
Abstract
Digestive cancers are the leading cause of cancer-related death worldwide and have high risks of morbidity and mortality. Histone methylation, which is mediated mainly by lysine methyltransferases, lysine demethylases, and protein arginine methyltransferases, has emerged as an essential mechanism regulating pathological processes in digestive cancers. Under certain conditions, aberrant expression of these modifiers leads to abnormal histone methylation or demethylation in the corresponding cancer-related genes, which contributes to different processes and phenotypes, such as carcinogenesis, proliferation, metabolic reprogramming, epithelial–mesenchymal transition, invasion, and migration, during digestive cancer development. In this review, we focus on the association between histone methylation regulation and the development of digestive cancers, including gastric cancer, liver cancer, pancreatic cancer, and colorectal cancer, as well as on its clinical application prospects, aiming to provide a new perspective on the management of digestive cancers.
Similar content being viewed by others
Introduction
Digestive cancers, including gastric cancer (GC), liver cancer, pancreatic cancer (PC), colorectal cancer (CRC), etc., are commonly observed malignancies in clinical practice. The latest epidemiological data indicate that an estimated 333,680 people will be diagnosed with digestive cancers in the United States in 2020 and that ~167,790 will die of these diseases; both of these estimates rank first among all cancers.1 Although progress has recently been made in the management of digestive cancers, the 5-year survival rate of some cancers remains unsatisfactory—for example, 18% for liver cancer and only 9% for PC.1
Therefore, further study of the molecular mechanism underlying digestive cancers is urgently needed. The interest in epigenetics has increased. Histone modifications, including acetylation, methylation, phosphorylation, and ubiquitylation, are a pivotal form of epigenetic information. Each of these modifications is associated with gene activity, gene silencing, or insulation between active and inactive gene regions.2 For example, the transcription factor FOXA1 drives large-scale enhancer reprogramming through H3K27ac (acetylation of lysine 27 of histone 3) modulation, thereby promoting PC progression.3 An increase in H3K9me2 in the promoter region of RARRES3 gene represses its transcription, thereby contributing to cancer cell migration in hepatocellular carcinoma (HCC).4
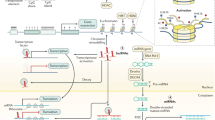
Since extensive studies have recently revealed the key role of histone methylation regulation in the development of digestive cancers, this review focuses on histone methylation. We highlight a cluster of histone methylation modifiers that play a vital role in digestive cancers—KMTs (lysine methyltransferases), KDMs (lysine demethylases), and PRMTs (protein arginine methyltransferases) (Fig. 1). These modifiers cause gene silencing or activation through the methylation or demethylation of related genomic histones, playing a crucial role in aspects of digestive cancer development, such as carcinogenesis, proliferation, metabolic reprogramming, epithelial–mesenchymal transition (EMT), invasion, and migration (Fig. 2). This review aims to discuss the association between the regulation of histone methylation and the development of digestive cancers, including GC, HCC, PC, and CRC, and to consider the underlying mechanism. This knowledge may provide a new perspective on the management of digestive cancers.

Activators or inhibitors of histone methylation modifiers in digestive cancers. During the development of digestive cancers, regulators activate or inhibit histone methylation modifiers (writers and erasers, which add methyl groups to or remove methyl groups from histone lysine residues, respectively), and ultimately regulate the expression of cancer-related genes

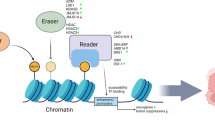
Regulation of the histone methylation of tumor-associated genes during digestive cancer development. Under certain conditions, aberrant methylation or demethylation of the corresponding cancer-related genes contributes to different processes and phenotypes during digestive cancer development, including carcinogenesis, proliferation, metabolic reprogramming, epithelial–mesenchymal transition, invasion, and migration. The blue circles indicate modifications that downregulate gene expression, while the red circles indicate modifications that upregulates gene expression
Main modifiers of histone methylation in digestive cancers
KMTs
KMTs are a group of enzymes that catalyze monomethylation, dimethylation, or trimethylation by adding one, two, or three methyl groups, respectively, from S-adenosyl-l-methionine to the ε-amino group of a histone lysine residue, thus regulating gene expression. For example, H3K27me3 (trimethylation of lysine 27 of histone 3) is often associated with transcriptionally repressed chromatin, and H3K4me3 is often linked to transcriptionally active chromatin. According to their defined protein domain or homologous sequence, KMTs are classified into eight distinct subfamilies: KMT1–8.5
A cluster of KMTs, including the H3K36 methyltransferase KMT3A,6 the H3K9/56 methyltransferase G9a (also called KMT1C),7 the H3K27 methyltransferase enhancer of zeste homolog 2 (EZH2, also called KMT6A),8,9 and the H3K4 methyltransferases KMT3E,10,11 KMT2A,12,13 KMT2D,14 etc., has been found to be ectopic expressed in digestive cancers. Among these KMTs, EZH2, a catalytic subunit of polycomb repressive complex 2 (PRC2),15 is one of the most commonly reported methyltransferases that represses gene expression in digestive cancers via H3K27me.16 EZH2 expression is elevated through the regulatory effects of various microRNAs and lncRNAs (long noncoding RNAs), thereby promoting the development of digestive cancers. For example, EZH2 is recruited by the lncRNA UCA1 and promotes direct interaction with the cyclin D1 promoter to activate the translation of cyclin D1, thereby fueling tumor growth in GC.17 Loss of miR-355 upregulates EZH2 expression by activating Sox4 in PC.18 In addition, loss of miR-26, miR-101, and miR-214 contributes to overexpression of EZH2, leading to the development of digestive cancers.19,20,21,22,23 Among the abovementioned microRNAs, the effect of miR-26 on EZH2 has been widely noted and has been identified in HCC, CRC, and GC.19,20,22 Another study identified that EZH2 can also bind to the lncRNA HOX-AS2 together with KDM1A to form a complex implicated in PC cell proliferation,24 and a similar function can also be performed by the lncRNA HOXA11-AS in GC.25 In addition to being modulated by microRNAs and lncRNAs, the expression of EZH2 is also promoted by cyclin-dependent kinase 4 (CDK4),26 Snail,27 Yin Yang-1 (YY-1),28 proviral integration site 1 (PIM-1) kinases,29 damage specific DNA-binding protein 2 (DDB2),30 nonacetylated homeobox B9 (HOXB9),31 and signal transducer and activator of transcription 3 (STAT3) signaling,32 thus contributing to the malignancy of digestive cancers. However, the activation of EZH2 may not be that straightforward; Battistelli et al. reported that the EMT-inducing transcription factor (EMT-TF) Snail recruits EZH2 to its target gene promoters in a HOTAIR-dependent manner,27 indicating that EZH2 activation is actually quite a complex process needing further exploration. Furthermore, the catalytic activity of EZH2 is not only determined by its own expression but also affected by its cofactors in PRC2. For example, activation of another core protein component of PRC2, embryonic ectoderm development (EED), promotes the catalytic function of EZH2, resulting in increased levels of H3K27me3 in PC cells.33 In addition, other studies have demonstrated that H3K27me3 levels in cancer cells are decreased concomitant with the reduction in the expression of suppressor of zeste 12 (SUZ12 protein homolog, a core component of PRC2), inhibiting the development of HCC and GC.34,35
KMT2A (also called mixed lineage leukemia 1, MLL1) is another histone methyltransferase attracting increasing attention in digestive cancers. KMT2A functions by forming the MWRAD complex with WD repeat domain 5 (WDR5), RBBP5, ASH2L, and DPY30,36 which possesses H3K4 methyltransferase activity and results in the installation of activating marks on chromatin at actively transcribed regions. Analysis of genomic and epigenomic features of HCC showed that p53 mutation led to independent transcriptional activation that could directly regulate a distinct set of chromatin regulators, including KMT2A.37 In addition, KMT2A expression is reduced by a decrease in the level of hPaf1/PD2 level, which decreases H3K4me2/3 and induces a corresponding decrease in the level of chromohelicase DNA-binding protein 1 (CHD1), an ATP-dependent chromatin remodeling enzyme that specifically binds to H3K4me2/3 marks.38 In addition to the influence of KMT2A targeting on MWRAD complex activity, the regulation of other components of the complex also affects its catalytic function.39 Studies have demonstrated that knockdown of WDR5 impacts H3K4 methylation in CRC cells and PC cells.40,41 Activation of WDR5 by the lncRNAs GCAWKR and GClnc1 facilitates H3K4me3 modification of the target genes protein tyrosine phosphatase type IVA (PTP4A1, member 1) and superoxide dismutase 2 (SOD2, mitochondrial) in GC.42,43 Furthermore, the lncRNA HOTTIP promotes HOXA9 expression by binding WDR5 to enhance Wnt/β-catenin pathway signaling, thus augmenting cancer stem cell (CSC) properties in human PC.44 Meanwhile, a study conducted by Mishra et al. has shown that KMT2A promotes leukemogenesis mainly through recruitment of MOF and forming H4K16ac (acetylation of lysine 16 of histone 4), not the intrinsic histone methyltransferase activity of KMT2A. However, the role of this mechanism in digestion cancers still needs further exploration.45
KDMs
KDMs are a class of enzymes that play a role in the demethylation of histone lysines. Similar to KMTs, KDMs are diverse, but recent whole-genome sequencing and copy number analysis identified KDM6A (lysine demethylase 6A) as one of the most frequently altered histone methylation modifiers in digestive cancers.13,46,47,48,49 KDM6A, also called ubiquitously transcribed X (UTX chromosome tetratricopeptide repeat protein), is a Jumonji C (JmjC) domain-containing demethylase that targets H3K27 in complex proteins associated with Set1 (COMPASS), contributing to transcriptional activation.50,51 In solid pseudopapillary neoplasms (SPNs), which are borderline pancreatic tumors, recruitment of KDM6A to the enhancer of the SCL2A1 gene is inhibited due to deletion of BRCA1 associated protein-1 (BAP1),52,53 thus activating SCL2A1 transcription and driving the malignancy of SPNs. In addition to performing its histone methylation function alone, KDM6A can also interact with other histone modification enzymes. For example, KDM6A interacts with the histone acetyltransferase protein CBP and recruits it to the CDH1 (E-cadherin) promoter in HCT-116 CRC cells, resulting in increased H3K27ac, a transcription-activating histone mark.54 Similarly, during PC, KDM6A cooperates with the histone acetyltransferase p300 to regulate gene expression, while loss of KDM6A results in decreased H3K27ac levels in the promoter regions of tumor suppressor genes such as CDKN1A, LOXL1, SASH1, TXNIP, and IGFBP2.55 In addition, another study showed that loss of KDM6A rewires the enhancers and/or activated super-enhancers (SEs) of several tumor-related genes,56 which are formed by clusters of H3K27ac-marked enhancers.57,58
In addition to KDM6A, other KDMs are activated or inhibited under certain conditions, thus playing a corresponding role in digestive cancers. PHD finger protein 5A (PHF5A) K29 acetylation enhances KDM3A expression by stabilizing its mRNA. PHF5A acetylation increases the level of the H3K9me1/2 demethylase KDM3A by reducing its aberrant alternative splicing of its mRNA in CRC, thereby modulating stress responses and carcinogenesis in CRC.59 KDM3A can also be activated by HIF1α via co-activators that bind to hypoxia response elements (HREs) in the doublecortin calmodulin-like kinase 1 (DCLK1) gene under hypoxic conditions in PC;60 DCLK1 is a marker of CSCs, and this process is promoted by protein inhibitor of activated STAT protein 4 (PIAS4).61 RUNX family transcription factor 3 (RUNX3)-mediated upregulation of miR-29b increases its targeting of the KDM2A 3′-UTR and decreases KDM2A expression, which suppresses the proliferation and migration of GC cells.62 Furthermore, KDM4A is activated by loss of miR-137,63 and KDM4C is activated by CircZMYM2 via its sponging of miR-335-5p in PC.64 Both KDM4A and KDM4C target H3K9me2/3, H3K36me2/3, and H3K56me2/3. Sun et al. also found that the lncRNA HOXA11-AS can recruit KDM1A (an H3K4me1/2 and H3K9me1/2 demethylase) and functions as a scaffold for KDM1A, EZH2, and DNA-methyltransferase 1 (DNMT1) in GC, resulting in the progression of cancer development.25 Moreover, KDMs are closely related both to each other and to other histone modification enzymes. For example, Tzatsos et al. found that KDM2B contributes to overexpression of KDM5A and EZH2, while KDM2B interacts with KDM5A and MYC to activate metabolic and ribosomal gene expression and binds EZH2 to silence the expression of lineage specification genes.65
PRMTs
Histone arginine methylation, which is catalyzed mainly by PRMTs, is also important in digestive cancers, but reports on histone arginine methylation are more scarce than those on histone lysine methylation to date. PRMTs can be divided into three categories according to their catalytic activity: type I (PRMT1, PRMT2, PRMT3, PRMT4, PRMT6, and PRMT8), type II (PRMT5 and PRMT9) and type III (PRMT7). All PRMTs use S-adenosylmethionine (SAM) as the methyl donor to catalyze three major forms of arginine methylation on histones: monomethylarginine (MMA), asymmetric dimethylarginine (aDMA), and symmetric dimethylarginine (sDMA).66
Multiple studies have demonstrated that several PRMTs, such as the histone H3 arginine 8 (H3R8) and H4R3 methyltransferase PRMT5,67,68 and histone H3 arginine 2 (H3R2) methyltransferase PRMT6,69,70 are highly expressed in digestive cancer cells. During the development of colon cancer, intestine-specific peroxisome proliferator-activated receptor alpha (PPARa) deficiency promotes PRMT6 expression via the RB1/E2F pathway and increases the enrichment of H3R2 asymmetric dimethylation (H3R2me2a) in the promoter of p27.70 Another study demonstrated that PRMT9 promotes HCC invasion and metastasis by activating PI3K/Akt/GSK‐3β/Snail signaling.71 However, the study did not explain whether this activation was caused by histone methylation of related genes, and this possibility needs further investigation. In addition, PRMTs not only play a role through histone arginine methylation but can also be associated with other histone methyltransferases. For example, recent studies have demonstrated that upregulated PRMT5 can epigenetically silence the expression of the E3 ubiquitin ligase F-box and WD repeat domain-containing 7 (FBW7) by increasing H4R3me2s and H3K9me3 and decreasing H3K9ac in PC,72 indicating that PRMT5 can recruit histone KMTs and histone deacetylases to promote tumor development. This process results in c-Myc stabilization,72 and fibroblast growth factor-binding protein 1 (FGFBP1) is a downstream target of the FBW7/c-Myc axis; thus, the FGFBP1-mediated FGF signaling pathway may be involved in the process described above.73 However, in addition to the histone methylation function of PRMTs, their non-histone methylation function has also received increasing attention. For instance, PRMT3-mediated methylation of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and PRMT4-mediated methylation of malate dehydrogenase 1 (MDH1) are involved in the metabolic reprogramming and proliferation of PC cells.74,75
Histone methylation mediates the development of digestive cancers
Carcinogenesis
Under stimulation by some external or internal factors, the regulation of histone methylation of cancer-related genes in normal digestive system cells may lead to abnormal cell growth and differentiation and thus to the occurrence of digestive cancer.
Hepatitis caused by viruses, alcohol, or fat intake is a major cause of HCC, and histone methylation plays an important role in its progression. Fan et al. demonstrated that in mice, KMT1B-mediated H3K9me3 contributes to nonalcoholic steatohepatitis, which accelerates hepatitis-induced hepatocarcinogenesis. In hepatocytes, KMT1B-mediated H3K9me3 represses Sirt1 transcription, while in macrophages, KMT1B-mediated repression of PPARγ transcription favors the proinflammatory M1 phenotype over the anti-inflammatory M2 phenotype, thereby elevating hepatic inflammation.76 Moreover, chromatin immunoprecipitation (ChIP) assays showed that during hepatitis B virus-induced hepatocarcinogenesis, HBV X protein (HBx) expression decreases the level of the silencing modification H3K27me3, while the level of the activating histone modification H3K4me1 is increased in the promoter of EpCAM, a host gene involved in HBV-mediated hepatocarcinogenesis.34 Similar conclusions were reached in a subsequent study conducted by Zhang et al.77
Regarding gastrointestinal carcinogenesis, researchers have found that KDM4B physically interacts with c-Jun on the promoters of IL-8, MMP1, and ITGAV via its demethylation activity, while infection with Helicobacter pylori significantly increases the occupancy of KDM4B and c-Jun, resulting in a significantly diminished H3K9me3 signal.78 Furthermore, another study identified three H3K27me modifier genes (EZH2, KDM6A, and KDM6B) that are individually associated with GC susceptibility via a synergistic triad interaction.46 During human colorectal carcinogenesis, mutations in Wnt/β-catenin signaling mediators may be one of the earliest events that initiates and drives tumor progression.79 Upregulation of disheveled segment polarity protein 22 (DVL), resulting from a significant decrease in H3K36me3 in the absence of KMT3A, leads to enhancement of Wnt/β-catenin signaling pathway activity and thereby drives colorectal carcinogenesis. Moreover, another study revealed the decrease in H3K27me3 and the increase in H3K4me3 in the WNT3 promoter region, indicating that histone methylation directly activates the Wnt/β-catenin signaling pathway to promote the initiation of CRC.80
In the pancreas, histone methylation plays a crucial role in the growth and differentiation of pancreatic cells even under physiological conditions. Studies have shown that KDM6A/B are crucial in endoderm differentiation in the pancreas.81,82 At the early stage of definitive endoderm differentiation, KDM6A/B promote mesoderm differentiation by demethylating H3K27me3, which upregulates WNT3 expression and activates the WNT signaling pathway. At the late stage, KDM6A/B demethylates H3K27me3 to activate the WNT antagonist DKK1 in order to suppress the WNT signaling pathway and promote endoderm differentiation over mesoderm differentiation.82 However, under some conditions, such as injury to the pancreas or chronic inflammation, loss of EZH2 results in impaired pancreatic regeneration and accelerates KrasG12D-driven neoplasia.83 Further studies showed that EZH2 is upregulated in injured pancreatic cells, while the presence of KRAS reverses the function of EZH2 during late-stage regeneration, thus sustaining the expression of nuclear factor of activated T cells 1 (NFATc1), a crucial driver of pancreatic carcinogenesis and progression, through a decrease in H3K27me3 and increase in H3K4me3.84 This event integrates signals from inflammatory networks to control cellular plasticity and counteracts TGFβ-mediated apoptosis induction.85,86,87
Acinar-to-ductal metaplasia (ADM), which is essential for PC carcinogenesis, is a reprogramming event in which acinar cells display marked cellular plasticity and transiently transform into progenitor-like cells exhibiting a ductal morphology in pancreatitis.88,89 This process, in conjunction with the expression of oncogenes such as K-ras, may trigger the development of PC.90 ChIP-seq data showed a significant gain of H3K27me3 and H2AK119ub (ubiquitination of lysine 119 of histone 2) at the regulatory acinar cell fate genes RBPJL, PTF1A, NR5A2, and BHLHA15 in pancreatic cells that had undergone ADM, and additional significant loss of H3K4me3 on these genes was found in PC cells.91 In addition, H3K4me3 on the Sox9 promoter increases as a result of NFATc1 activation during the ADM process, resulting in elevated expression of Sox9, a downstream effector of oncogenic Kras, and accelerates the formation of premalignant lesions in the pancreas.92,93
Proliferation
Uncontrolled proliferation is an important characteristic of tumor cells. Under appropriate conditions, cancer cells can multiply indefinitely and become immortalized.94 Histone methylation plays a key role in the uncontrolled proliferation of digestive cancer cells. For example, repressive H3K27me3 marks installed by EZH2 are enriched in the Kruppel-like factor 2 (KLF2) promoter, leading to a growth advantage in the tumor cell population in GC and CRC,95,96 via the HIF-1α/Notch-1 signaling pathway and the Hedgehog pathway.97,98 In addition, the proliferation of CRC cells is significantly accelerated by the EZH2-mediated H3K27me3 modification of the dual specificity phosphatase 5 (DUSP5) gene,99 a negative regulator of the mitogen-activated protein kinase (MAPK)-signaling pathway.100
Cell cycle dysregulation
Abnormalities in cell cycle network are closely related to the proliferation of tumor cells. During the development of digestive cancers, histone methylation can regulate cell cycle by regulating cyclins, CDKs cyclin-dependent kinase inhibitors (CKIs), thus causing cell cycle dysregulation and uncontrolled cell proliferation. Previous experiments with human pancreatic cell lines showed that KMT2D silencing leads to a decrease in the number and proportion of cells in G0/G1, accompanied by a global reduction in H3K4me1/2/3,101 suggesting that histone methylation is indeed involved in cell cycle regulation. Further studies have focused mainly on the regulation of CKIs.
The genes encoding P15 and P21, two commonly mentioned CKIs, exhibit increased levels of H3K27me3 and H3K9me3 and decreased levels of H3K4me2/3 in digestive cancers, including GC,102,103,104 CRC,105,106 HCC,107,108 and PC.109 This process, triggered by upstream lncRNAs such as BLACAT1, SNHG17 and CASC15, can inhibit the expression of P15 and P21 and lead to G0/G1 checkpoint deficiency.110,111,112 In addition, Zhang et al. found that EZH2-mediated H3K27me3 downregulates the expression of the p57 gene in addition to p15 gene, contributing to the regulation of the GC cell cycle and GC cell proliferation.113 In addition, TWIST recruits EZH2 to induce H3K27me3, which represses the transcription of the p16 gene by binding to the E-box in its promoter in PC cells under hypoxic conditions,114 and the same modification can be initiated by the lncRNA PVT1 in GC.115 P27kip1 is similarly inhibited by EZH2-mediated H3K27me3; the only difference is that EZH2 binds and modifies exon 1 rather than the promoter region.116 Another study suggested that EZH2 induced by KRAS mutation installs H3K27me3 in the promoter of miR-34a, thus epigenetically inactivating miR-34a expression to suppress the effects of p53 transactivation.117
Similarly, histone methylation also regulates cyclin and CDK in digestive cancer cells. According to recent studies, Jumonji domain-containing protein Jumonji domain-containing protein 6 (6JMJD6), a histone arginine demethylase, activates CDK4 expression by decreasing the H4R3me2s level in the CDK4 promoter in HCC.118 In addition, the transcription of CDK2 is promoted by KMT3E-mediated increases in H3K4me3 enrichment at the corresponding promoter sites.119 Regarding cyclins, the level of cyclin D1 is significantly increased due to decreases in repressive H3K9me2 marks and increases in activating H3K4me3 marks installed by KDMA/B and KMT2A, respectively.120 In addition, upregulation of Forkhead box transcription factor M1 (FOXM1) by KMT4-induced H3K79me2 in PC significantly increases the expression of its classical target genes cyclin A2 and cyclin B1 through the Wnt5a signaling pathway.121
Immune escape
In addition to the uncontrolled proliferation caused by cell cycle dysregulation, abnormal negative regulatory mechanisms of cell proliferation, especially immune escape, can also cause abnormal proliferation during the development of digestive cancers. For example, researchers have demonstrated that during PC, H3K4me3 in the BCL2L1, CFLAR, and MCL-1 gene promoters upregulates the expression levels of the anti-apoptotic proteins Bcl-x, FLIP, and Mcl-1 and that H3K9me3 in the BAK1, BAX, and BCL2L11 gene promoters downregulates the expression levels of the pro-apoptotic proteins Bak, Bax, and Bim.122 All six of these apoptosis-regulating genes are involved in PC growth and progression.123,124,125,126 In addition, a genome-wide ChIP-sequencing analysis identified that H3K9me3 is enriched in the FAS promoter in metastatic human colon carcinoma cells; this enrichment inhibits the expression of Fas and results in resistance to Fas-mediated apoptosis.127 Furthermore, EZH2-mediated H3K27me3 by the lncRNA HOTAIR in the death receptor 5 (DR5) gene promoter results in resistance to TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis;128 the same modification is found in the proapoptotic genes growth differentiation factor 15 (GDF15) and KLF2.129,130
In addition to apoptosis resistance, the production of the Th1-type chemokines CXC chemokine ligand 9 (CXCL9) and CXCL10, mediators of effector T cell trafficking, is suppressed by H3K27me3 in their gene promoters in colon cancer.131 However, the expression of CXC chemokine receptor 4 (CXCR4) is promoted by EZH2-mediated loss of miR-622,132 and CXCR4 can facilitate the evasion of immune surveillance by binding to CXCL12.133 Another experiment conducted by Bugide et al. showed that overexpressed EZH2 associates directly with the promoters of natural killer (NK) cell ligands, such as ULBP1 and MICA in HCC cells and then catalyzes the installation of repressive H3K27me3 marks on these promoters.134 In addition, Zhou et al. found that in PC, upregulation of FOXM1 by KMT4-induced H3K79me2 significantly attenuates antitumor responses, including bone marrow-derived dendritic cell (BMDC) maturation, cytokine secretion, and T cell activation, through the Wnt5a signaling pathway.121 Moreover, in PC cells, KMT2A overexpression increases the H3K4me3 level in the CD274 promoter region and upregulates the expression of PD-L1,135 a T cell inhibitory receptor ligand that results in potent immunosuppression.136
Metabolic reprogramming
To support the ability for uncontrolled proliferation, tumor cells have adapted many aspects of their metabolic patterns. One of the most well-known examples is the Warburg effect, through which the rate of aerobic glycolysis is increased in cancer cells, thus leading to decreased glucose oxidation and enhanced flux through the anabolic side branches of glycolysis.137 Similarly, the Warburg effect can be induced in digestive cancer cells through histone methylation or demethylation of related genes. Glucose transporter 1 (GLUT1), one of the major components regulating glucose homeostasis,138 can be activated by p-ERK/KDM4B-mediated removal of the repressive H3K9me3 mark, thus contributing to glucose uptake in colon cancer cells under glucose deprivation conditions.139 KMT2D inhibition mediates the loss of H3K4me3 in metabolic pathway genes, which results in alterations in aerobic glycolysis and lipid levels via GLUT3-mediated processes.140 As GLUT3 exhibits a high affinity for glucose, thus ensuring efficient glucose uptake into cancer cells,141 this process induces aerobic glycolysis and increases the levels of lipids, such as docosadienoic acid, docosatrienoic acid, and docosatetraenoic acid, which harbors oncogenic properties in PC cells.140 In addition, Sakamoto et al. demonstrated that H3K4me2 demethylation mediated by KDM1A overexpression suppresses the expression of two metabolism-related genes, PGC-1α and LCAD, which are involved in the transcriptional control of genes related to mitochondrial oxidative metabolism and fatty acid oxidation, respectively.142 Similar modifications can occur on the glyceronephosphate O-acyltransferase (GNPAT) gene to activate its transcription; the only difference is that this process is triggered by c-Myc. GNPAT recruits the enzyme USP30, which deubiquitylates and stabilizes dynamin-related protein 1 (DRP1), thus promoting dysregulation of mitochondrial morphology and lipid metabolism in HCC.143 Moreover, KDM5A-mediated H3K4me3 demethylation of the mitochondrial pyruvate carrier 1 (MPC-1) gene transcriptionally inhibits its expression, leading to elevated mitochondrial pyruvate metabolism and inhibition of glycolysis. As a result, a series of metabolic disorders occur, such as hyperpyruvatemia and lactic acidosis.144 In addition, another study confirmed that cooperative binding of KDM2B, KDM5A, and/or MYC activates the expression of genes involved in metabolic homeostasis and protein synthesis, such as ATP5G2 and RPL3/7/24/30, via enrichment of H3K4me3 on these genes.65
Regulation of histone methylation can affect metabolism in digestive cancer cells, and the metabolites in digestive cancer cells can reciprocally affect histone methylation. For example, FH (fumarase), which catalyzes the reversible hydration and dehydration of fumarate, can bind to the c-Jun gene promoter, inhibiting KDM2A activity through local fumarate production and promoting H3K36me2.145 In addition, Oliver et al. demonstrated that 6-aminonicotinamide (6AN), a 6-phosphogluconate dehydrogenase (PGD) inhibitor, induces broad enrichment of H3K9me2 across the CDH2 (N-cadherin) and topoisomerase 2β (TOP2B) genes, causing distant metastasis of PC.146
Interestingly, collectively, these studies suggest that histone methylation and metabolic reprogramming may form a positive feedback loop to promote digestive cancer progression. For instance, Yi et al. reported that HIF1a stabilization under hypoxic conditions leads to KDM1A upregulation at the mRNA and protein levels in PC,147 while progression of PC induces a hypoxic microenvironment through the Warburg effect and results in the production of a large amount of HIF1.148,149 A recent study conducted by Jiang et al. in CRC also supported this conclusion. O-linked N-acetylglucosamine transferase (OGT), which is negatively regulated by miR-101, enhances EZH2 protein stability and function and promotes the enrichment of repressive H3K27me3 marks on the miR-101 promoter region, resulting in the upregulation of OGT.150 This process creates a vicious cycle and further exacerbates tumor progression.
EMT
EMT is an important pathophysiological event that results in the loss of cell–cell adhesion, abnormal apical–basal polarity, and cytoskeletal reorganization, thereby enabling polarized, immotile epithelial cells to acquire mesenchymal abilities such as invasiveness and motility.151 Accumulating data have indicated that EMT leads to enhanced cell invasion and migration in digestive cancers.152,153 Moreover, the results of a tag and track experiment in pancreatic epithelial cells conducted by Rhim et al. indicated that EMT can occur even before the formation of cancer, showing that pancreatic epithelial cells can invade and enter the bloodstream to become circulating epithelial cells (CECs), maintaining the mesenchymal phenotype in pancreatic intraepithelial neoplasia (PanIN), a common precancerous lesion of PC.154 Several histone methylation modifications are involved in EMT in digestive cancers, especially EZH2-mediated H3K27me3 on the E-cadherin gene promoter. For example, Zhou et al. found that loss of ten-eleven translocation 1 (TET1) activates EZH2, which represses E-cadherin gene transcription by catalyzing H3K27me3 modification in the E-cadherin promoter in CRC cells.155 This modification leads to a decline in the expression of E-cadherin, a well-known hallmark of EMT.156 The same CDH1 gene promoter modification in CRC was also found in two other studies; one study showed that this modification was triggered by the lncRNA SNHG622 and the other showed that it was induced by the EMT-TF Snail2.157 During PC, EZH2 can also be recruited by TWIST and loss of microRNA-101, which binds to one or more E-boxes in the E-cadherin gene promoter and increases repressive H3K27me3 marks.114,158 In addition, EZH2-mediated H3K27me3 can regulate EMT indirectly through microRNAs and lncRNAs. EZH2 epigenetically silences miR-34a through H3K27me3 enrichment in the miR-34a promoter region. Downregulation of miR-34a in turn activates c-Met and thus results in transcriptional activation of Snail, thus contributing to the EMT process in GC cells and accelerating tumor metastasis.159 Another study demonstrated that miR-34a, along with miR-203, is repressed by EZH2-mediated H3K27me3, thereby activating the Snail1 and Snail2 EMT-TFs to downregulate E-cadherin.160 MiR-139-5p transcription is inhibited by EZH2 through an increase in H3K27me3; therefore, the levels of the EMT-TFs ZEB1 and ZEB2 increase, while the E-cadherin level decreases.161
In addition to EZH2-mediated H3K27me3, other histone methylation modifications are involved in regulating EMT in digestive cancers. Loss of KDM6A negatively regulates E-cadherin expression by coordinating the regulation of H3K27 methylation in the E-cadherin promoter in HCT-116 colon cancer cells, switching the transcriptionally active state to a transcriptionally repressive state.54 In addition, G9a-mediated upregulation of H3K9me2, a heterochromatic histone methylation marker, has been observed at the same site in HCC cells and results in further inhibition of E-cadherin expression.162,163 Moreover, recent studies showed that ChIP assays can detect decreases in H3K9me3 and increases in H3K4me3, which are catalyzed by KDM4B and KMT7, respectively, in the ZEB1 promoter region.164,165 Meanwhile, KMT5C silences expression of the mesenchymal-to-epithelial transition (MET)-promoting transcription factors FOXA1, OVOL1, and OVOL2 via its repressive mark H4K20me3 in PC.166 Furthermore, PRMT1 can bind to the promoter region of CTNNB1 and increase the β-catenin protein level in PC cells,167 but whether this effect is related to histone methylation needs further exploration.
Invasion and migration
Invasion and migration of cancer cells into the surrounding tissue and vasculature is an essential initial step in cancer metastasis, which contributes to the terminal stages of cancer. During this process, digestive cancer cells invade deep tissue and enter lymphatic and blood vessels for dissemination into the circulation via regulation of histone methylation. This dissemination enables the cancer cells to colonize distant organs.
For example, decreases in H3K27me3 in the MMP7 and HMGA2 promoter regions promote the progression of PC in Brg1-depleted PC cells,168 as MMP7 facilitates tumor cell invasion and HMGA2 promotes invasion and migration through EMT.169,170 Moreover, two other MMP family proteins, MMP9 and MMP14, are also activated during the development of digestive cancers. The level of H3K9me3 in the MMP9 promoter is markedly decreased,171 while MMP14 exhibits robust accumulation of H3K4me3 and loss of H3K27me3.91
In addition to MMP family proteins, researchers have also found that ectopic expression of a cluster of genes that participate in tumor invasion and metastasis in the digestive system is associated with abnormal regulation of histone methylation. For instance, the invasion and migration abilities of cancer cells are promoted by H3K9me3-mediated epigenetic silencing of numerous related genes, such as IGFBP3, CXCL3, NOS3, and SLIT1.172,173 The opposite modification, demethylation of H3K9me3, occurs in the promoter of MALAT1, thereby upregulating the expression of MALAT1 and enhancing the activity of the β-catenin signaling pathway.174 In addition to H3K9 modification, decreases in H3K4me3 in BMP7, WIF1, and TIMP2/3 also significantly enhance the invasion and migration abilities of cancer cells in HCC and CRC,175,176 thus accelerating tumor progression. H3K27me3 also participates in this process. Tang et al. proved that aberrant KDM6B expression decreases H3K27me3 in the SLUG gene promoter and activates the transcription of SLUG, which promotes migration, invasion, and stem cell-like behaviors in HCC.177 Similar modifications can be detected in the Wnt10b promoter region in HCC; these modifications activate the Wnt/β-catenin signaling pathway and reinforce cell proliferation, migration, and invasion.178 Moreover, PRMT6-overexpressing GC cells also acquire invasiveness through direct transcriptional inhibition of PCDH7 by increasing H3R2me2as level.179 Another study conducted by Li et al. demonstrated that H3K27me3 and H3K9me2, mediated directly and indirectly by EZH2, are enriched in the miR-218-2 promoter, thus leading to downregulation of gene expression in PC. Loss of miR-218 increases invasion and migration through the effect of the UDP-glycosyltransferase 8 (UGT8) protein.180 In addition to EZH2, KDM6B is also involved in the processes of invasion and migration. Loss of this histone demethylase enhances the invasion and migration of PC cells through downregulation of CCAAT-enhancer-binding protein alpha (CEBPA); mechanistically, loss of KDM6B induces H3K27me3 in the region upstream of the CEBPA transcription start site.181
However, conflicting results have been obtained. For example, inconsistent with the above results, GDF15, a migration-promoting factor,182 was found to be downregulated in PC cells via a lncRNA HOTAIR-induced increase in the H3K27me3 level in its promoter.129 Moreover, researchers have proven that EZH2-mediated H3K27me3 suppresses RUNX3 gene expression in PC and GC.183,184 Notably, RUNX3 has traditionally been considered a tumor suppressor, but an article published in Cell in 2015 reported that it had a reinforcing effect on metastatic seeding and colonization via extracellular matrix (ECM) remodeling in PC.185 Therefore, in this case, inhibition of this suppressor reduces the malignant behavior of digestive cancer cells rather than promoting tumor development, indicating that EZH2 can function as both an oncogene and a tumor suppressor.
Clinical application of histone methylation in digestive cancers
As mentioned above, the regulation of histone methylation has substantial clinical potential, especially in treatment strategies for digestive cancers. Currently, radical surgical resection is the most effective treatment. However, the prognosis of digestive cancers is still not very optimistic. In PC, for example, fewer than 20% of patients have the opportunity for surgery, and of those who undergo resection followed by adjuvant therapies, more than 80% relapse and ultimately die of their disease.186 Moreover, the lack of effective drugs for chemotherapy and targeted therapy and resistance to existing drugs are other important reasons for the dismal prognosis. For these reasons, discovering effective drug therapies for affected patients is of paramount importance. As discussed above, regulation of histone methylation plays a pivotal role in digestive cancer development. Thus, strategies that regulate histone methylation modifiers and thus control histone methylation may constitute potential treatments for digestive cancers.
Histone methylation as an adjuvant to reverse drug resistance
Chemotherapy and targeted therapy are still the first-line treatment modalities for digestive cancers, including HCC,187 GC,188 CRC, and PC,189,190,191 but their therapeutic effect differs widely across patients. Studies have revealed that regulation of histone methylation plays an important role in determining the efficacy of and resistance to chemotherapy and targeted therapy;192,193 EZH2-mediated H3K27 methylation and KDM1A-mediated H3K4/9 demethylation16,194 are especially important. Lima-Fernandes et al. demonstrated that inhibition of the H3K27 methyltransferase EZH2 by UNC1999 resulted in increased sensitivity to 5-fluorouracil in CRC cells, accompanied by downregulation of H3K27me3 in the promoter of Indian Hedgehog and decreased self-renewal of CRC-initiating cells.195 In addition, metformin decreases the gene expression of EZH2 in pancreatospheres derived from gemcitabine-resistant PC cells, thus increasing the sensitivity of PC cells to gemcitabine via targeted killing of CSCs.196 Two other types of agents, lactate dehydrogenase A (LDHA) inhibitors and curcumin, have also recently been shown to display synergistic cytotoxic activity with gemcitabine in PC; the underlying mechanism is at least partially mediated by inhibition of EZH2.197,198
Suppression of KDM1A might be an attractive target for regorafenib sensitization and clinical HCC therapy, but an in-depth mechanistic investigation is lacking.199 Further research proved that KDM1A inhibitors, such as pargyline and GSK2879552, dramatically suppress the stem-like properties of sorafenib-resistant HCC cells, including resensitization to sorafenib. Mechanistically, these KDM1A inhibitors negatively regulate the Wnt/β-catenin pathway through enrichment of activating H3K4me1/2 marks in the promoter of the Wnt antagonists Prickle1, APC and Sfrp5.200 This finding is consistent with the results reported by Lei et al., which indicated that knockdown of KDM1A sensitizes HCC cancer cells to cisplatin and sorafenib by increasing the expression of several suppressors of β-catenin signaling, especially Prickle1 and APC.201
In addition to studies on targeting EZH2 and KDM1A, recent studies have demonstrated that small molecule inhibitors of KMT-4 (EPZ5676) combined with 5-fluorouracil or PARP inhibitors, which are used to treat CRCs, show additive effects accompanied by decreased H3K79me3 levels.202 Moreover, the G9a inhibitor UNC0638 notably enhances the cytotoxicity of topoisomerase-based treatment in CRC; mechanistically, H3K9me2 in the PP2A promoter is decreased, thus activating the PP2A–RPA axis.203 Verticillin A is a selective HMTase inhibitor that inhibits SUV39H1, SUV39H2, G9a, GLP, NSD2, and KMT2A, which decreases the H3K9me3 level in the FAS promoter and restores Fas expression, thus alleviating 5-fluorouracil resistance in CRC. Verticillin A is less toxic but exhibits greater efficacy than decitabine and vorinostat.127 Lu et al. proved that a sublethal dose of verticillin A effectively overcomes the resistance of human PC cells to gemcitabine and suppresses tumor growth.122 Furthermore, verticillin A was recently found to have a synergistic effect with anti-PD-L1 therapy in PC.135
Histone methylation as a therapeutic target
In addition to reversing the effects of chemotherapeutic drug resistance, histone methylation can also constitute a direct therapeutic target in digestive cancers. In this regard, inhibition of EZH2-mediated H3K27me and G9a-mediated H3K9me/H3K56me are the most widely reported strategies. In fact, EZH2 inhibitors have even been identified as cancer prevention drugs. A recent study showed that suppressing EZH2 activity with GSK343 ameliorated experimental intestinal inflammation and delayed the onset of colitis-associated cancer accompanied by a gradual loss of H3K27me3 expression.204 DZNep (3-deazaneplanocin A), a potent chemical inhibitor of S-adenosylhomocysteine hydrolase that modulates chromatin accessibility through inhibition of histone methyltransferases, including EZH2,205 can lead to a significant reduction in H3K27me3 with a marked reduction in cell proliferation and migration in CRC. Similar effects can also occur in PC, decreasing the global H3K27me3 level and subsequently causing reexpression of miR-218, thus inhibiting cell proliferation, promoting apoptosis, and inducing cell cycle arrest in PC cells.180 A later study found that DZNep significantly alters miR-663a and miR-4787-5p expression and suppresses TGFb1-induced EMT signaling in PC.206 GSK126 is another EZH2 inhibitor that leads to epigenetic reprogramming in cancer cells, thus successfully promoting the infiltration of functional CD8þ T cells and substantially suppressing HCC growth.207 In addition, Huang et al. reported that suppressing EZH2-mediated H3K27me3 with GSK126 results in increased numbers of myeloid-derived suppressor cells (MDSCs) and decreased numbers of CD4+ and IFN-γ+ CD8+ T cells, which are closely related to antitumor immunity in CRC.208 UNC1999, an EZH2-specific inhibitor, not only reduced the aberrant H3K27 methylation that characterizes PC cells but also slowed the proliferation of cancer cells in three model systems.209 In addition, chaetospirolactone has been demonstrated to suppress the activity of the epigenetic regulator EZH2 and consistently decrease H3K27me3 to allow the transcription of DR4,210 which binds to TRAIL and leads to activation of the initiator caspase-8 and assembly of the death-inducing signaling complex (DISC).211 Subsequently, diosgenin, garcinol, FBW7, and the curcumin analog CDF were also identified as potential agents targeting EZH2 and thus hindering the development of PC.212,213,214,215
Inhibition of not only EZH2 but also G9a counteracts the development of digestive cancers. For example, pharmacological knockdown of G9a activity triggers an increase in autophagy by decreasing histone H3K9me2 and increasing histone H3K9ac, which leads to elevated expression of autophagy genes, such as LC3B, WIPI1, DOR, and p62, in PC cells, ultimately decreasing cell viability.7 Another study conducted by Kim et al. showed similar results in GC cells, while G9a inhibition in GC was mediated by kaempferol.216 A novel G9a inhibitor, BRD4770, was discovered to reduce cellular levels of H3K9me2 and H3K9me3 without inducing apoptosis, to induce senescence through ATM pathway activation, and to inhibit proliferation in the PC cell line PANC-1.217,218 Moreover, recent research has led to innovations in drug administration. Nanodiamond-mediated delivery of the G9a inhibitor UNC0646 maintained the biological functionality of UNC0646, with improved efficacy in reducing H3K9 methylation as well as enhancing the suppression of invasion in HCC cells,219 laying the foundation for future administration of small molecule histone methylation-regulating drugs. Similarly, dual targeting of G9a and DNMT1 by compounds such as CM-272 has also been proposed, thus providing a new perspective on HCC treatment.220
In addition to EZH2 and G9a, the novel curcumin analog L48H37 downregulates the histone methyltransferase KMT2D, as shown in a recent study, and KMT2D deficiency augments L48H37-induced apoptosis and blocks migration,221 thus forming a positive feedback loop. Moreover, combination treatment with the pan-H3K9me HMT inhibitor chaetocin and an aurora kinase A (AURKA) inhibitor diminishes H3K9 methylation at centromeres, induces mitotic aberrations, triggers an abnormal mitotic checkpoint response, and ultimately leads to mitotic catastrophe in PC.222 At the same time, the prospect of arginine methylation as a therapeutic target has been revealed gradually. For example, a recent study found that targeting PRMT5 activity by DW47800 inhibits the malignancy of HCC by downregulating the binding of H4R3me2s to the HNF4α promoter.223 Meanwhile, AMI-1, a small molecule inhibitor of PRMTs, was demonstrated to strongly inhibit proliferation and migratory activity in HCC and GC, along with a decreased expression levels of H4R3me2s and H3R8me2s.224,225
Conclusion
As stated above, increasing attention has been focused on the role of histone methylation in the development of digestive cancers and its potential in clinical application. However, current studies still have limitations. For example, EZH2 has been found to act as both an oncogene and a tumor suppressor, since it maintains, rather than specifies, the transcriptional repression state of thousands of target genes. EZH2 acts most often as a mediator in tumor development; thus, it may play different roles under different conditions.226 Furthermore, the mechanism by which histone methylation regulates gene expression is still controversial. In contrast to the traditional understanding, some researchers believe that H3K4me3 may be not the “cause” but rather the “effect” of upregulated gene expression. Demethylation of CpG islands may shape the distribution of H3K4me3, and, in turn, H3K4me3 may influence the chromatin landscape at CpG islands.227,228 Therefore, the detailed mechanism linking H3K4me3 to upregulation of gene expression remains to be explored.
In addition, most current studies have focused on histone lysine methylation, and the role of histone arginine methylation has been relatively neglected. The crosstalk between histone arginine methylation and lysine methylation is also important, and may play important roles in the development of digestive cancers. Since histone methylation can regulate gene expression, histone lysine methylation may occur on histone arginine methyltransferase genes to regulate histone arginine methylation, and vice versa. Demetriadou et al. reported that activating H3K4me3 marks are reduced but repressive H3K27me3 marks are significantly enriched in the PRMT5 gene promoter in HCT116 cells with depletion of N-alpha-acetyltransferase 40 (NAA40).229 Another study proved upregulation of PRMT5 induces increasing H4R3me2s and H3K9me3 in FBW7 gene in PC, indicating that PRMT5 can recruit histone KMTs and histone deacetylases to promote tumor development. These patterns suggest that interaction indeed occurs between histone arginine methylation and lysine methylation. Such interaction and internal mechanisms should be further investigated, such as the event upstream of this action and explanation of how histone modifiers are recruited. To broaden the research scope, the interactions among histone methylation, histone acetylation, DNA methylation, and other epigenetic modifications should also be given full attention.
Moreover, although new therapeutic targets and molecular mechanisms are being revealed, only a modest number of clinical trials have been conducted (Table 1). Furthermore, to date, clinical trials have focused primarily on the effects of histone methylation modifier inhibitors on hematologic malignancies, such as B-cell lymphoma and non-Hodgkin lymphoma. Most of the relevant clinical trials on digestive cancers are in the initial stages, most commonly phase 1 and phase 2. Moreover, clinical trials of EZH2 antagonists are far more numerous than those of other histone modification modifier inhibitors, such as tazemetostat and CPI-1205. Future studies should devote more attention to these aspects to identify novel treatment options for digestive cancers.
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer statistics, 2020. CA: Cancer J. Clin.70, 7–30 (2020).
Feinberg, A. P. The key role of epigenetics in human disease prevention and mitigation. N. Engl. J. Med.378, 1323–1334 (2018).
Roe, J. S. et al. Enhancer reprogramming promotes pancreatic cancer metastasis. Cell170, 875–888 e820 (2017).
Wei, L. et al. Histone methyltransferase G9a promotes liver cancer development by epigenetic silencing of tumor suppressor gene RARRES3. J. Hepatol.67, 758–769 (2017).
Allis, C. D. et al. New nomenclature for chromatin-modifying enzymes. Cell131, 633–636 (2007).
Vargas-Parra, G. M. et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic analysis. Int. J. Cancer141, 1365–1380 (2017).
Artal-Martinez de Narvajas, A. et al. Epigenetic regulation of autophagy by the methyltransferase G9a. Mol. Cell. Biol.33, 3983–3993 (2013).
Chen, Y. et al. RNAi targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Cancer Lett.297, 109–116 (2010).
Gan, L. et al. The polycomb group protein EZH2 induces epithelial–mesenchymal transition and pluripotent phenotype of gastric cancer cells by binding to PTEN promoter. J. Hematol. Oncol.11, 9 (2018).
Fu, L. N., Tan, J., Chen, Y. X. & Fang, J. Y. Genetic variants in the histone methylation and acetylation pathway and their risks in eight types of cancers. J. Dig. Dis.19, 102–111 (2018).
Wang, T. et al. SMYD3 controls a Wnt-responsive epigenetic switch for ASCL2 activation and cancer stem cell maintenance. Cancer Lett.430, 11–24 (2018).
Sausen, M. et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun.6, 7686 (2015).
Waddell, N. et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature518, 495–501 (2015).
Chakrabarty, S. et al. Targeted sequencing-based analyses of candidate gene variants in ulcerative colitis-associated colorectal neoplasia. Br. J. Cancer117, 136–143 (2017).
Qi, W. et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol.13, 381–388 (2017).
van Vlerken, L. E. et al. EZH2 is required for breast and pancreatic cancer stem cell maintenance and can be used as a functional cancer stem cell reporter. Stem Cells Transl. Med.2, 43–52 (2013).
Wang, Z. Q. et al. Long noncoding RNA UCA1 induced by SP1 promotes cell proliferation via recruiting EZH2 and activating AKT pathway in gastric cancer. Cell Death Dis.8, e2839 (2017).
Hasegawa, S. et al. A crucial epithelial to mesenchymal transition regulator, Sox4/Ezh2 axis is closely related to the clinical outcome in pancreatic cancer patients. Int. J. Oncol.48, 145–152 (2016).
Zhang, X. et al. MicroRNA-26a is a key regulon that inhibits progression and metastasis of c-Myc/EZH2 double high advanced hepatocellular carcinoma. Cancer Lett.426, 98–108 (2018).
Rossi, T. et al. E-cadherin downregulation and microRNAs in sporadic intestinal-type gastric cancer. Int. J. Mol. Sci.20, https://doi.org/10.3390/ijms20184452 (2019).
Chen, D. L. et al. Long non-coding RNA XIST regulates gastric cancer progression by acting as a molecular sponge of miR-101 to modulate EZH2 expression. J. Exp. Clin. Cancer Res.35, 142 (2016).
Xu, M. et al. lncRNA SNHG6 regulates EZH2 expression by sponging miR-26a/b and miR-214 in colorectal cancer. J. Hematol. Oncol.12, 3 (2019).
Nakahara, O. et al. Carcinogenesis of intraductal papillary mucinous neoplasm of the pancreas: loss of microRNA-101 promotes overexpression of histone methyltransferase EZH2. Ann. Surg. Oncol.19, 565–571 (2011).
Lian, Y. et al. The lncRNA-HOXA-AS2/EZH2/LSD1 oncogene complex promotes cell proliferation in pancreatic cancer. Am. J. Transl. Res.9, 5496–5506 (2017).
Sun, M. et al. LncRNA HOXA11-AS promotes proliferation and invasion of gastric cancer by scaffolding the chromatin modification factors PRC2, LSD1, and DNMT1. Cancer Res.76, 6299–6310 (2016).
Le Duff, M. et al. Regulation of senescence escape by the cdk4-EZH2-AP2M1 pathway in response to chemotherapy. Cell Death Dis.9, 199 (2018).
Battistelli, C. et al. The Snail repressor recruits EZH2 to specific genomic sites through the enrollment of the lncRNA HOTAIR in epithelial-to-mesenchymal transition. Oncogene36, 942–955 (2017).
Tsang, D. P. et al. Yin Yang 1-mediated epigenetic silencing of tumour-suppressive microRNAs activates nuclear factor-kappaB in hepatocellular carcinoma. J. Pathol.238, 651–664 (2016).
Xu, J. et al. PIM-1 contributes to the malignancy of pancreatic cancer and displays diagnostic and prognostic value. J. Exp. Clin. Cancer Res.35, 133 (2016).
Huang, S. et al. DDB2 is a novel regulator of Wnt signaling in colon cancer. Cancer Res.77, 6562–6575 (2017).
Song, J. et al. HOXB9 acetylation at K27 is responsible for its suppression of colon cancer progression. Cancer Lett.426, 63–72 (2018).
Pan, Y. -M. et al. STAT3 signaling drives EZH2 transcriptional activation and mediates poor prognosis in gastric cancer. Mol. Cancer15, https://doi.org/10.1186/s12943-016-0561-z (2016).
Pandya, P. et al. PICOT binding to chromatin-associated EED negatively regulates cyclin D2 expression by increasing H3K27me3 at the CCND2 gene promoter. Cell Death Dis.10, 685 (2019).
Zhang, H. et al. PLK1 and HOTAIR accelerate proteasomal degradation of SUZ12 and ZNF198 during Hepatitis B virus-induced liver carcinogenesis. Cancer Res.75, 2363–2374 (2015).
Wu, X. et al. Ubiquitin-specific protease 3 promotes cell migration and invasion by interacting with and deubiquitinating SUZ12 in gastric cancer. J. Exp. Clin. Cancer Res.38, 277, https://doi.org/10.1186/s13046-019-1270-4 (2019).
Smith, E., Lin, C. & Shilatifard, A. The super elongation complex (SEC) and MLL in development and disease. Genes Dev.25, 661–672 (2011).
Ding, X. et al. Genomic and epigenomic features of primary and recurrent hepatocellular carcinomas. Gastroenterology157, 1630–1645 e1636 (2019).
Dey, P., Ponnusamy, M. P., Deb, S. & Batra, S. K. Human RNA polymerase II-association factor 1 (hPaf1/PD2) regulates histone methylation and chromatin remodeling in pancreatic cancer. PLoS ONE6, e26926 (2011).
Shen, S. M. et al. PTENα and PTENβ promote carcinogenesis through WDR5 and H3K4 trimethylation. Nat. Cell Biol.21, 1436–1448 (2019).
Carugo, A. et al. In vivo functional platform targeting patient-derived xenografts identifies WDR5-Myc association as a critical determinant of pancreatic cancer. Cell Rep.16, 133–147 (2016).
Tan, X. et al. PI3K/AKT-mediated upregulation of WDR5 promotes colorectal cancer metastasis by directly targeting ZNF407. Cell Death Dis.8, e2686 (2017).
Ma, M. et al. lncRNA GCAWKR promotes gastric cancer development by scaffolding the chromatin modification factors WDR5 and KAT2A. Mol. Ther.26, 2658–2668 (2018).
Sun, T. T. et al. LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov.6, 784–801 (2016).
Fu, Z. et al. LncRNA HOTTIP modulates cancer stem cell properties in human pancreatic cancer by regulating HOXA9. Cancer Lett.410, 68–81 (2017).
Mishra, B. P. et al. The histone methyltransferase activity of MLL1 is dispensable for hematopoiesis and leukemogenesis. Cell Rep.7, 1239–1247 (2014).
Lee, S. W., Park, D. Y., Kim, M. Y. & Kang, C. Synergistic triad epistasis of epigenetic H3K27me modifier genes, EZH2, KDM6A, and KDM6B, in gastric cancer susceptibility. Gastric Cancer22, 640–644 (2019).
Jung, S. H. et al. Clonal structures of regionally synchronous gastric adenomas and carcinomas. Clin. Cancer Res.24, 4715–4725 (2018).
Viel, A. et al. A specific mutational signature associated with DNA 8-oxoguanine persistence in MUTYH-defective colorectal cancer. EBioMedicine20, 39–49 (2017).
Kodama, T. et al. Transposon mutagenesis identifies genes and cellular processes driving epithelial-mesenchymal transition in hepatocellular carcinoma. Proc. Natl Acad. Sci. USA113, E3384–E3393 (2016).
Hong, S. et al. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl Acad. Sci. USA104, 18439–18444 (2007).
Piunti, A. & Shilatifard, A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Science352, aad9780 (2016).
Amato, E. et al. Molecular alterations associated with metastases of solid pseudopapillary neoplasms of the pancreas. J. Pathol.247, 123–134 (2019).
Wang, L. et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat. Med.24, 758–769 (2018).
Zha, L. et al. Epigenetic regulation of E-cadherin expression by the histone demethylase UTX in colon cancer cells. Med. Oncol. (Northwood, Lond., Engl.)33, 21 (2016).
Watanabe, S. et al. Loss of KDM6A characterizes a poor prognostic subtype of human pancreatic cancer and potentiates HDAC inhibitor lethality. Int. J. Cancer145, 192–205 (2019).
Andricovich, J. et al. Loss of KDM6A activates super-enhancers to induce gender-specific squamous-like pancreatic cancer and confers sensitivity to BET inhibitors. Cancer Cell33, 512–526 e518 (2018).
Hnisz, D. et al. Super-enhancers in the control of cell identity and disease. Cell155, 934–947 (2013).
Zhang, X. et al. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet.48, 176–182 (2016).
Wang, Z. et al. Acetylation of PHF5A modulates stress responses and colorectal carcinogenesis through alternative splicing-mediated upregulation of KDM3A. Mol. Cell74, 1250–1263.e1256 (2019).
Dandawate, P. et al. The histone demethylase KDM3A, increased in human pancreatic tumors, regulates expression of DCLK1 and promotes tumorigenesis in mice. Gastroenterology157, 1646–1659 e1611 (2019).
Chien, W. et al. PIAS4 is an activator of hypoxia signalling via VHL suppression during growth of pancreatic cancer cells. Br. J. Cancer109, 1795–1804 (2013).
Kong, Y. et al. RUNX3-mediated up-regulation of miR-29b suppresses the proliferation and migration of gastric cancer cells by targeting KDM2A. Cancer Lett.381, 138–148 (2016).
Neault, M., Mallette, Frédérick, A. & Richard, S. miR-137 modulates a tumor suppressor network-inducing senescence in pancreatic cancer cells. Cell Rep.14, 1966–1978 (2016).
An, Y. et al. circZMYM2 competed endogenously with miR-335-5p to regulate JMJD2C in pancreatic cancer. Cell. Physiol. Biochem.51, 2224–2236 (2018).
Tzatsos, A. et al. KDM2B promotes pancreatic cancer via polycomb-dependent and -independent transcriptional programs. J. Clin. Invest.123, 727–739 (2013).
Blanc, R. S. & Richard, S. Arginine methylation: the coming of age. Mol. Cell65, 8–24 (2017).
Ge, L. et al. PRMT5 promotes epithelial–mesenchymal transition via EGFR-beta-catenin axis in pancreatic cancer cells. J. Cell Mol. Med.24, 1969–1979 (2020).
Liu, M. et al. PRMT5-dependent transcriptional repression of c-Myc target genes promotes gastric cancer progression. Theranostics10, 4437–4452 (2020).
Hsu, M. C. et al. Protein arginine methyltransferase 3 enhances chemoresistance in pancreatic cancer by methylating hnRNPA1 to increase ABCG2 expression. Cancers11, https://doi.org/10.3390/cancers11010008 (2018).
Luo, Y. et al. Intestinal PPARalpha protects against colon carcinogenesis via regulation of methyltransferases DNMT1 and PRMT6. Gastroenterology157, 744–759 e744 (2019).
Jiang, H. et al. PRMT9 promotes hepatocellular carcinoma invasion and metastasis via activating PI3K/Akt/GSK-3β/Snail signaling. Cancer Sci.109, 1414–1427 (2018).
Qin, Y. et al. PRMT5 enhances tumorigenicity and glycolysis in pancreatic cancer via the FBW7/cMyc axis. J. Cell Commun. Signal.17, 30 (2019).
Zhang, Z. et al. FGFBP1, a downstream target of the FBW7/c-Myc axis, promotes cell proliferation and migration in pancreatic cancer. Am. J. Cancer Res.9, 2650–2664 (2019).
Hsu, M. C. et al. Protein arginine methyltransferase 3-induced metabolic reprogramming is a vulnerable target of pancreatic cancer. J. Hematol. Oncol.12, 79 (2019).
Wang, Y. P. et al. Arginine methylation of MDH1 by CARM1 inhibits glutamine metabolism and suppresses pancreatic cancer. Mol. Cell64, 673–687 (2016).
Fan, Z. et al. The histone methyltransferase Suv39h2 contributes to nonalcoholic steatohepatitis in mice. Hepatology65, 1904–1919 (2017).
Zhang, H. et al. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology64, 1033–1048 (2016).
Wu, M. C. et al. KDM4B is a coactivator of c-Jun and involved in gastric carcinogenesis. Cell Death Dis.10, 68 (2019).
Davies, R. J., Miller, R. & Coleman, N. Colorectal cancer screening: prospects for molecular stool analysis. Nat. Rev. Cancer5, 199–209 (2005).
McCleland, M. L. et al. Cdk8 deletion in the Apc(Min) murine tumour model represses EZH2 activity and accelerates tumourigenesis. J. Pathol.237, 508–519 (2015).
Shpargel, K. B., Starmer, J., Yee, D., Pohlers, M. & Magnuson, T. KDM6 demethylase independent loss of histone H3 lysine 27 trimethylation during early embryonic development. PLoS Genet.10, e1004507 (2014).
Jiang, W., Wang, J. & Zhang, Y. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res.23, 122–130 (2013).
Mallen-St Clair, J. et al. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev.26, 439–444 (2012).
Chen, N. M. et al. Context-dependent epigenetic regulation of nuclear factor of activated T cells 1 in pancreatic plasticity. Gastroenterology152, 1507–1520 e1515 (2017).
Hasselluhn, M. C., Schmidt, G. E., Ellenrieder, V., Johnsen, S. A. & Hessmann, E. Aberrant NFATc1 signaling counteracts TGFbeta-mediated growth arrest and apoptosis induction in pancreatic cancer progression. Cell Death Dis.10, 446 (2019).
Singh, S. K. et al. Antithetical NFATc1-Sox2 and p53-miR200 signaling networks govern pancreatic cancer cell plasticity. EMBO J.34, 517–530 (2015).
Baumgart, S. et al. Inflammation-induced NFATc1–STAT3 transcription complex promotes pancreatic cancer initiation by KrasG12D. Cancer Discov.4, 688–701 (2014).
Liou, G. Y. et al. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J. Cell Biol.202, 563–577 (2013).
Strobel, O. et al. In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology133, 1999–2009 (2007).
Guerra, C. et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell11, 291–302 (2007).
Benitz, S. et al. Ring1b-dependent epigenetic remodelling is an essential prerequisite for pancreatic carcinogenesis. Gut68, 2007–2018 (2019).
Kopp, J. L. et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell22, 737–750 (2012).
Tsuda, M. et al. The BRG1/SOX9 axis is critical for acinar cell-derived pancreatic tumorigenesis. J. Clin. Invest.128, 3475–3489 (2018).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell144, 646–674 (2011).
Huang, M. et al. Long noncoding RNA LINC00673 is activated by SP1 and exerts oncogenic properties by interacting with LSD1 and EZH2 in gastric cancer. Mol. Ther.25, 1014–1026 (2017).
Xu, M. et al. The long noncoding RNA SNHG1 regulates colorectal cancer cell growth through interactions with EZH2 and miR-154-5p. Mol. Cancer17, 141 (2018).
Lin, J. et al. Krüppel-like factor 2 inhibits hepatocarcinogenesis through negative regulation of the Hedgehog pathway. Cancer Sci.110, 1220–1231 (2019).
Wang, H. G. et al. KLF2 inhibits cell growth via regulating HIF-1α/Notch-1 signal pathway in human colorectal cancer HCT116 cells. Oncol. Rep.38, 584–590 (2017).
Ding, J. et al. Long noncoding RNA CRNDE promotes colorectal cancer cell proliferation via epigenetically silencing DUSP5/CDKN1A expression. Cell Death Dis.8, e2997 (2017).
Yan, X. et al. Dual specificity phosphatase 5 is a novel prognostic indicator for patients with advanced colorectal cancer. Am. J. cancer Res.6, 2323–2333 (2016).
Dawkins, J. B. et al. Reduced expression of histone methyltransferases KMT2C and KMT2D correlates with improved outcome in pancreatic ductal adenocarcinoma. Cancer Res.76, 4861–4871 (2016).
Liu, Z. et al. Over-expressed long noncoding RNA HOXA11-AS promotes cell cycle progression and metastasis in gastric cancer. Mol. Cancer16, 82 (2017).
Liu, Y. W. et al. LincRNAFEZF1-AS1 represses p21 expression to promote gastric cancer proliferation through LSD1-Mediated H3K4me2 demethylation. Mol. Cancer16, 39 (2017).
Wu, Q. et al. Long non-coding RNA CASC15 regulates gastric cancer cell proliferation, migration and epithelial mesenchymal transition by targeting CDKN1A and ZEB1. Mol. Oncol.12, 799–813 (2018).
Su, J. et al. Long noncoding RNA BLACAT1 indicates a poor prognosis of colorectal cancer and affects cell proliferation by epigenetically silencing of p15. Cell Death Dis.8, e2665 (2017).
Sha, M. Q. et al. EZH2 mediates lidamycin-induced cellular senescence through regulating p21 expression in human colon cancer cells. Cell Death Dis.7, e2486 (2016).
Huo, Q. et al. Dysfunction of IKZF1/MYC/MDIG axis contributes to liver cancer progression through regulating H3K9me3/p21 activity. Cell Death Dis.8, e2766 (2017).
Wang, D. et al. Depletion of histone demethylase KDM5B inhibits cell proliferation of hepatocellular carcinoma by regulation of cell cycle checkpoint proteins p15 and p27. J. Exp. Clin. Cancer Res.35, 37 (2016).
Batchu, R. B. et al. EZH2-shRNA-mediated upregulation of p21waf1/cip1 and its transcriptional enhancers with concomitant downmodulation of mutant p53 in pancreatic ductal adenocarcinoma. Surgery154, 739–746 (2013). discussion 737–746.
Matsuura, I. et al. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature430, 226–231 (2004).
Kato, S. et al. Cyclin-dependent kinase pathway aberrations in diverse malignancies: clinical and molecular characteristics. Cell Cycle14, 1252–1259 (2015).
Feng, X. H., Liang, Y. Y., Liang, M., Zhai, W. & Lin, X. Direct Interaction of c-Myc with Smad2 and Smad3 to Inhibit TGF-beta-Mediated Induction of the CDK Inhibitor p15(Ink4B). Mol. Cell62, 152 (2016).
Zhang, G. et al. LncRNA SNHG17 promotes gastric cancer progression by epigenetically silencing of p15 and p57. J. Cell. Physiol.234, 5163–5174 (2019).
Chen, S. et al. Hypoxia induces TWIST-activated epithelial–mesenchymal transition and proliferation of pancreatic cancer cells in vitro and in nude mice. Cancer Lett.383, 73–84 (2016).
Kong, R. et al. Long noncoding RNA PVT1 indicates a poor prognosis of gastric cancer and promotes cell proliferation through epigenetically regulating p15 and p16. Mol. Cancer14, 82 (2015).
Ougolkov, A. V., Bilim, V. N. & Billadeau, D. D. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin. Cancer Res.14, 6790–6796 (2008).
Li, C. H. et al. EZH2 coupled with HOTAIR to silence MicroRNA-34a by the induction of heterochromatin formation in human pancreatic ductal adenocarcinoma. Int. J. Cancer140, 120–129 (2017).
Wan, J. et al. JMJD6 promotes hepatocellular carcinoma carcinogenesis by targeting CDK4. Int. J. Cancer144, 2489–2500 (2019).
Wang, Y. et al. Amplification of SMYD3 promotes tumorigenicity and intrahepatic metastasis of hepatocellular carcinoma via upregulation of CDK2 and MMP2. Oncogene38, 4948–4961 (2019).
Li, J. et al. KDM3 epigenetically controls tumorigenic potentials of human colorectal cancer stem cells through Wnt/beta-catenin signalling. Nat. Commun.8, 15146 (2017).
Zhou, Z. et al. Epigenetically modulated FOXM1 suppresses dendritic cell maturation in pancreatic cancer and colon cancer. Mol. Oncol.13, 873–893 (2019).
Lu, C. et al. Contrasting roles of H3K4me3 and H3K9me3 in regulation of apoptosis and gemcitabine resistance in human pancreatic cancer cells. BMC Cancer18, 149 (2018).
Tan, N. et al. Bcl-2/Bcl-xL inhibition increases the efficacy of MEK inhibition alone and in combination with PI3 kinase inhibition in lung and pancreatic tumor models. Mol. Cancer Ther.12, 853–864 (2013).
Huang, S. & Sinicrope, F. A. BH3 mimetic ABT-737 potentiates TRAIL-mediated apoptotic signaling by unsequestering Bim and Bak in human pancreatic cancer cells. Cancer Res.68, 2944–2951 (2008).
Haag, C. et al. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut60, 225–237 (2011).
Abulwerdi, F. et al. A novel small-molecule inhibitor of mcl-1 blocks pancreatic cancer growth in vitro and in vivo. Mol. Cancer Ther.13, 565–575 (2014).
Paschall, A. V. et al. H3K9 trimethylation silences Fas expression to confer colon carcinoma immune escape and 5-fluorouracil chemoresistance. J. Immunol.195, 1868–1882 (2015).
Yang, S. Z. et al. The long non-coding RNA HOTAIR enhances pancreatic cancer resistance to TNF-related apoptosis-inducing ligand. J. Biol. Chem.292, 10390–10397 (2017).
Kim, K. et al. HOTAIR is a negative prognostic factor and exhibits pro-oncogenic activity in pancreatic cancer. Oncogene32, 1616–1625 (2013).
Huang, M. D. et al. Long non-coding RNA TUG1 is up-regulated in hepatocellular carcinoma and promotes cell growth and apoptosis by epigenetically silencing of KLF2. Mol. Cancer14, 165 (2015).
Nagarsheth, N. et al. PRC2 epigenetically silences Th1-Type chemokines to suppress effector T-cell trafficking in colon cancer. Cancer Res.76, 275–282 (2016).
Liu, H., Liu, Y., Liu, W., Zhang, W. & Xu, J. EZH2-mediated loss of miR-622 determines CXCR4 activation in hepatocellular carcinoma. Nat. Commun.6, 8494 (2015).
Ghanem, I. et al. Insights on the CXCL12–CXCR4 axis in hepatocellular carcinoma carcinogenesis. Am. J. Transl. Res.6, 340–352 (2014).
Bugide, S., Green, M. R. & Wajapeyee, N. Inhibition of enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc. Natl Acad. Sci. USA115, E3509–E3518 (2018).
Lu, C. et al. The MLL1–H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. J. Natl Cancer Inst.109, https://doi.org/10.1093/jnci/djw283 (2017).
Winograd, R. et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol. Res.3, 399–411 (2015).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science324, 1029–1033 (2009).
Wu, N. et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via GLUT1. Mol. Cell49, 1167–1175 (2013).
Fu, L. N. et al. Role of JMJD2B in colon cancer cell survival under glucose-deprived conditions and the underlying mechanisms. Oncogene37, 389–402 (2018).
Koutsioumpa, M. et al. Lysine methyltransferase 2D regulates pancreatic carcinogenesis through metabolic reprogramming. Gut68, 1271–1286 (2019).
Simpson, I. A. et al. The facilitative glucose transporter GLUT3: 20 years of distinction. Am. J. Physiol. Endocrinol. Metab.295, E242–E253 (2008).
Sakamoto, A. et al. Lysine demethylase LSD1 coordinates glycolytic and mitochondrial metabolism in hepatocellular carcinoma cells. Cancer Res.75, 1445–1456 (2015).
Gu, L. et al. Amplification of glyceronephosphate O-acyltransferase and recruitment of USP30 stabilize DRP1 to promote hepatocarcinogenesis. Cancer Res.78, 5808–5819 (2018).
Cui, J. et al. A novel KDM5A/MPC-1 signaling pathway promotes pancreatic cancer progression via redirecting mitochondrial pyruvate metabolism. Oncogene39, 1140–1151 (2019).
Wang, T. et al. O-GlcNAcylation of fumarase maintains tumour growth under glucose deficiency. Nat. Cell Biol.19, 833–843 (2017).
McDonald, O. G. et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat. Genet.49, 367–376 (2017).
Qin, Y. et al. LSD1 sustains pancreatic cancer growth via maintaining HIF1alpha-dependent glycolytic process. Cancer Lett.347, 225–232 (2014).
Vaupel, P., Hockel, M. & Mayer, A. Detection and characterization of tumor hypoxia using pO2 histography. Antioxid. Redox Signal.9, 1221–1235 (2007).
Masoud, G. N. & Li, W. HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm. Sin. B5, 378–389 (2015).
Jiang, M. et al. O-GlcNAcylation promotes colorectal cancer metastasis via the miR-101-O-GlcNAc/EZH2 regulatory feedback circuit. Oncogene38, 301–316 (2019).
Kalluri, R. & Weinberg, R. A. The basics of epithelial–mesenchymal transition. J. Clin. Invest.119, 1420–1428 (2009).
Yamada, S. et al. Epithelial-to-mesenchymal transition predicts prognosis of pancreatic cancer. Surgery154, 946–954 (2013).
Ning, X. et al. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett.359, 198–205 (2015).
Rhim, A. D. et al. EMT and dissemination precede pancreatic tumor formation. Cell148, 349–361 (2012).
Zhou, Z. et al. Loss of TET1 facilitates DLD1 colon cancer cell migration via H3K27me3-mediated down-regulation of E-cadherin. J. Cell. Physiol.233, 1359–1369 (2018).
Thiery, J. P., Acloque, H., Huang, R. Y. & Nieto, M. A. Epithelial–mesenchymal transitions in development and disease. Cell139, 871–890 (2009).
Hu, Y. et al. Epigenetic suppression of E-cadherin expression by Snail2 during the metastasis of colorectal cancer. Clin. Epigenet.10, 154 (2018).
Qazi, A. M. et al. Restoration of E-cadherin expression in pancreatic ductal adenocarcinoma treated with microRNA-101. Surgery152, 704–713 (2012).
Liu, Y. W. et al. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis.6, e1802 (2015).
Terashima, M., Ishimura, A., Wanna-Udom, S. & Suzuki, T. MEG8 long noncoding RNA contributes to epigenetic progression of the epithelial-mesenchymal transition of lung and pancreatic cancer cells. J. Biol. Chem.293, 18016–18030 (2018).
Ma, J., Zhang, J., Weng, Y. C. & Wang, J. C. EZH2-mediated microRNA-139-5p regulates epithelial–mesenchymal transition and lymph node metastasis of pancreatic cancer. Mol. Cells41, 868–880 (2018).
Song, W. et al. LncRNA TRERNA1 facilitates hepatocellular carcinoma metastasis by dimethylating H3K9 in the CDH1 promoter region via the recruitment of the EHMT2/SNAI1 complex. Cell Prolif.52, e12621 (2019).
Hu, Y. et al. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci.110, 3442–3452 (2019).
Li, S., Wu, L., Wang, Q., Li, Y. & Wang, X. KDM4B promotes epithelial-mesenchymal transition through up-regulation of ZEB1 in pancreatic cancer. Acta Biochim. Biophys. Sin.47, 997–1004 (2015).
Guo, T. et al. ISL1 predicts poor outcomes for patients with gastric cancer and drives tumor progression through binding to the ZEB1 promoter together with SETD7. Cell Death Dis.10, 33 (2019).
Viotti, M. et al. SUV420H2 is an epigenetic regulator of epithelial/mesenchymal states in pancreatic cancer. J. Cell Biol.217, 763–777 (2018).
Song, C. et al. PRMT1 promotes pancreatic cancer growth and predicts poor prognosis. Cell. Oncol.43, 51–62 (2020).
von Figura, G. et al. The chromatin regulator Brg1 suppresses formation of intraductal papillary mucinous neoplasm and pancreatic ductal adenocarcinoma. Nat. Cell Biol.16, 255–267 (2014).
Jakubowska, K. et al. Expressions of matrix metalloproteinases 2, 7, and 9 in carcinogenesis of pancreatic ductal adenocarcinoma. Dis. Mark.2016, 9895721 (2016).
Gong, J., Wang, Y., Jiang, B., Xu, B. & Hu, C. Impact of high-mobility-group A2 overexpression on epithelial–mesenchymal transition in pancreatic cancer. Cancer Manag. Res.11, 4075–4084 (2019).
Peng, K. et al. Histone demethylase JMJD2D interacts With β-catenin to induce transcription and activate colorectal cancer cell proliferation and tumor growth in mice. Gastroenterology156, 1112–1126 (2019).
Wong, C. M. et al. Up-regulation of histone methyltransferase SETDB1 by multiple mechanisms in hepatocellular carcinoma promotes cancer metastasis. Hepatology63, 474–487 (2016).
Shuai, W. et al. SUV39H2 promotes colorectal cancer proliferation and metastasis via tri-methylation of the SLIT1 promoter. Cancer Lett.422, 56–69 (2018).
Wu, X. et al. JMJD2C promotes colorectal cancer metastasis via regulating histone methylation of MALAT1 promoter and enhancing beta-catenin signaling pathway. J. Exp. Clin. Cancer Res38, 435 (2019).
Xia, L. et al. CHD4 has oncogenic functions in initiating and maintaining epigenetic suppression of multiple tumor suppressor genes. Cancer Cell31, 653–668 e657 (2017).
Ji, X. et al. Lysine-specific demethylase 5C promotes hepatocellular carcinoma cell invasion through inhibition BMP7 expression. BMC Cancer15, 801 (2015).
Tang, B. et al. Aberrant JMJD3 expression upregulates slug to promote migration, invasion, and stem cell-like behaviors in hepatocellular carcinoma. Cancer Res.76, 6520–6532 (2016).
Zhang, S. et al. CRISPR/Cas9-mediated knockout of NSD1 suppresses the hepatocellular carcinoma development via the NSD1/H3/Wnt10b signaling pathway. J. Exp. Clin. Cancer Res.38, 467 (2019).
Okuno, K. et al. Asymmetric dimethylation at histone H3 arginine 2 by PRMT6 in gastric cancer progression. Carcinogenesis40, 15–26 (2019).
Li, C. H. et al. Enhancer of zeste homolog 2 silences microRNA-218 in human pancreatic ductal adenocarcinoma cells by inducing formation of heterochromatin. Gastroenterology144, 1086–1097 e1089 (2013).
Yamamoto, K. et al. Loss of histone demethylase KDM6B enhances aggressiveness of pancreatic cancer through downregulation of C/EBPalpha. Carcinogenesis35, 2404–2414 (2014).
Kalli, M. et al. Solid stress-induced migration is mediated by GDF15 through Akt pathway activation in pancreatic cancer cells. Sci. Rep.9, 978 (2019).
Fujii, S., Ito, K., Ito, Y. & Ochiai, A. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. J. Biol. Chem.283, 17324–17332 (2008).
Deng, M. et al. TET-mediated sequestration of miR-26 drives EZH2 expression and gastric carcinogenesis. Cancer Res.77, 6069–6082 (2017).
Whittle, M. C. et al. RUNX3 controls a metastatic switch in pancreatic ductal adenocarcinoma. Cell161, 1345–1360 (2015).
Kleeff, J. et al. Pancreatic cancer. Nat. Rev. Dis. Prim.2, 16022 (2016).
Benson, A. B. 3rd et al. NCCN guidelines insights: hepatobiliary cancers, version 1.2017. J. Natl Compr. Cancer Netw.15, 563–573 (2017).
Ajani, J. A. et al. Gastric cancer, version 3.2016, NCCN Clinical Practice Guidelines in Oncology. J. Natl Compr. Cancer Netw.14, 1286–1312 (2016).
Tempero, M. A. et al. Pancreatic adenocarcinoma, version 2.2017, NCCN Clinical Practice Guidelines in Oncology. J. Natl Compr. Cancer Netw.15, 1028–1061 (2017).
Benson, A. B. et al. Rectal cancer, version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J. Natl Compr. Cancer Netw.16, 874–901 (2018).
Benson, A. B. et al. NCCN guidelines insights: colon cancer, version 2.2018. J. Natl Compr. Cancer Netw.16, 359–369 (2018).
Manuyakorn, A. et al. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J. Clin. Oncol.28, 1358–1365 (2010).
Maftouh, M. et al. A polymorphism in the promoter is associated with EZH2 expression but not with outcome in advanced pancreatic cancer patients. Pharmacogenomics15, 609–618 (2014).
Liu, C. et al. LSD1 stimulates cancer-associated fibroblasts to drive Notch3-dependent self-renewal of liver cancer stem-like cells. Cancer Res.78, 938–949 (2018).
Lima-Fernandes, E. et al. Targeting bivalency de-represses Indian Hedgehog and inhibits self-renewal of colorectal cancer-initiating cells. Nat. Commun.10, 1436 (2019).
Bao, B. et al. Metformin inhibits cell proliferation, migration and invasion by attenuating CSC function mediated by deregulating miRNAs in pancreatic cancer cells. Cancer Prev. Res.5, 355–364 (2012).
Maftouh, M. et al. Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer110, 172–182 (2014).
Yoshida, K., Toden, S., Ravindranathan, P., Han, H. & Goel, A. Curcumin sensitizes pancreatic cancer cells to gemcitabine by attenuating PRC2 subunit EZH2, and the lncRNA PVT1 expression. Carcinogenesis38, 1036–1046 (2017).
Wu, L. W. et al. Suppression of LSD1 enhances the cytotoxic and apoptotic effects of regorafenib in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun.512, 852–858 (2019).
Huang, M. et al. Targeting KDM1A attenuates Wnt/beta-catenin signaling pathway to eliminate sorafenib-resistant stem-like cells in hepatocellular carcinoma. Cancer Lett.398, 12–21 (2017).
Lei, Z. J. et al. Lysine-specific demethylase 1 promotes the stemness and chemoresistance of Lgr5(+) liver cancer initiating cells by suppressing negative regulators of β-catenin signaling. Oncogene34, 3188–3198 (2015).
Kari, V. et al. The histone methyltransferase DOT1L is required for proper DNA damage response, DNA repair, and modulates chemotherapy responsiveness. Clin. Epigenetics11, 4 (2019).
Luo, C. W. et al. G9a governs colon cancer stem cell phenotype and chemoradioresistance through PP2A-RPA axis-mediated DNA damage response. Radiother. Oncol.124, 395–402 (2017).
Zhou, J. et al. Targeting EZH2 histone methyltransferase activity alleviates experimental intestinal inflammation. Nat. Commun.10, 2427 (2019).
Cheng, L. L. et al. TP53 genomic status regulates sensitivity of gastric cancer cells to the histone methylation inhibitor 3-deazaneplanocin A (DZNep). Clin. Cancer Res.18, 4201–4212 (2012).
Mody, H. R., Hung, S. W., AlSaggar, M., Griffin, J. & Govindarajan, R. Inhibition of S-adenosylmethionine-dependent methyltransferase attenuates TGF 1-induced EMT and Metastasis in pancreatic cancer: putative roles of miR-663a and miR-4787-5p. Mol. Cancer Res.14, 1124–1135 (2016).
Wei, Y. et al. Plasma cell polarization to the immunoglobulin G phenotype in hepatocellular carcinomas involves epigenetic alterations and promotes hepatoma progression in mice. Gastroenterology156, 1890–1904 e1816 (2019).
Huang, S. et al. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res.79, 2009–2020 (2019).
Huang, L. et al. Ductal pancreatic cancer modeling and drug screening using human pluripotent stem cell- and patient-derived tumor organoids. Nat. Med.21, 1364–1371 (2015).
Hu, W., Jia, X., Gao, Y. & Zhang, Q. Chaetospirolactone reverses the apoptotic resistance towards TRAIL in pancreatic cancer. Biochem. Biophys. Res. Commun.495, 621–628 (2018).
Ashkenazi, A. Directing cancer cells to self-destruct with pro-apoptotic receptor agonists. Nat. Rev. Drug Discov.7, 1001–1012 (2008).
Guo, W. et al. Diosgenin exhibits tumor suppressive function via down-regulation of EZH2 in pancreatic cancer cells. Cell Cycle18, 1745–1758 (2019).
Huang, C. C. et al. Garcinol downregulates Notch1 signaling via modulating miR-200c and suppresses oncogenic properties of PANC-1 cancer stem-like cells. Biotechnol. Appl. Biochem.64, 165–173 (2017).
Jin, X. et al. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J. Biol. Chem.292, 6269–6280 (2017).
Bao, B. et al. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res.72, 335–345 (2012).
Kim, T. W., Lee, S. Y., Kim, M., Cheon, C. & Ko, S. G. Kaempferol induces autophagic cell death via IRE1-JNK-CHOP pathway and inhibition of G9a in gastric cancer cells. Cell Death Dis.9, 875 (2018).
Yuan, Y. et al. A small-molecule probe of the histone methyltransferase G9a induces cellular senescence in pancreatic adenocarcinoma. ACS Chem. Biol.7, 1152–1157 (2012).
Yuan, Y. et al. Gossypol and an HMT G9a inhibitor act in synergy to induce cell death in pancreatic cancer cells. Cell Death Dis.4, e690 (2013).
Gu, M. et al. Nanodiamond-mediated delivery of a G9a inhibitor for hepatocellular carcinoma therapy. ACS Appl Mater. Interfaces11, 45427–45441 (2019).
Barcena-Varela, M. et al. Dual targeting of histone methyltransferase G9a and DNA-methyltransferase 1 for the treatment of experimental hepatocellular carcinoma. Hepatology69, 587–603 (2019).
Li, S. S. et al. KMT2D deficiency enhances the anti-cancer activity of L48H37 in pancreatic ductal adenocarcinoma. World J. Gastrointest. Oncol.11, 599–621 (2019).
Mathison, A. et al. Combined AURKA and H3K9 methyltransferase targeting inhibits cell growth by inducing mitotic catastrophe. Mol. Cancer Res.15, 984–997 (2017).
Zheng, B. N. et al. Targeting PRMT5 activity inhibits the malignancy of hepatocellular carcinoma by promoting the transcription of HNF4α. Theranostics9, 2606–2617 (2019).
Zhang, B. et al. Targeting protein arginine methyltransferase 5 inhibits human hepatocellular carcinoma growth via the downregulation of beta-catenin. J. Transl. Med.13, 349 (2015).
Zhang, B. et al. Arginine methyltransferase inhibitor 1 inhibits gastric cancer by downregulating eIF4E and targeting PRMT5. Toxicol. Appl. Pharmacol.336, 1–7 (2017).
Comet, I., Riising, E. M., Leblanc, B. & Helin, K. Maintaining cell identity: PRC2-mediated regulation of transcription and cancer. Nat. Rev. Cancer16, 803–810 (2016).
Hughes, A. L., Kelley, J. R. & Klose, R. J. Understanding the interplay between CpG island-associated gene promoters and H3K4 methylation. Biochim. Biophys. Acta Gene Regul. Mech.1863, 194567 (2020).
Howe, F. S., Fischl, H., Murray, S. C. & Mellor, J. Is H3K4me3 instructive for transcription activation? BioEssays39, 1–12 (2017).
Demetriadou, C. et al. NAA40 contributes to colorectal cancer growth by controlling PRMT5 expression. Cell Death Dis.10, 236 (2019).
Acknowledgements
This study was supported by the National Nature Science Foundation of China (2019, 81972321 to L.Y.; 2019, 81974376 to Y.P.Z.).
Author information
Authors and Affiliations
Contributions
Study concept and design: Y.C., B.R., J.Y., H.W., G.Y., R.X., L.Y., and Y.Z. Drafting of the manuscript: Y.C. Critical revision of the manuscript for important intellectual content: B.R., J.Y., H.W., G.Y., R.X., L.Y., Y.Z. Obtained funding: L.Y. and Y.Z. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Y., Ren, B., Yang, J. et al. The role of histone methylation in the development of digestive cancers: a potential direction for cancer management. Sig Transduct Target Ther 5, 143 (2020). https://doi.org/10.1038/s41392-020-00252-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41392-020-00252-1
