, Lin D. Neural circuits of social behaviors: Innate yet flexible. Neuron. 2021;109:1600–20.
Chen P, Hong W. Neural circuit mechanisms of social behavior. Neuron. 2018;98:16–30.
Kohl J, Dulac C. Neural control of parental behaviors. Curr Opin Neurobiol. 2018;49:116–22.
Goodwin NL, Nilsson SRO, Golden SA. Rage against the machine: advancing the study of aggression ethology via machine learning. Psychopharmacology. 2020;237:2569–88.
Anderson DJ, Adolphs R. A framework for studying emotions across species. Cell. 2014;157:187–200.
Froemke RC, Young LJ. Oxytocin, neural plasticity, and social behavior. Annu Rev Neurosci. 2021;44:359–81.
Young LJ, Lim MM, Gingrich B, Insel TR. Cellular mechanisms of social attachment. Horm Behav. 2001;40:133–8.
Klawonn AM, Malenka RC. Nucleus accumbens modulation in reward and aversion. Cold Spring Harb Symp Quant Biol. 2018;83:119–29.
Russo SJ, Nestler EJ. The brain reward circuitry in mood disorders. Nat Rev Neurosci. 2013;14:609–25.
Insel TR, Shapiro LE. Oxytocin receptor distribution reflects social organization in monogamous and polygamous voles. Proc Natl Acad Sci USA. 1992;89:5981–5.
de Waal FBM. What is an animal emotion? Ann N Y Acad Sci. 2011;1224:191–206.
Adolphs R, Anderson D. Social and emotional neuroscience. Curr Opin Neurobiol. 2013;23:291–3.
Peters KZ, Cheer JF, Tonini R. Modulating the neuromodulators: dopamine, serotonin, and the endocannabinoid system. Trends Neurosci. 2021;44:464–77.
Niederkofler V, Asher TE, Dymecki SM. Functional interplay between dopaminergic and serotonergic neuronal systems during development and adulthood. ACS Chem Neurosci. 2015;6:1055–70.
Brown S-L, Praag HMv. The role of serotonin in psychiatric disorders. New York: Brunner/Mazel; 1991.
Tritsch NX, Sabatini BL. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron. 2012;76:33–50.
Müller CP, Cunningham KA. Handbook of the behavioral neurobiology of serotonin. San Diego, CA: Elsevier; 2020.
Steinberg EE, Christoffel DJ, Deisseroth K, Malenka RC. Illuminating circuitry relevant to psychiatric disorders with optogenetics. Curr Opin Neurobiol 2015;30:9–16.
Lerner TN, Ye L, Deisseroth K. Communication in neural circuits: tools, opportunities, and challenges. Cell. 2016;164:1136–50.
Moy SS, Nadler JJ, Young NB, Perez A, Holloway LP, Barbaro RP, et al. Mouse behavioral tasks relevant to autism: phenotypes of 10 inbred strains. Behav Brain Res. 2007;176:4–20.
Silverman JL, Yang M, Lord C, Crawley JN. Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci. 2010;11:490–502.
Urai AE, Doiron B, Leifer AM, Churchland AK. Large-scale neural recordings call for new insights to link brain and behavior. Nat Neurosci. 2022;25:11–19.
Pereira TD, Shaevitz JW, Murthy M. Quantifying behavior to understand the brain. Nat Neurosci. 2020;23:1537–49.
Mathis A, Schneider S, Lauer J, Mathis MW. A primer on motion capture with deep learning: principles, pitfalls, and perspectives. Neuron. 2020;108:44–65.
Pereira TD, Tabris N, Matsliah A, Turner DM, Li J, Ravindranath S, et al. SLEAP: a deep learning system for multi-animal pose tracking. Nat Methods. 2022;19:486–95.
Graving JM, Chae D, Naik H, Li L, Koger B, Costelloe BR, et al. DeepPoseKit, a software toolkit for fast and robust animal pose estimation using deep learning. Elife. 2019;8:47994–48035.
Lauer J, Zhou M, Ye S, Menegas W, Schneider S, Nath T, et al. Multi-animal pose estimation, identification and tracking with DeepLabCut. Nat Methods. 2022;19:496–504.
Nilsson SR, Goodwin NL, Choong JJ, Hwang S, Wright HR, Norville ZC, et al. Simple behavioral analysis (SimBA)—an open source toolkit for computer classification of complex social behaviors in experimental animals. bioRxiv. 2020. https://doi.org/10.1101/2020.04.19.049452.
Chen Z, Zhang R, Eva Zhang Y, Zhou H, Fang H-S, Rock RR, et al. AlphaTracker: a multi-animal tracking and behavioral analysis tool. bioRxiv. 2020. https://doi.org/10.1101/2020.12.04.405159.
Wiltschko AB, Tsukahara T, Zeine A, Anyoha R, Gillis WF, Markowitz JE, et al. Revealing the structure of pharmacobehavioral space through motion sequencing. Nat Neurosci. 2020;23:1433–43.
Segalin C, Williams J, Karigo T, Hui M, Zelikowsky M, Sun JJ, et al. The mouse action recognition system (MARS) software pipeline for automated analysis of social behaviors in mice. Elife. 2021;10:63720–54.
Goodwin NL, Nilsson SRO, Choong JJ, Golden SA. Toward the explainability, transparency, and universality of machine learning for behavioral classification in neuroscience. Curr Opin Neurobiol. 2022;73:102544.
Mathis MW, Mathis A. Deep learning tools for the measurement of animal behavior in neuroscience. Curr Opin Neurobiol. 2020;60:1–11.
Moy SS, Nadler JJ, Perez A, Barbaro RP, Johns JM, Magnuson TR, et al. Sociability and preference for social novelty in five inbred strains: an approach to assess autistic-like behavior in mice. Genes Brain Behav. 2004;3:287–302.
Yang M, Silverman JL, Crawley JN. Automated three-chambered social approach task for mice. Curr Protoc Neurosci. 2011;56:8.26.1–8.26.16.
Tzschentke TM. Measuring reward with the conditioned place preference (CPP) paradigm: update of the last decade. Addict Biol. 2007;12:227–462.
Panksepp JB, Lahvis GP. Social reward among juvenile mice. Genes Brain Behav. 2007;6:661–71.
Dolen G, Darvishzadeh A, Huang KW, Malenka RC. Social reward requires coordinated activity of nucleus accumbens oxytocin and serotonin. Nature. 2013;501:179–84.
Nardou R, Lewis EM, Rothhaas R, Xu R, Yang A, Boyden E, et al. Oxytocin-dependent reopening of a social reward learning critical period with MDMA. Nature. 2019;569:116–20.
Ferguson JN, Aldag JM, Insel TR, Young LJ. Oxytocin in the medial amygdala is essential for social recognition in the mouse. J Neurosci. 2001;21:8278–85.
Kiser D, Steemers B, Branchi I, Homberg JR. The reciprocal interaction between serotonin and social behaviour. Neurosci Biobehav Rev. 2012;36:786–98.
Muller CL, Anacker AMJ, Veenstra-VanderWeele J. The serotonin system in autism spectrum disorder: from biomarker to animal models. Neuroscience. 2016;321:24–41.
Okaty BW, Commons KG, Dymecki SM. Embracing diversity in the 5-HT neuronal system. Nat Rev Neurosci. 2019;20:397–424.
Walsh JJ, Christoffel DJ, Heifets BD, Ben-Dor GA, Selimbeyoglu A, Hung LW, et al. 5-HT release in nucleus accumbens rescues social deficits in mouse autism model. Nature. 2018;560:589–94.
Green AR, Mechan AO, Elliott JM, O’Shea E, Colado MI. The pharmacology and clinical pharmacology of 3,4-methylenedioxymethamphetamine (MDMA, “ecstasy”). Pharm Rev. 2003;55:463–508.
Rothman RB, Baumann MH. Therapeutic and adverse actions of serotonin transporter substrates. Pharm Ther. 2002;95:73–88.
Kamilar-Britt P, Bedi G. The prosocial effects of 3,4-methylenedioxymethamphetamine (MDMA): Controlled studies in humans and laboratory animals. Neurosci Biobehav Rev. 2015;57:433–46.
Heifets BD, Salgado JS, Taylor MD, Hoerbelt P, Cardozo Pinto DF, Steinberg EE, et al. Distinct neural mechanisms for the prosocial and rewarding properties of MDMA. Sci Transl Med. 2019;11:1–11.
Gunaydin LA, Grosenick L, Finkelstein JC, Kauvar IV, Fenno LE, Adhikari A, et al. Natural neural projection dynamics underlying social behavior. Cell. 2014;157:1535–51.
Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, Janak PH. A causal link between prediction errors, dopamine neurons and learning. Nat Neurosci. 2013;16:966–73.
Lammel S, Lim BK, Malenka RC. Reward and aversion in a heterogeneous midbrain dopamine system. Neuropharmacology. 2014;76:351–9.
Liu Z, Lin R, Luo M. Reward contributions to serotonergic functions. Annu Rev Neurosci. 2020;43:141–62.
Venniro M, Shaham Y. An operant social self-administration and choice model in rats. Nat Protoc. 2020;15:1542–59.
Solie C, Girard B, Righetti B, Tapparel M, Bellone C. VTA dopamine neuron activity encodes social interaction and promotes reinforcement learning through social prediction error. Nat Neurosci. 2022;25:86–97.
Hu RK, Zuo Y, Ly T, Wang J, Meera P, Wu YE, et al. An amygdala-to-hypothalamus circuit for social reward. Nat Neurosci. 2021;24:831–42.
Hung LW, Neuner S, Polepalli JS, Beier KT, Wright M, Walsh JJ, et al. Gating of social reward by oxytocin in the ventral tegmental area. Science. 2017;357:1406–11.
Xiao L, Priest MF, Nasenbeny J, Lu T, Kozorovitskiy Y. Biased Oxytocinergic modulation of midbrain dopamine systems. Neuron. 2017;95:368–84.e5.
Song Z, Borland JM, Larkin TE, O’Malley M, Albers HE. Activation of oxytocin receptors, but not arginine-vasopressin V1a receptors, in the ventral tegmental area of male Syrian hamsters is essential for the reward-like properties of social interactions. Psychoneuroendocrinology. 2016;74:164–72.
Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215.
Sabatini BL, Tian L. Imaging neurotransmitter and neuromodulator dynamics in vivo with genetically encoded indicators. Neuron. 2020;108:17–32.
Mathur BN, Capik NA, Alvarez VA, Lovinger DM. Serotonin induces long-term depression at corticostriatal synapses. J Neurosci. 2011;31:7402–11.
Christoffel DJ, Walsh JJ, Hoerbelt P, Heifets BD, Llorach P, Lopez RC, et al. Selective filtering of excitatory inputs to nucleus accumbens by dopamine and serotonin. Proc Natl Acad Sci USA. 2021;118:1–10.
Folkes OM, Baldi R, Kondev V, Marcus DJ, Hartley ND, Turner BD, et al. An endocannabinoid-regulated basolateral amygdala-nucleus accumbens circuit modulates sociability. J Clin Invest. 2020;130:1728–42.
Murugan M, Jang HJ, Park M, Miller EM, Cox J, Taliaferro JP, et al. Combined social and spatial coding in a descending projection from the prefrontal cortex. Cell. 2017;171:1663–77.e16.
Nakajima M, Gorlich A, Heintz N. Oxytocin modulates female sociosexual behavior through a specific class of prefrontal cortical interneurons. Cell. 2014;159:295–305.
Li K, Nakajima M, Ibanez-Tallon I, Heintz N. A cortical circuit for sexually dimorphic oxytocin-dependent anxiety behaviors. Cell. 2016;167:60–72.e11.
Lukas M, Toth I, Veenema AH, Neumann ID. Oxytocin mediates rodent social memory within the lateral septum and the medial amygdala depending on the relevance of the social stimulus: male juvenile versus female adult conspecifics. Psychoneuroendocrinology. 2013;38:916–26.
Kogan JH, Frankland PW, Silva AJ. Long-term memory underlying hippocampus-dependent social recognition in mice. Hippocampus. 2000;10:47–56.
Hitti FL, Siegelbaum SA. The hippocampal CA2 region is essential for social memory. Nature. 2014;508:88–92.
Piskorowski RA, Nasrallah K, Diamantopoulou A, Mukai J, Hassan SI, Siegelbaum SA, et al. Age-dependent specific changes in area CA2 of the hippocampus and social memory deficit in a mouse model of the 22q11.2 deletion syndrome. Neuron. 2016;89:163–76.
Alexander GM, Farris S, Pirone JR, Zheng C, Colgin LL, Dudek SM. Social and novel contexts modify hippocampal CA2 representations of space. Nat Commun. 2016;7:10300.
Leroy F, Brann DH, Meira T, Siegelbaum SA. Input-timing-dependent plasticity in the hippocampal CA2 region and its potential role in social memory. Neuron. 2017;95:1089–102.e5.
Dominguez S, Rey CC, Therreau L, Fanton A, Massotte D, Verret L, et al. Maturation of PNN and ErbB4 signaling in area CA2 during adolescence underlies the emergence of PV interneuron plasticity and social memory. Cell Rep. 2019;29:1099–112.e4.
Tirko NN, Eyring KW, Carcea I, Mitre M, Chao MV, Froemke RC, et al. Oxytocin transforms firing mode of CA2 hippocampal neurons. Neuron. 2018;100:593–608.e3.
Raam T, McAvoy KM, Besnard A, Veenema AH, Sahay A. Hippocampal oxytocin receptors are necessary for discrimination of social stimuli. Nat Commun. 2017;8:2001.
Lin YT, Hsieh TY, Tsai TC, Chen CC, Huang CC, Hsu KS. Conditional deletion of hippocampal CA2/CA3a oxytocin receptors impairs the persistence of long-term social recognition memory in mice. J Neurosci. 2018;38:1218–31.
Smith AS, Williams Avram SK, Cymerblit-Sabba A, Song J, Young WS. Targeted activation of the hippocampal CA2 area strongly enhances social memory. Mol Psychiatry. 2016;21:1137–44.
Okuyama T, Kitamura T, Roy DS, Itohara S, Tonegawa S. Ventral CA1 neurons store social memory. Science. 2016;353:1536–41.
Meira T, Leroy F, Buss EW, Oliva A, Park J, Siegelbaum SA. A hippocampal circuit linking dorsal CA2 to ventral CA1 critical for social memory dynamics. Nat Commun. 2018;9:4163.
Phillips ML, Robinson HA, Pozzo-Miller L. Ventral hippocampal projections to the medial prefrontal cortex regulate social memory. Elife. 2019;8:44182–213.
Stevenson EL, Caldwell HK. Lesions to the CA2 region of the hippocampus impair social memory in mice. Eur J Neurosci. 2014;40:3294–301.
Wu X, Morishita W, Beier KT, Heifets BD, Malenka RC. 5-HT modulation of a medial septal circuit tunes social memory stability. Nature. 2021;599:96–101.
Oliva A, Fernandez-Ruiz A, Leroy F, Siegelbaum SA. Hippocampal CA2 sharp-wave ripples reactivate and promote social memory. Nature. 2020;587:264–69.
de Waal FB. Putting the altruism back into altruism: the evolution of empathy. Annu Rev Psychol. 2008;59:279–300.
Panksepp JB, Lahvis GP. Rodent empathy and affective neuroscience. Neurosci Biobehav Rev. 2011;35:1864–75.
Smith ML, Hostetler CM, Heinricher MM, Ryabinin AE. Social transfer of pain in mice. Sci Adv. 2016;2:e1600855.
Baptista-de-Souza D, Nunciato AC, Pereira BC, Fachinni G, Zaniboni CR, Canto-de-Souza A. Mice undergoing neuropathic pain induce anxiogenic-like effects and hypernociception in cagemates. Behav Pharmacol. 2015;26:664–72.
Carnevali L, Montano N, Tobaldini E, Thayer JF, Sgoifo A. The contagion of social defeat stress: insights from rodent studies. Neurosci Biobehav Rev. 2020;111:12–18.
Galef BG. A case study in behavioral analysis, synthesis and attention to detail: social learning of food preferences. Behav Brain Res. 2012;231:266–71.
Burkett JP, Andari E, Johnson ZV, Curry DC, de Waal FB, Young LJ. Oxytocin-dependent consolation behavior in rodents. Science. 2016;351:375–8.
Ueno H, Suemitsu S, Murakami S, Kitamura N, Wani K, Matsumoto Y, et al. Helping-like behaviour in mice towards conspecifics constrained inside tubes. Sci Rep. 2019;9:5817.
Church RM. Emotional reactions of rats to the pain of others. J Comp Physiol Psychol. 1959;52:132–4.
Langford DJ, Crager SE, Shehzad Z, Smith SB, Sotocinal SG, Levenstadt JS, et al. Social modulation of pain as evidence for empathy in mice. Science. 2006;312:1967–70.
Smith ML, Walcott AT, Heinricher MM, Ryabinin AE. Anterior cingulate cortex contributes to alcohol withdrawal-induced and socially transferred hyperalgesia. eNeuro. 2017;4:1–10.
Smith ML, Asada N, Malenka RC. Anterior cingulate inputs to nucleus accumbens control the social transfer of pain and analgesia. Science. 2021;371:153–59.
Singer T, Seymour B, O’Doherty J, Kaube H, Dolan RJ, Frith CD. Empathy for pain involves the affective but not sensory components of pain. Science. 2004;303:1157–62.
Timmers I, Park AL, Fischer MD, Kronman CA, Heathcote LC, Hernandez JM, et al. Is empathy for pain unique in its neural correlates? A meta-analysis of neuroimaging studies of empathy. Front Behav Neurosci. 2018;12:289.
Li Z, Lu YF, Li CL, Wang Y, Sun W, He T, et al. Social interaction with a cagemate in pain facilitates subsequent spinal nociception via activation of the medial prefrontal cortex in rats. Pain. 2014;155:1253–61.
Allsop SA, Wichmann R, Mills F, Burgos-Robles A, Chang CJ, Felix-Ortiz AC, et al. Corticoamygdala transfer of socially derived information gates observational learning. Cell. 2018;173:1329–42.e18.
Jeon D, Kim S, Chetana M, Jo D, Ruley HE, Lin SY, et al. Observational fear learning involves affective pain system and Cav1.2 Ca2+ channels in ACC. Nat Neurosci. 2010;13:482–8.
Morozov A. Behavioral modulation by social experiences in rodent models. Curr Protoc Neurosci. 2018;84:e50.
Debiec J, Olsson A. Social fear learning: from animal models to human function. Trends Cogn Sci. 2017;21:546–55.
Leblanc H, Ramirez S. Linking social cognition to learning and memory. J Neurosci. 2020;40:8782–98.
Rogers-Carter MM, Varela JA, Gribbons KB, Pierce AF, McGoey MT, Ritchey M, et al. Insular cortex mediates approach and avoidance responses to social affective stimuli. Nat Neurosci. 2018;21:404–14.
Terranova JI, Yokose J, Osanai H, Marks WD, Yamamoto J, Ogawa SK, et al. Hippocampal-amygdala memory circuits govern experience-dependent observational fear. Neuron. 2022;110:1416–31.
Twining RC, Vantrease JE, Love S, Padival M, Rosenkranz JA. An intra-amygdala circuit specifically regulates social fear learning. Nat Neurosci. 2017;20:459–69.
Ferretti V, Maltese F, Contarini G, Nigro M, Bonavia A, Huang H, et al. Oxytocin signaling in the central amygdala modulates emotion discrimination in mice. Curr Biol. 2019;29:1938–53.e6.
Horie K, Inoue K, Nishimori K, Young LJ. Investigation of Oxtr-expressing neurons projecting to nucleus accumbens using Oxtr-ires-Cre Knock-in prairie Voles (Microtus ochrogaster). Neuroscience. 2020;448:312–24.
Kim SW, Kim M, Shin HS. Affective empathy and prosocial behavior in rodents. Curr Opin Neurobiol. 2021;68:181–89.
Warren BL, Vialou VF, Iniguez SD, Alcantara LF, Wright KN, Feng J, et al. Neurobiological sequelae of witnessing stressful events in adult mice. Biol Psychiatry. 2013;73:7–14.
Warren BL, Sial OK, Alcantara LF, Greenwood MA, Brewer JS, Rozofsky JP, et al. Altered gene expression and spine density in nucleus accumbens of adolescent and adult male mice exposed to emotional and physical stress. Dev Neurosci. 2014;36:250–60.
Cooper SE, Kechner M, Caraballo-Perez D, Kaska S, Robison AJ, Mazei-Robison MS. Comparison of chronic physical and emotional social defeat stress effects on mesocorticolimbic circuit activation and voluntary consumption of morphine. Sci Rep. 2017;7:8445.
Hernandez-Lallement J, Gomez-Sotres P, Carrillo M. Towards a unified theory of emotional contagion in rodents—a meta-analysis. Neurosci Biobehav Rev. 2022;132:1229–48.
Association AP. Diagnostic and statistical manual of mental disorders. 5th ed. Washington, DC: American Psychiatric Association; 2013.
Takumi T, Tamada K, Hatanaka F, Nakai N, Bolton PF. Behavioral neuroscience of autism. Neurosci Biobehav Rev. 2020;110:60–76.
Arakawa H. From multisensory assessment to functional interpretation of social behavioral phenotype in transgenic mouse models for autism spectrum disorders. Front Psychiatry. 2020;11:592408.
Veenstra-VanderWeele J, Muller CL, Iwamoto H, Sauer JE, Owens WA, Shah CR, et al. Autism gene variant causes hyperserotonemia, serotonin receptor hypersensitivity, social impairment and repetitive behavior. Proc Natl Acad Sci USA. 2012;109:5469–74.
O’Reilly KC, Anacker AMJ, Rogers TD, Forsberg CG, Wang J, Zhang B, et al. A social encounter drives gene expression changes linked to neuronal function, brain development, and related disorders in mice expressing the serotonin transporter Ala56 variant. Neurosci Lett. 2020;730:135027.
Nakai N, Nagano M, Saitow F, Watanabe Y, Kawamura Y, Kawamoto A, et al. Serotonin rebalances cortical tuning and behavior linked to autism symptoms in 15q11-13 CNV mice. Sci Adv. 2017;3:e1603001.
Luo J, Feng Q, Wei L, Luo M. Optogenetic activation of dorsal raphe neurons rescues the autistic-like social deficits in Shank3 knockout mice. Cell Res. 2017;27:950–53.
Walsh JJ, Llorach P, Cardozo Pinto DF, Wenderski W, Christoffel DJ, Salgado JS, et al. Systemic enhancement of serotonin signaling reverses social deficits in multiple mouse models for ASD. Neuropsychopharmacology. 2021;46:2000–10.
Rubenstein JL, Merzenich MM. Model of autism: increased ratio of excitation/inhibition in key neural systems. Genes Brain Behav. 2003;2:255–67.
Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, et al. Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci Transl Med. 2017;9:1–10.
Lazaro MT, Taxidis J, Shuman T, Bachmutsky I, Ikrar T, Santos R, et al. Reduced prefrontal synaptic connectivity and disturbed oscillatory population dynamics in the CNTNAP2 model of autism. Cell Rep. 2019;27:2567–78.e6.
Lee DK, Li SW, Bounni F, Friedman G, Jamali M, Strahs L, et al. Reduced sociability and social agency encoding in adult Shank3-mutant mice are restored through gene re-expression in real time. Nat Neurosci. 2021;24:1243–55.
Guo B, Chen J, Chen Q, Ren K, Feng D, Mao H, et al. Anterior cingulate cortex dysfunction underlies social deficits in Shank3 mutant mice. Nat Neurosci. 2019;22:1223–34.
Stoppel LJ, Kazdoba TM, Schaffler MD, Preza AR, Heynen A, Crawley JN, et al. R-baclofen reverses cognitive deficits and improves social interactions in two lines of 16p11.2 deletion mice. Neuropsychopharmacology. 2018;43:513–24.
Antoine MW, Langberg T, Schnepel P, Feldman DE. Increased excitation-inhibition ratio stabilizes synapse and circuit excitability in four autism mouse models. Neuron. 2019;101:648–61.e4.
Xu B, Roos JL, Levy S, van Rensburg EJ, Gogos JA, Karayiorgou M. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–5.
Malhotra D, Sebat J. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell. 2012;148:1223–41.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nat Rev Dis Prim. 2015;1:15071.
Donegan ML, Stefanini F, Meira T, Gordon JA, Fusi S, Siegelbaum SA. Coding of social novelty in the hippocampal CA2 region and its disruption and rescue in a 22q11.2 microdeletion mouse model. Nat Neurosci. 2020;23:1365–75.
Kirov G, Rees E, Walters JT, Escott-Price V, Georgieva L, Richards AL, et al. The penetrance of copy number variations for schizophrenia and developmental delay. Biol Psychiatry. 2014;75:378–85.
Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017;49:27–35.
Bristow GC, Thomson DM, Openshaw RL, Mitchell EJ, Pratt JA, Dawson N, et al. 16p11 duplication disrupts hippocampal-orbitofrontal-amygdala connectivity, revealing a neural circuit endophenotype for schizophrenia. Cell Rep. 2020;31:107536.
Ross RS, LoPresti ML, Schon K, Stern CE. Role of the hippocampus and orbitofrontal cortex during the disambiguation of social cues in working memory. Cogn Affect Behav Neurosci. 2013;13:900–15.
Becker JAJ, Kieffer BL, Le Merrer J. Differential behavioral and molecular alterations upon protracted abstinence from cocaine versus morphine, nicotine, THC and alcohol. Addict Biol. 2017;22:1205–17.
Goeldner C, Lutz PE, Darcq E, Halter T, Clesse D, Ouagazzal AM, et al. Impaired emotional-like behavior and serotonergic function during protracted abstinence from chronic morphine. Biol Psychiatry. 2011;69:236–44.
Valentinova K, Tchenio A, Trusel M, Clerke JA, Lalive AL, Tzanoulinou S, et al. Morphine withdrawal recruits lateral habenula cytokine signaling to reduce synaptic excitation and sociability. Nat Neurosci. 2019;22:1053–56.
Young KA, Liu Y, Gobrogge KL, Wang H, Wang Z. Oxytocin reverses amphetamine-induced deficits in social bonding: evidence for an interaction with nucleus accumbens dopamine. J Neurosci. 2014;34:8499–506.
Bardo MT, Neisewander JL, Kelly TH. Individual differences and social influences on the neurobehavioral pharmacology of abused drugs. Pharm Rev. 2013;65:255–90.
Venniro M, Zhang M, Caprioli D, Hoots JK, Golden SA, Heins C, et al. Volitional social interaction prevents drug addiction in rat models. Nat Neurosci. 2018;21:1520–29.
Venniro M, Russell TI, Zhang M, Shaham Y. Operant social reward decreases incubation of heroin craving in male and female rats. Biol Psychiatry. 2019;86:848–56.
Golden SA, Covington HE 3rd, Berton O, Russo SJ. A standardized protocol for repeated social defeat stress in mice. Nat Protoc. 2011;6:1183–91.
Fox ME, Lobo MK. The molecular and cellular mechanisms of depression: a focus on reward circuitry. Mol Psychiatry. 2019;24:1798–815.
Walsh JJ, Friedman AK, Sun H, Heller EA, Ku SM, Juarez B, et al. Stress and CRF gate neural activation of BDNF in the mesolimbic reward pathway. Nat Neurosci. 2014;17:27–9.
Tye KM, Mirzabekov JJ, Warden MR, Ferenczi EA, Tsai HC, Finkelstein J, et al. Dopamine neurons modulate neural encoding and expression of depression-related behaviour. Nature. 2013;493:537–41.
Pena CJ, Kronman HG, Walker DM, Cates HM, Bagot RC, Purushothaman I, et al. Early life stress confers lifelong stress susceptibility in mice via ventral tegmental area OTX2. Science. 2017;356:1185–88.
Shin S, Pribiag H, Lilascharoen V, Knowland D, Wang XY, Lim BK. Drd3 signaling in the lateral septum mediates early life stress-induced social dysfunction. Neuron. 2018;97:195–208.e6.
Knowland D, Lilascharoen V, Pacia CP, Shin S, Wang EH, Lim BK. Distinct ventral pallidal neural populations mediate separate symptoms of depression. Cell. 2017;170:284–97.e18.
Matthews GA, Nieh EH, Vander Weele CM, Halbert SA, Pradhan RV, Yosafat AS, et al. Dorsal raphe dopamine neurons represent the experience of social isolation. Cell. 2016;164:617–31.
Musardo S, Contestabile A, Knoop M, Baud O, Bellone C. Oxytocin neurons mediate the effect of social isolation via the VTA circuits. Elife. 2022;11:73421–42.
Chaudhury D, Walsh JJ, Friedman AK, Juarez B, Ku SM, Koo JW, et al. Rapid regulation of depression-related behaviours by control of midbrain dopamine neurons. Nature. 2013;493:532–6.
Hultman R, Ulrich K, Sachs BD, Blount C, Carlson DE, Ndubuizu N, et al. Brain-wide electrical spatiotemporal dynamics encode depression vulnerability. Cell. 2018;173:166–80.e14.
Koo JW, Chaudhury D, Han MH, Nestler EJ. Role of mesolimbic brain-derived neurotrophic factor in depression. Biol Psychiatry. 2019;86:738–48.
Christoffel DJ, Golden SA, Russo SJ. Structural and synaptic plasticity in stress-related disorders. Rev Neurosci. 2011;22:535–49.
Christoffel DJ, Golden SA, Walsh JJ, Guise KG, Heshmati M, Friedman AK, et al. Excitatory transmission at thalamo-striatal synapses mediates susceptibility to social stress. Nat Neurosci. 2015;18:962–4.
Franklin TB, Silva BA, Perova Z, Marrone L, Masferrer ME, Zhan Y, et al. Prefrontal cortical control of a brainstem social behavior circuit. Nat Neurosci. 2017;20:260–70.
Li Q, Zhang B, Cao H, Liu W, Guo F, Shen F, et al. Oxytocin exerts antidepressant-like effect by potentiating dopaminergic synaptic transmission in the mPFC. Neuropharmacology. 2020;162:107836.
Cheng J, Umschweif G, Leung J, Sagi Y, Greengard P. HCN2 channels in cholinergic interneurons of nucleus accumbens shell regulate depressive behaviors. Neuron. 2019;101:662–72.e5.
Sulzer D, Cragg SJ, Rice ME. Striatal dopamine neurotransmission: regulation of release and uptake. Basal Ganglia. 2016;6:123–48.
Higley MJ, Soler-Llavina GJ, Sabatini BL. Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat Neurosci. 2009;12:1121–8.
Yamamuro K, Bicks LK, Leventhal MB, Kato D, Im S, Flanigan ME, et al. A prefrontal-paraventricular thalamus circuit requires juvenile social experience to regulate adult sociability in mice. Nat Neurosci. 2020;23:1240–52.
Felix-Ortiz AC, Beyeler A, Seo C, Leppla CA, Wildes CP, Tye KM. BLA to vHPC inputs modulate anxiety-related behaviors. Neuron. 2013;79:658–64.
Felix-Ortiz AC, Tye KM. Amygdala inputs to the ventral hippocampus bidirectionally modulate social behavior. J Neurosci. 2014;34:586–95.
Felix-Ortiz AC, Burgos-Robles A, Bhagat ND, Leppla CA, Tye KM. Bidirectional modulation of anxiety-related and social behaviors by amygdala projections to the medial prefrontal cortex. Neuroscience. 2016;321:197–209.
King BH, Hollander E, Sikich L, McCracken JT, Scahill L, Bregman JD, et al. Lack of efficacy of citalopram in children with autism spectrum disorders and high levels of repetitive behavior: citalopram ineffective in children with autism. Arch Gen Psychiatry. 2009;66:583–90.
Williams K, Brignell A, Randall M, Silove N, Hazell P. Selective serotonin reuptake inhibitors (SSRIs) for autism spectrum disorders (ASD). Cochrane Database Syst Rev. 2013;8:CD004677.
Ford CL, Young LJ. Refining oxytocin therapy for autism: context is key. Nat Rev Neurol. 2022;18:67–8.
Aishworiya R, Valica T, Hagerman R, Restrepo B. An update on psychopharmacological treatment of autism spectrum disorder. Neurotherapeutics. 2022;19:248–62.
Parker KJ, Oztan O, Libove RA, Sumiyoshi RD, Jackson LP, Karhson DS, et al. Intranasal oxytocin treatment for social deficits and biomarkers of response in children with autism. Proc Natl Acad Sci USA. 2017;114:8119–24.
Parker KJ, Oztan O, Libove RA, Mohsin N, Karhson DS, Sumiyoshi RD, et al. A randomized placebo-controlled pilot trial shows that intranasal vasopressin improves social deficits in children with autism. Sci Transl Med. 2019;11:1–26.
Oztan O, Garner JP, Constantino JN, Parker KJ. Neonatal CSF vasopressin concentration predicts later medical record diagnoses of autism spectrum disorder. Proc Natl Acad Sci USA. 2020;117:10609–13.
Heifets BD, Malenka RC. MDMA as a probe and treatment for social behaviors. Cell. 2016;166:269–72.
Danforth AL, Grob CS, Struble C, Feduccia AA, Walker N, Jerome L, et al. Reduction in social anxiety after MDMA-assisted psychotherapy with autistic adults: a randomized, double-blind, placebo-controlled pilot study. Psychopharmacology. 2018;235:3137–48.
Heifets BD, Malenka RC. Better living through chemistry: MDMA’s prosocial mechanism as a starting point for improved therapeutics. Neuropsychopharmacology. 2021;46:261.
Henderson C, Wijetunge L, Kinoshita MN, Shumway M, Hammond RS, Postma FR, et al. Reversal of disease-related pathologies in the fragile X mouse model by selective activation of GABAB receptors with arbaclofen. Sci Transl Med. 2012;4:152ra28.
DeFilippis M, Wagner KD. Treatment of autism spectrum disorder in children and adolescents. Psychopharmacol Bull. 2016;46:18–41.
Berry-Kravis E, Hagerman R, Visootsak J, Budimirovic D, Kaufmann WE, Cherubini M, et al. Arbaclofen in fragile X syndrome: results of phase 3 trials. J Neurodev Disord. 2017;9:3.
Penton T, Catmur C, Banissy MJ, Bird G, Walsh V. Non-invasive stimulation of the social brain: the methodological challenges. Soc Cogn Affect Neurosci. 2022;17:15–25.
Boggio PS, Asthana MK, Costa TL, Valasek CA, Osorio AA. Promoting social plasticity in developmental disorders with non-invasive brain stimulation techniques. Front Neurosci. 2015;9:294.
Sturm V, Fricke O, Buhrle CP, Lenartz D, Maarouf M, Treuer H, et al. DBS in the basolateral amygdala improves symptoms of autism and related self-injurious behavior: a case report and hypothesis on the pathogenesis of the disorder. Front Hum Neurosci. 2012;6:341.
Anderson DJ. Circuit modules linking internal states and social behaviour in flies and mice. Nat Rev Neurosci. 2016;17:692–704.
Wu H, Miller KJ, Blumenfeld Z, Williams NR, Ravikumar VK, Lee KE, et al. Closing the loop on impulsivity via nucleus accumbens delta-band activity in mice and man. Proc Natl Acad Sci USA. 2018;115:192–97.
Mahajan UV, Ojukwu DI, Azagury DE, Safer DL, Cunningham T, Halpern CH. Can responsive deep brain stimulation be a cost-effective treatment for severe obesity? Obesity. 2022;30:338–46.
Anpilov S, Shemesh Y, Eren N, Harony-Nicolas H, Benjamin A, Dine J, et al. Wireless optogenetic stimulation of oxytocin neurons in a semi-natural setup dynamically elevates both pro-social and agonistic behaviors. Neuron. 2020;107:644–55.e7.
Funding
Authors’ and certain work discussed in this review are supported by grants from the NIH (P50 DA042012 to RCM; F32 MH103949 to JJW; F32 MH106206 and K99 DK115985 to DJC).
Author information
Authors and Affiliations
Contributions
JJW, DJC, and RCM wrote and edited this manuscript.
Corresponding authors
Ethics declarations
Competing interests
RCM is on the scientific advisory board of MapLight Therapeutics, MindMed, Bright Minds Biosciences, AZ Therapies, and Cyclerion.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Walsh, J.J., Christoffel, D.J. & Malenka, R.C. Neural circuits regulating prosocial behaviors. Neuropsychopharmacol. 48, 79–89 (2023). https://doi.org/10.1038/s41386-022-01348-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-022-01348-8
This article is cited by
-
Therapeutic mechanisms of psychedelics and entactogens
Neuropsychopharmacology (2024)
-
Detection, processing and reinforcement of social cues: regulation by the oxytocin system
Nature Reviews Neuroscience (2023)
-
Current Status of Psychopharmacological, Neuromodulation, and Oxytocin Treatments for Autism: Implications for Clinical Practice
Advances in Neurodevelopmental Disorders (2023)
-
Early life sleep disruption potentiates lasting sex-specific changes in behavior in genetically vulnerable Shank3 heterozygous autism model mice
Molecular Autism (2022)
-
Early resource scarcity alters motivation for natural rewards in a sex- and reinforcer-dependent manner
Psychopharmacology (2022)
