
Abstract
Background
Preterm infants with bronchopulmonary dysplasia (BPD) have lifelong increased risk of respiratory morbidities associated with environmental pathogen exposure and underlying mechanisms are poorly understood. The resident immune cells of the lung play vital roles in host defense. However, the effect of perinatal events associated with BPD on pulmonary-specific immune cells is not well understood.
Methods
We used a double-hit model of BPD induced by prenatal chorioamnionitis followed by postnatal hyperoxia, and performed a global transcriptome analysis of all resident pulmonary immune cells.
Results
We show significant up-regulation of genes involved in chemokine-mediated signaling and immune cell chemotaxis, and down-regulation of genes involved in multiple T lymphocyte functions. Multiple genes involved in T cell receptor signaling are downregulated and Cd8a gene expression remains downregulated at 2 months of age in spite of recovery in normoxia for 6 weeks. Furthermore, the proportion of CD8a+CD3+ pulmonary immune cells is decreased.
Conclusions
Our study has highlighted that perinatal lung inflammation in a double-hit model of BPD results in short- and long-term dysregulation of genes associated with the pulmonary T cell receptor signaling pathway, which may contribute to increased environmental pathogen-associated respiratory morbidities seen in children and adults with BPD.
Impact
-
In a translationally relevant double-hit model of BPD induced by chorioamnionitis and postnatal hyperoxia, we identified pulmonary immune cell-specific transcriptomic changes and showed that T cell receptor signaling genes are downregulated in short term and long term.
-
This is the first comprehensive report delineating transcriptomic changes in resident immune cells of the lung in a translationally relevant double-hit model of BPD.
-
Our study identifies novel resident pulmonary immune cell-specific targets for potential therapeutic modulation to improve short- and long-term respiratory health of preterm infants with BPD.
Similar content being viewed by others


Introduction
Bronchopulmonary dysplasia (BPD) is a serious complication of preterm birth characterized by arrest of alveolar development.1 Clinical BPD results from a combination of prenatal and postnatal environmental insults, with prenatal insults likely priming the developing lung to postnatal insults.2,3 Chorioamnionitis, an inflammatory disorder of the chorioamniotic membranes, is the most common identifiable cause of preterm birth, and is closely linked to the pathogenesis of BPD.4,5 Epidemiological data indicate increased risk of respiratory infections and worsened outcomes following respiratory infections in preterm infants with BPD and confirmed histological chorioamnionitis.6,7 Furthermore, preterm infants with BPD and chorioamnionitis have higher incidence and severity of respiratory morbidities such as asthma and chronic obstructive pulmonary disease (COPD) as adults.8,9 The lungs are a major portal of entry for pathogens and environmental toxins, and a major site of immune regulation. Therefore, a key determinant of disease susceptibility and severity of respiratory infections, asthma, and COPD is the host pulmonary immune system.10 However, mechanisms governing the altered pulmonary immune function in preterm infants with BPD remain elusive.
The lung is a complex system consisting of many different cell types with distinct function. The immune cells constitute nearly a third of all pulmonary cells. A growing body of literature implicates that inflammatory environmental exposures during pregnancy and early postnatal period result in lifelong alteration in immune cell health.11 Since many critical steps involved in the formation of immune cell lineages occur during pregnancy and early postnatal period, inflammatory insults during this critical developmental window are thought to induce immune programming and result in immune dysfunction.12 Importantly, longitudinal studies of peripheral blood immune cells demonstrate dissimilar immune profiles between term-corrected preterm infants and term infants at birth.12,13 These data suggest an important role for perinatal conditions surrounding preterm birth in programming the immune cells.
Many studies have demonstrated that tissue-specific immune cells differ from circulating immune cells.14,15,16 Respiratory viruses, especially influenza A and coronaviruses such as SARS-CoV, represent continuing global threats to human health with higher mortality and morbidities in those with pre-existing pulmonary diseases.17 Pulmonary-specific immune cells play crucial roles in clearance of such viruses during acute respiratory infections.18,19,20 Although a significant body of literature has demonstrated abnormal inflammatory processes in the developing lung due to perinatal exposure to environmental toxins, the potential short- and long-term consequences on developing pulmonary immune cells are not well understood. Importantly, the combined effects of prenatal inflammation induced by chorioamnionitis, which is the most common antecedent of preterm birth, and postnatal inflammation induced by hyperoxia, which is the most common therapy given to preterm infants, on pulmonary immune cells have not been previously examined.
Therefore, to address the knowledge gaps in understanding the effect of consecutive exposure to prenatal chorioamnionitis followed by postnatal hyperoxia on pulmonary immune cells, our objective was to examine global transcriptomic changes of all resident pulmonary immune cells in a clinically relevant double-hit animal model of BPD induced by prenatal chorioamnionitis and postnatal hyperoxia. We hypothesized that a double-hit injury to the developing lung induced by prenatal chorioamnionitis and postnatal hyperoxia will result in short- and long-term dysregulation of resident pulmonary immune cell-specific gene expression.
Methods
Animal model
Timed-pregnant Sprague Dawley rats were purchased from Charles River laboratories and were injected via an intra-amniotic approach at embryonic day 20 with 1 µg of lipopolysaccharide (LPS) (Sigma-Aldrich; O111:B4) or normal saline (NS) after a laparotomy under general anesthesia with isoflurane. Following natural delivery, pups were placed in normoxia (RA) or in a hyperoxia chamber with 85% oxygen (O2). Continuous hyperoxia was maintained for 2 weeks during which time the dams were rotated every 24–48 h. Pups were then euthanized at 2 weeks of age for 2-week analyses or allowed to grow in room air for 6 weeks for 2-month analyses. All animals used in the study and experimental protocols were approved by the Nemours Institutional Animal Care and Use Committee and followed guideline set forth in the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
Lung histology and morphometry
For quantification of alveolarization, lungs were infused with 2% paraformaldehyde via a tracheal catheter at a uniform pressure of 25 cm H2O for 3 min using a 25G or 21G catheter for 2-week old and 2-month old rats respectively. Hematoxylin and eosin-stained tissue sections were used to obtain 10 random pictures of each lung at ×20 magnification. The images were viewed under a field of equally spaced horizontal lines using ImageJ and mean linear intercept (MLI) was calculated as the average of total length of lines divided by the total intercepts of alveolar septa from each lung. Image analysis was performed by a blinded observer.
Lung single-cell preparation and isolation of immune cells
Animals were euthanized by CO2 inhalation followed by exsanguination. Lungs were perfused with phosphate-buffered saline (PBS) through the right ventricle to flush out all blood from the lungs. Lungs were dissected and minced into small pieces with a razor blade. The minced lung was suspended in a prewarmed digestive solution containing dispase (50 U/ml; Collaborative Biosciences), collagenase (480 U/ml; Life Technologies), and DNase (0.33 U/ml; Promega).21 The suspension was incubated at 37 °C for 30 min for 2-week-old lungs, and 60 min for 2-month-old lungs. Digestion was stopped by adding 1 ml of fetal bovine serum (FBS) at the end of incubation period and cells were then filtered through a 100 µm cell strainer. Red blood cells were lysed by suspending the filtered cells in 1 ml of ACK lysis buffer for 5 min and washed with PBS containing 1% FBS and 1 mM EDTA. Cells were stained with DAPI, anti-rat CD45-FITC (ebiosciences), anti-rat CD326-APC (RayBiotech), and anti-rat CD31-PECy7 (ebiosciences) fluorochrome-conjugated antibodies. Cells were negatively gated for DAPI to exclude dead cells. CD45+ immune cells, CD45−CD31−CD326+ epithelial cells, CD45−CD326−CD31+ endothelial cells, and CD45−CD31−CD326− mesenchymal cells were identified and sorted using a FACSAriaIII cell sorter. To confirm the purity and viability of the sorted populations, an aliquot of the sorted cells was run through the cell sorter a second time, and cells were found to be greater than 95% viable (DAPI−) and pure according to the cell-surface markers described above. Cells were directly sorted into aliquots of 500 μl of RLT lysis buffer (QIAgen), homogenized in a QIA-shredder (QIAgen), and stored in −80 °C.
Flow cytometry
To compare the frequency of pulmonary immune cell types, aliquots of lung single-cell suspensions were stained for DAPI, anti-rat CD45-FITC (ebiosciences), and the following fluorochrome-labeled antibodies to known rat-specific immune cell-surface markers: anti-rat CD3-APC (Biolegend), anti-rat CD8a-PECy7 (Thermo Fisher), anti-rat CD4-PECy7 (Biolegend), anti-rat CD45R (B220)-PECy7 (Thermo Fisher), and anti-rat CD43-PerCpCy5.5 (Biolegend).22 Flow cytometric compensation was performed using fluorescent compensation beads (UltraComp eBeadsTM Compensation beads, Thermo Fisher). Positive and negative populations within the DAPI-negative CD45-positive immune cells were defined using unstained and fluorescence minus one controls. A minimum of 10,000 events per sample was acquired using FACSAriaIII cell sorter and analyzed (BD FACS Diva software version 8.0.1).
RNA isolation and quantitative real-time qPCR
RNA was extracted using Qiagen All prep kit or RNeasy mini kit. Reverse transcription was performed using SuperScriptTM IV First-Strand Synthesis System (Invitrogen). Reverse transcribed cDNA, TaqMan Gene Expression Assay (Supplementary Table 1) and TaqMan Fast Advanced Master Mix were combined and quantitative PCR was performed on a Quantstudio 12K Flex Real-Time PCR system (Thermo Fisher Scientific) using a 384-well plate. Beta-actin was used as the housekeeping gene. Data analysis was performed with the comparative Ct (ddCt) method using Thermo Fisher Expression Suite Software v1.1.
Microarray
Fragmented biotin-labeled cDNA (from 50 ng of RNA) was synthesized using the GeneChip WT Plus kit. Affymetrix gene chips (rat Clariom S) were hybridized with 2.5 μg fragmented and biotin-labeled cDNA in 100 μl of hybridization cocktail. The rat Affymetrix Clariom S gene chips were used for microarray, which provides a coverage of >22,900 annotated genes. Target denaturation was performed at 99 °C for 5 min and then 45 °C for 5 min, followed by hybridization with rotation 60 r.p.m. for 16–18 h at 45 °C. Arrays were then washed and stained with Gene chip Fluidic Station 450, using Affymetrix GeneChip hybridization wash & stain kit. Chips were scanned on an Affymetrix Gene Chip Scanner 3000 7G, using Command Console Software. Quality Control of the experiment was performed by TAC 4.0 software. sst-RMA normalization was performed from Affymetrix CEL files by using TAC 4.0 Software. Experimental group (LPS + O2) were compared to control group (NS + RA) by using Transcriptome array console software 4.0. Twofold differentially expressed genes with statistically significant p values <0.05 were used for further analysis. Gene expression data are deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE161836.
Gene enrichment analysis
The data obtained from the TAC were utilized to generate gene lists of upregulated and downregulated genes. Biological processes and gene ontology (GO) terms that were enriched among upregulated and downregulated genes with false discovery rate (FDR) p value < 0.01 were determined using the DAVID v6.8 software (The Database for Annotation, Visualization and Integrated Discovery).23,24
Statistical analysis
A minimum of five biological replicates per experimental group were used for histological, flow cytometric, and real-time qPCR analyses. Three biological replicates per experimental group were used for microarray analysis. For biological validation of microarray results, experiments were repeated and real-time qPCR assays were performed using immune cells isolated from the second cohort of rat pups. Both males and females were included and sex was determined by ano-genital exam. Sex-specific differences were assessed for a minimum of two biological replicates per experimental group. Statistical analysis was performed using Prism software version 8 (GraphPad Software, San Diego, CA). All data are represented as mean and standard deviation. Statistical significance was set at a p value <0.05 and calculated using Mann–Whitney non-parametric t-test for comparison of two groups, and two-way ANOVA followed by Holm–Sidak’s multiple comparisons test for assessment of sex-specific differences.
Results
Combined prenatal exposure to LPS and postnatal hyperoxia impairs alveolarization
First, we assessed the effect of combined prenatal LPS and postnatal hyperoxia exposure by analyzing the lung morphology. Lungs of rat pups harvested at 2 weeks of age following exposure to prenatal LPS and postnatal hyperoxia exhibited significant arrest of alveolarization as measured by MLI, compared to the prenatal NS and postnatal RA group (32 ± 2.3 vs. 25 ± 0.8; LPS + O2 vs. NS + RA, p = 0.0079, n = 5 per group) (Fig. 1a–c). There were no sex-specific differences (Fig S1A). Following recovery in normoxia for 6 weeks, the MLI of lungs from pups exposed to prenatal LPS and postnatal hyperoxia remained significantly higher than those exposed to prenatal NS and postnatal RA (37 ± 8.1 vs. 27 ± 2.6; LPS + O2 vs. NS + RA, p = 0.002, n = 6 per group) (Fig. 1d–f). Thus, prenatal exposure to LPS and postnatal exposure to hyperoxia results in significant arrest of alveolarization that persists into adulthood in spite of long-term recovery in room air.

a, b Hematoxylin and eosin (H and E)-stained lung tissue section from 2-week-old rat pups exposed to prenatal normal saline and postnatal normoxia (NS + RA) and prenatal LPS and postnatal hyperoxia (LPS + O2). c Mean linear intercept (MLI) to assess alveolarization is significantly increased in the LPS + O2 group compared to the NS + RA group, N = 5 per group. Scale bar = 200 μm. d, e H and E-stained lung tissue section from 2-month-old rat pups exposed to NS + RA and LPS + O2. f MLI is significantly increased in the LPS + O2 group compared to the NS + RA group in spite of recovery in room air for 6 weeks. N = 6 per group. Scale bar = 200 μm.
Isolation and characterization of resident pulmonary immune cells
Non-recirculating resident pulmonary immune cells play a vital role in maintaining the delicate balance between immunity and tolerance.25 Therefore, in this study we evaluated the effect of combined prenatal LPS and postnatal hyperoxia exposure on the resident pulmonary immune cells. We excluded the circulating immune cells by perfusing the lungs with PBS and identified the resident pulmonary cells by positive selection for CD45, a pan-immune cell-surface marker (Fig. 2a). As expected, the CD45+ sorted pulmonary immune cells had high Ptprc gene expression compared to the non-immune pulmonary cells (Fig. 2b). We further confirmed the purity of sorted pulmonary immune cells by performing real-time qPCR for epithelial, endothelial, and mesenchymal cell-specific marker genes. The CD45+ pulmonary immune cells had significantly low expression of Nkx2-1, Pecam-1, and Acta2 compared to the epithelial (CD45−CD326+), endothelial (CD45−CD31+), and mesenchymal (CD45−CD326−CD31−) cells, respectively, that were also isolated from the same lungs (Fig. 2c–e). Thus, we confirmed that isolated pulmonary immune cells were devoid of contamination with non-immune pulmonary cells.

a Flow cytometric gating used for isolation of pulmonary immune cells. b Isolated pulmonary immune cells validated by demonstration of high expression of immune cell-specific gene (Ptprc) compared to non-immune pulmonary cells by qPCR. c Isolated pulmonary immune cells have low expression of epithelial cell-specific gene Nkx-2 compared to pulmonary epithelial cells, d low expression of endothelial cell-specific gene Pecam-1 compared to pulmonary endothelial cells, and e low expression of mesenchymal cell-specific gene Acta2 compared to pulmonary mesenchymal cells isolated from the same lung, N = 5 per group. f, g Quantification of pulmonary immune cells as a percentage of all viable pulmonary cells isolated at 2 weeks and 2 months of age respectively. N = 6–12 per group.
To determine whether combined prenatal LPS and postnatal hyperoxia exposure resulted in a change in the number of resident pulmonary immune cells, we quantified the percentage of resident pulmonary immune cells as a fraction of all viable pulmonary cells. The percentage of CD45+ pulmonary immune cells were similar between the NS + RA and LPS + O2 groups at both 2 weeks (23 ± 5.1% vs. 27.5 ± 12.2%; NS + RA vs. LPS + O2, p = 0.6, n = 11–12 per group) (Fig. 2f) and 2 months of age (37 ± 11% vs. 50.6 ± 12.8%; NS + RA vs. LPS + O2, p = 0.2, n = 6–7 per group) (Fig. 2g).
Consecutive exposure to prenatal chorioamnionitis and postnatal hyperoxia induces differential gene expression in resident pulmonary immune cells
Microarray was performed on RNA extracted from pulmonary immune cells isolated from 2-week-old rat pups exposed to prenatal LPS and postnatal hyperoxia. Rat pups injected with intrauterine NS and postnatally exposed to normoxia were used as controls. Principal component analysis performed to analyze the correlation between data from different samples demonstrated good separation between the NS + RA and LPS + O2 groups (Fig. 3a). Differentially expressed genes with a p value <0.05 and fold change ≥2 were used for further analysis. In total, 648 genes were upregulated and 665 genes were downregulated (Fig. 3b and Tables 1 and 2). We next analyzed GO terms of most significantly represented biological processes associated with top differentially up- or downregulated genes with a FDR p value <0.01. GO enrichment analysis of upregulated genes revealed significant enrichment of biological processes involved in the chemokine-mediated signaling pathway, lymphocyte chemotaxis, neutrophil chemotaxis, monocyte chemotaxis, inflammatory responses, cellular response to interferon-gamma, and cellular responses to tumor necrosis factor (Fig. 3c and Table 3). Among the downregulated genes, the most significantly overrepresented ones were related to several key T cell-related biological processes such as T cell co-stimulation, positive regulation of T cell proliferation, T cell activation, negative thymic T cell selection, and T cell receptor V(D)J recombination (Fig. 3d and Table 4). In summary, the GO analyses for both up- and downregulated genes indicated multiple important transcriptomic differences in the resident pulmonary immune cells induced by consecutive exposure to prenatal chorioamnionitis and postnatal hyperoxia.

a Principal component analysis (PCA) of the microarray data obtained from pulmonary immune cells isolated from three pups in the NS + RA group represented in blue and three pups in LPS + O2 represented in red. b Volcano plot of differentially expressed genes in the LPS + O2 group compared to the NS + RA group (n = 3 per group) with p value <0.05 represented in colored dots. Upregulated genes with log2 fold change ≥1 and p value <0.05 represented in red and downregulated genes with negative log2 fold change ≥1 and p value <0.05 represented in dark blue. c Gene ontology terms enriched in upregulated genes with false discovery rate (FDR) < 0.01 and d gene ontology terms enriched in downregulated genes with FDR < 0.01. Red bar represents log2 fold enrichment and blue bar represents negative log10 of corrected p value.
Combined prenatal chorioamnionitis and postnatal hyperoxia downregulates key genes involved in T cell receptor signaling
Given that five out of seven overrepresented biological processes of top differentially downregulated genes were related to T cell function, we next focused on the differences in genes involved in T cell receptor signaling. As shown in Fig. 4a, many genes identified to be differentially expressed in the microarray analysis and known to be important in T cell receptor signaling were significantly downregulated in the LPS + O2 group, compared to the NS + RA group. We next confirmed and validated these results by repeating the experiments in a separate cohort of animals and performing real-time qPCR analysis of select genes in pulmonary immune cells of this second cohort of pups. qPCR assay confirmed down-regulation of five key T cell receptor signaling genes, namely Cd8a, Cd3d, Cd3e, Cd5, and Trat1, in the LPS + O2 group compared to the NS + RA group (Fig. 4b–f). There were no significant sex-specific differences in expression of Cd8a, Cd3d, Cd3e, Cd5, and Trat1 genes (Fig S1B–F).

a Heat map showing the changes of gene expression associated with the T cell receptor signaling pathway between NS + RA and LPS + O2 groups. b–e Real-time PCR validation of the genes associated with down-regulation of T cell receptor signaling identified by microarray analysis at 2 weeks of age. N = 5 per group. Data are presented as mean ± standard deviation of log2 fold change of the genes in the LPS + O2 group compared to the NS + RA group.
Down-regulation of Cd8a gene expression persists into adulthood in spite of recovery in normoxia
Since pulmonary T cell function is critical in maintaining balance between host immune defense and tolerance, we examined whether combined prenatal chorioamnionitis and postnatal hyperoxia-induced down-regulation of pulmonary T cell receptor signaling genes persists into adulthood. In spite of recovery in normoxia for 6 weeks, pulmonary immune cell Cd8a gene expression remained significantly lower in the LPS + O2 group compared to the NS + RA group (log2 fold change: −0.49 ± 0.3 vs. 0 ± 0.3; LPS + O2 vs. NS + RA, p = 0.017, n = 5–7 per group) (Fig. 5a). In contrast, gene expression of other perinatally downregulated T cell receptor signaling genes recovered to near normal levels. There was no significant difference in gene expression of Cd3d, Cd3e, Cd5, and Trat1 between the LPS + O2 and NS + RA groups (Fig. 5b–e).

a–e Real-time PCR validation of the genes associated with down-regulation of T cell receptor signaling at 2 months of age following recovery in room air for 6 weeks. N = 5–7 per group. Data are presented as mean ± standard deviation of log2 fold change of the genes in the LPS + O2 group compared to the NS + RA group.
Composition of pulmonary immune cell types is modified by combined prenatal chorioamnionitis and postnatal hyperoxia exposure
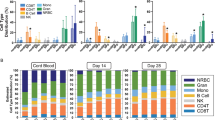
We next investigated if dysregulated expression of genes involved in T cell receptor signaling induced by combined prenatal chorioamnionitis and postnatal hyperoxia was associated with altered pulmonary T lymphocyte frequency at 2 weeks of age by flow cytometric analyses of lung single-cell suspensions stained with DAPI, CD45-FITC, CD3-APC, and either CD4-PECy7 or CD8 PECy7. The frequency of total pulmonary T lymphocytes (DAPI−CD45+CD3+ cells) was similar between the NS+RA and LPS+O2 groups (17.1 ± 2.3% vs. 18.3 ± 3.8%; NS + RA vs. LPS + O2, p = 0.69, n = 5 per group). In contrast, the proportion of CD8 T cells (DAPI−CD45+CD3+CD8a+ cells) was significantly lower in the LPS + O2 group compared to the NS + RA group (9 ± 1.1% vs. 6.8 ± 1.3%; NS + RA vs. LPS + O2, p = 0.0476, n = 5 per group). There was no difference in the frequency of CD4 T cells (DAPI−CD45+CD3+CD4+ cells) (12 ± 1.9% vs. 10.9 ± 3.4%; NS+RA vs. LPS+O2, p = 0.42, n = 5 per group) (Fig. 6b). We further investigated whether combined prenatal chorioamnionitis and postnatal hyperoxia modified frequencies of other pulmonary immune cell subpopulations, namely B cells and monocytes. There was no significant difference in the proportion of CD45R+B cells (DAPI−CD45+CD45R+ cells) (56.7 ± 8.7% vs. 52.5 ± 6.8%; NS + RA vs. LPS + O2, p = 0.59, n = 5 per group) (Fig. 6c), and CD43+ monocytes (DAPI−CD45+CD43+ cells) (48.5 ± 1.4% vs. 54.7 ± 6.5%; NS + RA vs. LPS + O2, p = 0.15, n = 5 per group) (Fig. 6d). There were no significant sex-specific differences in the proportions of immune cell types (Fig S2A–E).

a Flow cytometric gating used for defining pulmonary immune cell types. b–d Quantification of pulmonary immune cell types as a percentage of all viable pulmonary CD45+ immune cells at 2 weeks. N = 5 per group.
Discussion
In this study, we used a clinically relevant double-hit model of BPD induced by prenatal chorioamnionitis and postnatal hyperoxia to examine global transcriptomic changes in resident pulmonary immune cells. Our study highlights significant up-regulation of genes involved in chemokine-mediated signaling and immune cell chemotaxis and down-regulation of genes involved in various T lymphocyte functions. Specifically, genes related to T cell receptor signaling are downregulated, including long-term suppression of CD8a gene expression into adulthood in spite of recovery in room air for 6 weeks. Furthermore, these transcriptome changes were associated with a decrease in pulmonary CD8+ T lymphocyte frequencies at 2 weeks of age.
BPD is associated with high mortality and morbidity. Importantly, infants with BPD are at risk for adverse respiratory morbidities throughout the lifetime, such as increased risk of infections, asthma, susceptibility to smoking, and COPD.6,7,8,9 Pathogenic mechanisms that lead to BPD and its long-term complications have origins prior to birth and are related to adverse antenatal factors such as chorioamnionitis, pre-eclampsia, and gestational diabetes.3 Prenatal inflammation caused by chorioamnionitis is one of the most frequent identifiable causes of preterm births and clinical investigations demonstrate strong association between chorioamnionitis and BPD.4,5 Therefore, a rat model consisting of prenatal intra-amniotic LPS injection and postnatal hyperoxia exposure is a clinically relevant animal model of human infants who are prematurely born due to chorioamnionitis and require postnatal oxygen therapy due to lung immaturity.
Prenatal insults can alter hematopoiesis, induce epigenetic programming, alter immune cell fate commitment, or cause direct insults on differentiated immune cell types resulting in immunomodulation.11,12 Immunomodulation can skew the balance toward immunosuppression and subsequently enhance susceptibility to infections or chronic lung disease. Alternatively, the balance may be skewed toward immunoenhancement, subsequently leading to hypersensitivity diseases such as asthma or allergy. Majority of mechanistic information in the field of chorioamnionitis or BPD comes from immune cells isolated from peripheral blood or cord blood from human tissue, or from immune cell phenotyping from lung tissue of animals exposed to either prenatal chorioamnionitis or postnatal hyperoxia.15,26,27 However, mechanisms involved in the resident pulmonary immune cells as a result of combined prenatal LPS and postnatal hyperoxia-induced lung injury is not well understood. Furthermore, recent studies have shown that cord blood measurements are poor predictors of postnatal immune system states, due to inherent tissue differences.12 Lung tissue-resident memory CD4 T cells compartmentalized in the lung tissue and did not circulate or migrate from the lung.28 Lung infection with influenza induced lung-resident memory B cells that did not migrate or recirculate and were distinct from lymphoid cells.29 Ex vivo studies have shown suppressed transcriptional responses to Staph epidermidis in monocytes derived from preterm neonates with chorioamnionitis.30 Similarly, ex vivo analyses of fetal and neonatal monocytes derived from intrauterine LPS exposed sheep exhibited immune paralysis following stimulation with LPS or other Toll-like receptor ligands.31 However, the effect of consecutive injuries in pulmonary immune cells in vivo is not known. Contemporary perspectives have also highlighted the pressing need to evaluate pulmonary cell-specific molecular mechanisms of BPD using translationally relevant animal models which combine a background of prenatal insults and postnatal injuries.32 The lung is a unique organ that must constantly protect against inhaled pathogens and innocuous particles, without mounting a destructive response. Recent studies have shown that non-recirculating resident pulmonary immune cells play a vital role in maintaining the delicate balance between immunity and tolerance.25 Therefore, in this study we aimed to evaluate the transcriptome changes of resident pulmonary immune cells in vivo following a double-hit injury relevant to clinical BPD.
Our study highlights significant down-regulation of genes involved in multiple T lymphocytic biologic processes as a result of combined prenatal LPS and postnatal hyperoxia exposure. Since the lung is directly exposed to the external environment, the immune system of the lungs plays a critical role in host defense against pathogenic organisms. T lymphocytes play critical roles in host defense against pathogens that escape the innate defenses. Longitudinal phenotyping of peripheral blood T lymphocytes in preterm infants has shown that those with fewer CD31+IL8+CD4+ T cells at term-corrected gestational age compared to term infants have a 3.5-fold increased risk for respiratory morbidities in their first year after NICU discharge.13 It is well established that lung-resident T cells play important roles in pathogen detection and barrier tissue protection by acting as first-line defense to eliminate infected cells.33,34 T lymphocytes express T cell receptors (TCR) that recognize antigens from different pathogens and subsequently differentiate into different subsets that function uniquely to eliminate the pathogen. Th1 and CD8+ T cells provide host defense against viral pathogens such as influenza and respiratory syncytial virus (RSV), whereas Th2 and Th17 responses contribute to exacerbate diseases in influenza and RSV.35 Dysregulated T cell immune responses result in delayed clearance of pathogen and increased pathogen dissemination in the lung. Importantly, emerging data from single-cell RNA-sequencing from bronchoalveolar lavage fluid (BALF) from patients with COVID-19 suggest an important role for pulmonary CD8 T cell responses.19 Lower CD8 T cells were observed in BALF of patients with severe infection compared to patients with moderate infection.19 Whether the prolonged suppression of Cd8a gene expression induced by perinatal lung injury seen in our model contributes to increased infection severity in adulthood remains to be examined.
Animal and human studies have shown that the presence of IAV-specific CD4+ and CD8+ memory T cells correlated with better protection from subsequent infection. Importantly these studies demonstrated that memory T cells that remain resident in the lung provided the most potent protection against a second heterologous IAV infections, and not all subsets of memory T cells are equally protective.36,37 On the basis of this data, new vaccine protocols are being developed to specifically evoke memory T cells in the lung. Furthermore, worse outcome in response to viral respiratory infections and limited or delayed response to vaccines seen in infants compared to adults is attributed to impaired establishment of lung-resident memory T cells following infections in neonatal mice compared to adult mice.38 For these reasons, it is important to specifically understand changes in lung specific T cells induced by perinatal events in order to develop appropriate pulmonary T cell directed therapies to protect such infants from secondary viral infections. Furthermore, a recent study demonstrated that perinatal glucocorticoid exposure resulted in long-term diminished CD8 T cell response in adulthood to tumor growth and bacterial infection through a persistent alteration of the hypothalamic–pituitary–adrenal (HPA) axis. These effects were largely mediated by T cell intrinsic glucocorticoid receptor signaling and long-term epigenetic reprogramming of naïve CD8 T cells.39 Whether a persistent alteration in the HPA axis is induced in our double-hit model of BPD remains to be examined.
There are a few limitations to our study. First, we used a single injection of intra-amniotic LPS to induce chorioamnionitis. In addition to LPS-induced chorioamnionitis, preterm infants are often exposed to intrauterine infection due to urea plasma and mycoplasma which are known to modulate LPS-induced chorioamnionitis.40 Whether immune cell responses to postnatal hyperoxia will differ with respect to the organism causing chorioamnionitis remains to be examined. Furthermore, the animals in our study were not delivered preterm and our experimental design did not allow parsing the immune dysregulation due to prenatal LPS exposure vs. postnatal hyperoxia exposure, or the effect of other postnatal factors such as mechanical ventilation and sepsis on pulmonary immune cells. Finally, the consequences of transcriptome changes on the functional readouts of immune function were not measured.
Conclusion
In summary, our study is the first to specifically examine pulmonary-specific immune cell transcriptomics in a clinically relevant animal model of BPD induced by prenatal chorioamnionitis and postnatal hyperoxia. Our novel findings highlight the short- and long-term impacts of perinatal events on resident pulmonary immune cells that may predispose infants and adults with BPD at higher risk for respiratory morbidities.
References
Higgins, R. D. et al. Bronchopulmonary dysplasia: executive summary of a workshop. J. Pediatr. 197, 300–308 (2018).
Pryhuber, G. S. Postnatal infections and immunology affecting chronic lung disease of prematurity. Clin. Perinatol. 42, 697–718 (2015).
Taglauer, E., Abman, S. H. & Keller, R. L. Recent advances in antenatal factors predisposing to bronchopulmonary dysplasia. Semin. Perinatol. 42, 413–424 (2018).
Sarno, L. et al. Histological chorioamnionitis and risk of pulmonary complications in preterm births: a systematic review and meta-analysis. J. Matern. Fetal Neonatal Med. 1–10, https://doi.org/10.1080/14767058.2019.1689945 (2019).
Villamor-Martinez, E. et al. Association of chorioamnionitis with bronchopulmonary dysplasia among preterm infants: a systematic review, meta-analysis, and metaregression. JAMA Netw. Open 2, e1914611 (2019).
Chaw, P. S. et al. Respiratory syncytial virus-associated acute lower respiratory infections in children with bronchopulmonary dysplasia: systematic review and meta-analysis. J. Infect. Dis., https://doi.org/10.1093/infdis/jiz492 (2019).
McDowell, K. M. et al. Pulmonary morbidity in infancy after exposure to chorioamnionitis in late preterm infants. Ann. Am. Thorac. Soc. 13, 867–876 (2016).
Collaco, J. M. & McGrath-Morrow, S. A. Respiratory phenotypes for preterm infants, children, and adults: bronchopulmonary dysplasia and more. Ann. Am. Thorac. Soc. 15, 530–538 (2018).
Getahun, D. et al. Effect of chorioamnionitis on early childhood asthma. Arch. Pediatr. Adolesc. Med. 164, 187–192 (2010).
Lloyd, C. M. & Marsland, B. J. Lung homeostasis: influence of age, microbes, and the immune system. Immunity 46, 549–561 (2017).
Rychlik, K. A. & Sille, F. C. M. Environmental exposures during pregnancy: mechanistic effects on immunity. Birth Defects Res. 111, 178–196 (2019).
Olin, A. et al. Stereotypic immune system development in newborn children. Cell 174, 1277–1292.e1214 (2018).
Scheible, K. M. et al. T cell developmental arrest in former premature infants increases risk of respiratory morbidity later in infancy. JCI Insight 3, https://doi.org/10.1172/jci.insight.96724 (2018).
Gleditsch, D. D. et al. Maternal inflammation modulates infant immune response patterns to viral lung challenge in a murine model. Pediatr. Res. 76, 33–40 (2014).
Kallapur, S. G. et al. Intra-amniotic IL-1beta induces fetal inflammation in rhesus monkeys and alters the regulatory T cell/IL-17 balance. J. Immunol. 191, 1102–1109 (2013).
Connors, T. J. et al. Airway CD8(+) T cells are associated with lung injury during infant viral respiratory tract infection. Am. J. Respir. Cell Mol. Biol. 54, 822–830 (2016).
Bradley, B. T. & Bryan, A. Emerging respiratory infections: the infectious disease pathology of SARS, MERS, pandemic influenza, and Legionella. Semin Diagn. Pathol. 36, 152–159 (2019).
Zheng, J. & Perlman, S. Immune responses in influenza A virus and human coronavirus infections: an ongoing battle between the virus and host. Curr. Opin. Virol. 28, 43–52 (2018).
Liao, M. et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat. Med. https://doi.org/10.1038/s41591-020-0901-9 (2020).
Monticelli, L. A. et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 12, 1045–1054 (2011).
Alapati, D. et al. In utero gene editing for monogenic lung disease. Sci. Transl. Med. 11, https://doi.org/10.1126/scitranslmed.aav8375 (2019).
Barnett-Vanes, A., Sharrock, A., Birrell, M. A. & Rankin, S. A single 9-colour flow cytometric method to characterise major leukocyte populations in the rat: validation in a model of LPS-induced pulmonary inflammation. PLoS ONE 11, e0142520 (2016).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang da, W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Ardain, A., Marakalala, M. J. & Leslie, A. Tissue-resident innate immunity in the lung. Immunology 159, 245–256 (2020).
Dedja, A. et al. Lipopolysaccharide-induced chorioamnionitis and postnatal lung injury: the beneficial effects of L-citrulline in newborn rats. Exp. Lung Res. 44, 226–240 (2018).
Rosen, D. et al. Accelerated thymic maturation and autoreactive T cells in bronchopulmonary dysplasia. Am. J. Respir. Crit. Care Med. 174, 75–83 (2006).
Teijaro, J. R. et al. Cutting edge: tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol. 187, 5510–5514 (2011).
Allie, S. R. et al. The establishment of resident memory B cells in the lung requires local antigen encounter. Nat. Immunol. 20, 97–108 (2019).
de Jong, E. et al. Exposure to chorioamnionitis alters the monocyte transcriptional response to the neonatal pathogen Staphylococcus epidermidis. Immunol. Cell Biol. 96, 792–804 (2018).
Kramer, B. W. et al. Endotoxin-induced chorioamnionitis modulates innate immunity of monocytes in preterm sheep. Am. J. Respir. Crit. Care Med. 171, 73–77 (2005).
Ambalavanan, N. & Morty, R. E. Searching for better animal models of BPD: a perspective. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L924–L927 (2016).
Iijima, N. & Iwasaki, A. T cell memory. A local macrophage chemokine network sustains protective tissue-resident memory CD4 T cells. Science 346, 93–98 (2014).
Schenkel, J. M. et al. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98–101 (2014).
Chen, K. & Kolls, J. K. T cell-mediated host immune defenses in the lung. Annu. Rev. Immunol. 31, 605–633 (2013).
Pizzolla, A. et al. Resident memory CD8(+) T cells in the upper respiratory tract prevent pulmonary influenza virus infection. Sci. Immunol. 2, https://doi.org/10.1126/sciimmunol.aam6970 (2017).
Wu, T. et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J. Leukoc. Biol. 95, 215–224 (2014).
Zens, K. D. et al. Reduced generation of lung tissue-resident memory T cells during infancy. J. Exp. Med. 214, 2915–2932 (2017).
Hong, J. Y. et al. Long-term programming of CD8 T cell immunity by perinatal exposure to glucocorticoids. Cell 180, 847–861.e815 (2020).
Snyder, C. C. et al. Modulation of lipopolysaccharide-induced chorioamnionitis by Ureaplasma parvum in sheep. Am. J. Obstet. Gynecol. 208, 399.e391–398 (2013).
Acknowledgements
The authors thank the histology core, cell science core, and biomolecular core labs at Nemours Biomedical Research Center for their technical assistance and guidance. This study was supported by the Institutional Development Award (IDeA) (pilot award to D.A.) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number NIH COBRE P30GM114736 (PI: T.H.S.), NIH K08 HL151760 01 (to D.A.), American thoracic society foundation (to D.A.), Hemant Desai Fellow Research Grant (to D. Shrestha), and The Nemours Foundation.
Author information
Authors and Affiliations
Contributions
D. Shrestha performed experiments and drafted the manuscript; G.X.Y. performed experiments and data analysis; D. Stabley, S.G.B., Y.Z., L.G., J.H., A.H., and M.S. performed experiments and edited the manuscript, T.H.S. and Z.H.A. edited the manuscript; S.A. performed microarray experiments and analyses; and D.A. conceived and designed research, performed experiments, analyzed data, drafted the manuscript, and approved the final version of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplemental information
Rights and permissions
About this article

Cite this article
Shrestha, D., Ye, G.X., Stabley, D. et al. Pulmonary immune cell transcriptome changes in double-hit model of BPD induced by chorioamnionitis and postnatal hyperoxia. Pediatr Res 90, 565–575 (2021). https://doi.org/10.1038/s41390-020-01319-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41390-020-01319-z
This article is cited by
-
Leveraging transcriptomics to develop bronchopulmonary dysplasia endotypes: a concept paper
Respiratory Research (2023)
-
Omics approaches: interactions at the maternal–fetal interface and origins of child health and disease
Pediatric Research (2023)
-
Association of immune cell recruitment and BPD development
Molecular and Cellular Pediatrics (2022)
