
Abstract
Programmed cell death-1 (PD-1) belongs to an inhibitory signaling pathway capable of maintaining central and peripheral immune tolerance. Blockage of PD-1 has been identified as a promising immunotherapeutic approach for cancer and chronic infectious diseases. However, it is unknown whether PD-1 pathway regulates stem cell function. It is generally believed that mesenchymal stem cells (MSCs) produce PD-1 ligand, but fail to express PD-1. In this study, we show that neural crest-derived MSCs from dental pulp (MSC-DP), but not MSCs from bone marrow, expressed PD-1. Knocking down PD-1 expression in MSC-DP results in a significantly reduced capacity for cell proliferation and accelerated multipotential differentiation. Mechanistically, we show that PD-1 regulates a SHP2/ERK/Notch cascade to maintain proliferation and a SHP2/ERK/β-catenin cascade to inhibit osteo-/odontogenic differentiation. This study indicates that PD-1 is a key surface molecule controlling cell proliferation and multipotential differentiation of MSC-DP. Through regulating PD-1/SHP2/ERK signaling, we can significantly improve the quality and quantity of culture-expanded MSC-DP for potential clinical therapies.
Similar content being viewed by others


Introduction
Mesenchymal stem cells (MSCs) exist in a variety of organs and tissues, including bone marrow, umbilical cord, adipose tissue, skeletal muscle, and dental tissues. MSCs from dental pulp (MSC-DP) are a unique population of highly proliferative neural crest-derived stem cells [1, 2]. They can be isolated from the dental pulp of exfoliated deciduous teeth or permanent teeth [3,4,5]. MSC-DP are multipotent MSCs capable of differentiating into osteo-/odontogenic cells, adipocytes, chondrocytes and neural cells‚ and can regenerate dentin-pulp-like tissue in vivo [3,4,5,6]. MSC-DP also possess immunomodulatory properties that can regulate CD4+ T cells, CD3+ T cells, and regulatory T cells (Tregs). Systemic infusion of MSC-DP can ameliorate autoimmune disease phenotypes [7,8,9,10]. However, the underlying mechanisms that control MSC-DP self-renewal and differentiation are largely unknown.
Inhibitory receptor programmed cell death-1 (PD-1), a member of the CD28 family, is a key mediator for T cell response and immune tolerance [11]. PD-1 is expressed in various immune cells, including activated T cells, B cells, macrophages, dendritic cells, and natural killer cells [12]. PD-1-mediated negative immune signaling proceeds through engagement with two ligands, known as PD-L1 (B7-H1) and PD-L2 (B7-DC) [13, 14]. Upon activation, PD-1 suppresses exhausted CD4+ T cells in early phases of T cell activation as well as T cells effector functions, leading to immune tolerance [15]. PD-1 knockout mice develop lupus-like autoimmune disease with glomerulonephritis and cardiomyopathy [16, 17]. Moreover, PD-1 pathway plays an important role in cancer immunology by targeting tumor-infiltrating CD8+ T cells to induce CD8+ T cell apoptosis and inhibit CD8+ T cell function, leading to inhibition of tumor immune-surveillance [18]. Because of PD-1’s role as a negative immune checkpoint, immunotherapies targeting this pathway have shown significant potential for cancer therapy. However, it is largely unknown whether PD-1 pathway also contributes to non-immune cell function.
It is believed that MSCs produce PD-1 ligand without expression of PD-1. In this study, we show that MSC-DP, but not bone marrow MSCs (BMMSCs), expressed PD-1. PD-1 is required to maintain cell proliferation and inhibits multipotential differentiation of MSC-DP. In addition, PD-1 is a key surface molecule for MSC-DP selection and purification.
Results
MSC-DP express PD-1
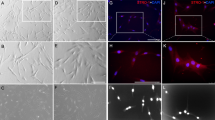
It is generally believed that MSCs expressed PD-1 ligand, but failed to produce PD-1 [19]. To assess whether MSC-DP express PD-1, we isolated MSC-DP from exfoliated deciduous teeth (which we refer to hereafter as stem cells from human exfoliated deciduous teeth or SHED) and permanent teeth (dental pulp stem cells; DPSCs), as described in our previous studies [3, 5]. We found that both SHED and DPSCs, but not BMMSCs, expressed PD-1 on the cell membrane, as assessed by Western blotting, qPCR, immunostaining, and flow cytometric analysis (Fig. 1a–c and Fig. S1A). However, SHED, DPSCs, and BMMSCs expressed PD-L1 in the cytoplasm (Fig. 1a). SHED expressed elevated levels of PD-1 when compared to DPSCs (Fig. 1a, b).

MSC-DP express PD-1. a Western blot analysis showed that SHED and DPSCs, but not BMMSCs, expressed PD-1 in the cytomembrane. SHED, DPSCs, and BMMSCs expressed PD-L1 in the cytoplasm. b RT-PCR analysis showed that SHED expressed higher levels of PD-1 mRNA than DPSCs. BMMSCs expressed higher levels of PD-L1 than SHED and DPSCs. c Immunocytofluorescent staining showed PD-1 (red) and PD-L1 (green) expression in SHED, DPSCs, and BMMSCs, scale bar = 20 μm. d Immunocytofluorescence staining showed co-localization of PD-1 (red) and MSC marker CD90 (green) in SHED, scale bar = 20 μm. e Flow cytometric analysis showed that 16.76% of SHED were double-positive for PD-1 and CD73, 17.55% of SHED were double-positive for PD-1 and CD90, and 11.86% of SHED were double-positive for PD-1 and CD105, n = 3 in each group; **P < 0.01; ***P < 0.005; error bars: mean ± SD
We next used immunocytofluorescent staining to show that PD-1 was co-expressed with MSC marker CD90 in SHED (Fig. 1d). Flow cytometric analysis was also used to show that 16.76% of SHED were PD-1+/CD73+, 17.55% PD-1+/CD90+, and 11.86% PD-1+/CD105+ double-positive (Fig. 1e). To further confirm that MSC-DP express PD-1, we collected the dental pulp from human exfoliated deciduous teeth and used immunofluorescent staining to show that PD-1 and CD90 double-positive cells were present in this tissue (Fig. S1B). These data suggest that PD-1 is a specific surface molecule of MSC-DP.
PD-1 is required to maintain cell proliferation and inhibit multipotential differentiation of SHED
PD-1 is a type I transmembrane protein and serves as a co-inhibitory receptor in immune cells. After binding with PD ligands, PD-1 signaling inhibits T cell function and induces immune tolerance [20, 21]. To explore the functional role of PD-1 in MSC-DP, we used recombinant human PD-L1 and PD-L2 to treat SHED and found no significant effect on the number of 7AAD+AnnexinV+ apoptotic SHED (Fig. 2a). Treatment with PD-L2, but not PD-L1, increased the proliferation rate and population doublings, as assessed by BrdU staining and continued culture assay, respectively (Fig. 2b, c). These data suggest that PD-1 ligands/pathway accelerate the SHED proliferation.

PD-1 maintains cell proliferation and inhibits differentiation in SHED. a Flow cytometric analysis showed that PD-L1 and PD-L2 treatment had no significant effects on the percentage of 7AAD+AnnexinV+ cells out of the total number of SHED when compared to untreated SHED. b BrdU labeling assay showed that PD-L2, but not PD-L1, treatment improved the proliferation rate of SHED when compared with the untreated group. c Continued culture assay showed that the number of population doublings was increased when treated with PD-L2 compared to the untreated SHED, while PD-L1 treatment had no significant effects. d BrdU labeling assay showed that PD-1 siRNA treatment downregulated proliferation rate in SHED. e Continued culture assay showed that PD-1 siRNA treatment reduced the number of population doublings in SHED. f PD-1 siRNA treatment elevated mineralized nodule formation when compared to the control SHED, as assessed by alizarin red staining. g Western blot analysis showed that PD-1 siRNA treatment upregulated the expression levels of Runx2 and OCN compared to untreated SHED when cultured under osteogenic inductive conditions. h When implanted into immunocompromised mice using HA/TCP as a carrier, PD-1 knockdown SHED exhibited an increased capacity to generate new bone when compared to the control SHED, scale bar = 200 μm. i Immunostaining and Western blot analysis showed that PD-1 siRNA treatment increased the expression levels of β-III tubulin and NeuN under neurogenic inductive condition, scale bar = 20 μm, n = 3 in each group; **P < 0.01; ***P < 0.005; error bars: mean ± SD. Vehicle: scrambled siRNA-treated SHED
To further determine the role of PD-1 in MSC proliferation and differentiation, we used PD-1 siRNA to knockdown PD-1 expression in SHED, DPSCs, and BMMSCs (Fig. S2A, F). We found that PD-1 siRNA treatment significantly reduced the proliferation rate and the number of population doublings of SHED compared to the control group (Fig. 2d, e). Moreover, PD-1 siRNA treatment markedly increased the osteo-/odontogenic differentiation of SHED to form mineralized nodules after 3 and 7 days of culture under osteogenic inductive conditions (Fig. 2f). Western blot analysis confirmed that the expression levels of Runx2 and OCN in PD-1 siRNA-treated SHED were markedly upregulated compared to the untreated group (Fig. 2g). When implanted into immunocompromised mice using HA/TCP as a carrier, PD-1 siRNA-treated SHED exhibited an increased capacity to form mineralized tissue when compared to the control SHED (Fig. 2h). We also used siRNA to knock down PD-1 expression in SHED and examined their neurogenic differentiation capacity. We found that PD-1 siRNA treatment significantly increased the expression levels of neurogenic markers β-III tubulin and NeuN under neurogenic inductive condition, as assessed by immunocytofluorescent staining and Western blotting (Fig. 2i). Furthermore, we confirmed that siRNA knockdown of PD-1 inhibited the proliferation rate and population doubling and promoted osteo-/odontogenic differentiation of DPSCs (Fig. S2B-E). However, PD-1 siRNA treatment had no significant effect on the proliferation rate, population doubling, or osteogenic differentiation of BMMSCs (Fig. S2G-J). These data indicate that PD-1 plays a critical role in maintaining proliferation and inhibiting differentiation in MSC-DP.
Isolation of PD-1+ SHED
We showed that a small fraction (19.02%) of SHED was positive for PD-1, assessed by Flow cytometric analysis (Fig. S1A). Since CD90 is a well-known MSC surface marker, we isolated PD-1+CD90+ SHED (16.84%) by flow cytometric cell sorting to ensure purity of isolated SHED. CD90+ SHED were gated and sorted into CD90+ PD-1+ (90.39% purity) and CD90+ PD-1− (100% purity) populations for the following experiments (Fig. 3a). qPCR analysis showed that PD-1+ SHED, but not PD-1− SHED, expressed PD-1 mRNA (Fig. 3b). PD-1+ SHED showed significantly elevated rates of cell proliferation and population doubling, as determined by BrdU labeling and continued culture assays, respectively, when compared with PD-1− SHED (Fig. 3c, d). To further clarify the role of PD-1 in SHED proliferation, we performed cell cycle analysis and found that 57.25% of PD-1+ SHED was in S/G2/M phases and only 8.58% of PD-1− SHED was in S/G2/M phases (Fig. 3e), suggesting that PD-1+ SHED have elevated proliferation capacity than PD-1− SHED. Under osteogenic culture conditions, PD-1+ SHED showed reduced osteo-/odontogenic differentiation, as indicated by a decreased ability to form mineralized nodules (Fig. 3f) and decreased expression of osteogenic markers Runx2 and OCN (Fig. 3g). When implanted into immunocompromised mice subcutaneously, PD-1+ SHED exhibited a significantly reduced capacity to generate new bone when compared to PD-1− SHED (Fig. 3h). Moreover, we found that PD-1+ SHED had a decreased neurogenic differentiation potential under neurogenic inductive condition, as indicated by decreased expression of neurogenic markers β-III tubulin and NeuN when compared with PD-1− SHED (Fig. 3i). These data suggest that PD-1+ SHED may represent a subset of highly proliferative cells with reduced capacity for differentiation.

Identification and characterization of PD-1+ SHED. a Flow cytometry analysis showed that 16.84% of SHED were double-positive for PD-1 and CD90. CD90+ cells were gated and sorted for PD-1+ (90.39% purity) and PD-1– (100% purity) populations for the following experiments. b RT-PCR analysis showed that CD90+PD-1+ SHED, but not CD90+PD-1– SHED, expressed PD-1 mRNA. c BrdU labeling assay showed that the proliferation rate of PD-1+ SHED was higher than that of PD-1– SHED. d Continued culture assay showed that PD-1+ SHED had an increased number of population doublings relative to PD-1− SHED. e Cell cycle analysis showed that the S/G2/M phases of PD-1+ SHED was much higher than that of PD-1− SHED. f Alizarin red staining showed that the capacity for mineralized nodule formation of PD-1+ SHED was lower than that of PD-1− SHED. g Western blot analysis showed that PD-1+ SHED expressed decreased levels of Runx2 and OCN compared to PD-1− SHED when cultured under osteogenic inductive conditions. h When implanted into immunocompromised mice using HA/TCP as a carrier, PD-1+ SHED exhibited less capacity to generate new bone than PD-l− SHED, scale bar = 200 μm. i Immunostaining and Western blot analysis showed that the expression levels of β-III tubulin and NeuN in PD-1+ SHED were lower than that in PD-1− SHED under neurogenic inductive condition, scale bar = 20 μm, n = 3 in each group; ***P < 0.005; error bars: mean ± SD. Vehicle: scrambled siRNA-treated SHED
PD-1 regulates proliferation and differentiation of SHED via SHP2/ERK pathway
Tyrosine phosphatase SHP2 is an intracellular domain responsible for transmission of inhibitory signals in PD-1/PD-L pathway [22]. In immune response, engagement with PD-L leads to the phosphorylation of PD-1 cytoplasmic tails and recruitment of SHP2, which dephosphorylates multiple downstream pathways, blocking effects of T cell activation [23]. However, it is unknown whether SHP2, as a downstream target of PD-1, is also involved in PD-1-controlled MSC-DP proliferation and differentiation. We used Western blot analysis to show that the expression level of p-SHP2, but not total SHP2, was decreased in PD-1 siRNA-treated SHED compared to the control SHED (Fig. 4a). We further showed that only SHED and DPSCs, but not BMMSCs, expressed p-SHP2 (Fig. 4b). Next, we used SHP2 siRNA to knock down SHP2 expression in SHED (Fig. S3A) and found that the proliferation rate and population doublings of SHED were significantly decreased, as assessed by BrdU labeling and continued culture assay, respectively (Fig. 4c, d). Under osteogenic culture conditions, SHP2 siRNA-treated SHED showed an increased ability to form mineralized nodules, as assessed by alizarin red staining (Fig. 4e), and increased expression levels of osteogenic markers Runx2 and OCN (Fig. 4f). Moreover, we used SHP2 inhibitor (NSC87877) treatment to assess the role of SHP2 in SHED proliferation and differentiation. We showed that NSC87877 treatment inhibited SHP2 signaling in a dose-dependent manner (Fig. S3B). We thus used 50 μM NSC87877, the most effective dose, to inhibit SHP2 signaling in this study. Also, we found a significantly reduced proliferation rate and number of population doublings (Fig. 4g, h), along with elevated osteogenic differentiation (Fig. 4i, j). These data indicate that SHP2 acts downstream of PD-1 to regulate proliferation and differentiation of MSC-DP.

PD-1 maintains proliferation and differentiation of SHED via SHP2. a Western blot analysis showed that PD-1 siRNA treatment downregulated the expression level of p-SHP2, but not total SHP2, in SHED. b Western blot analysis showed that the expression level of p-SHP2 in SHED was higher than in DPSCs, while BMMSCs failed to express p-SHP2. c BrdU labeling assay showed that the proliferation rate of SHED was downregulated after SHP2 siRNA treatment. d Continued culture assay showed that SHP2 siRNA treatment reduced the number of population doublings in SHED. e SHP2 siRNA treatment elevated mineralized nodule formation when compared to the control SHED, as assessed by alizarin red staining. f Western blot analysis showed that SHP2 siRNA treatment upregulated the expression levels of Runx2 and OCN compared to untreated SHED when cultured under osteogenic inductive conditions. g BrdU labeling assay showed that the proliferation rate of SHED was downregulated by SHP2 inhibitor (NSC87877) treatment. h Continued culture assay showed that NSC87877 treatment could reduce the population doublings of SHED. i NSC87877 treatment elevated mineralized nodule formation when compared to control SHED, as assessed by alizarin red staining. j Western blot analysis showed that NSC87877 treatment upregulated the expression levels of Runx2 and OCN compared to those of untreated SHED when cultured under osteogenic inductive conditions, n = 3 in each group; **P < 0.01; ***P < 0.005; error bars: mean ± SD. Vehicle: scrambled siRNA-treated SHED
Previous studies showed that SHP2 is required for the activation of Ras/ERK pathway in embryonic stem cells (ESCs) [24]. To identify the detailed downstream pathway of SHP2, we examined the expression levels of ERK in PD-1 and SHP2 siRNA-treated SHED and found that the expression level of p-ERK, but not total ERK, was significantly decreased when compared to the control group (Fig. 5a). Also, treatment with SHP2 inhibitor NSC87877 reduced the expression level of p-ERK in SHED (Fig. 5a). We next used an ERK inhibitor (PD98059) to reduce the expression level of ERK in SHED and found that this treatment decreased the proliferation rate and population doublings of SHED, as determined by BrdU labeling and continued culture assays, respectively (Fig. 5b, c and Fig. S4). Moreover, we showed that PD98059-treated SHED had increased capacity to form mineralized nodules, as assessed by alizarin red staining (Fig. 5d), and expressed osteogenic markers Runx2 and OCN (Fig. 5e). However, PD98059 treatment failed to affect PD-1 or SHP2 expression (Fig. 5f). Next, we used an ERK activator (PEITC) to treat SHP2 siRNA-treated SHED and found that this treatment significantly elevated their proliferation rate and the number of population doublings, along with decreased mineralized nodule formation and reduced expression levels of Runx2 and OCN (Fig. 5g–j and Fig. S4). Collectively, these data suggest that PD-1 regulates a SHP2/ERK cascade to control proliferation and differentiation of MSC-DP.

SHP2 modulates proliferation and differentiation of SHED via ERK pathway. a Western blot analysis showed that the expression level of p-ERK, but not total ERK, in SHED was significantly downregulated when treated by PD-1 siRNA, SHP2 siRNA, or SHP2 inhibitor (NSC87877). b BrdU labeling assay showed that the proliferation rate of SHED was downregulated by ERK inhibitor (PD98059) treatment. c Continued culture assay showed that PD98059 treatment reduced the number of population doublings in SHED. d PD98059 treatment elevated mineralized nodule formation when compared to control SHED, as assessed by alizarin red staining. e Western blot analysis showed that PD98059 treatment upregulated the expression levels of Runx2 and OCN compared to untreated SHED when cultured under osteogenic inductive conditions. f Western blot analysis showed that PD98059 treatment had no significant effects on the expression levels of PD-1 or p-SHP2 in SHED. g BrdU labeling assay showed that ERK activator (PEITC) treatment rescued the decreased proliferation rate of SHP2 siRNA-treated SHED. h Continued culture assay showed that PEITC treatment rescued the decreased population doublings of SHP2 siRNA-treated SHED. i siSHP2-treated SHED showed elevated mineralized nodule formation, as assessed by alizarin red staining, while PEITC treatment could inhibit the elevated mineralized nodule formation. j Western blot analysis showed that when cultured under osteogenic inductive conditions, SHP2 siRNA treatment upregulated the expression levels of Runx2 and OCN in SHED, while these increases were blocked by PEITC treatment, n = 3 in each group; **P < 0.01; ***P < 0.005; error bars: mean ± SD. Vehicle: scrambled siRNA-treated SHED
PD-1/SHP2/ERK modulate proliferation via Notch signaling and suppress differentiation via Wnt/β-catenin signaling in SHED
It is well known that Notch signaling plays an important role in regulating stem cell self-renewal/proliferation [25]. To explore the detailed mechanisms of how PD-1 may interact with Notch signaling in regulating SHED proliferation, we examined the expression levels of Notch1, Notch2, and NICD in PD-1 and SHP2 siRNA-treated SHED. We found that the expression levels of Notch1 and NICD, but not Notch2, were significantly decreased in both PD-1 and SHP2 siRNA-treated SHED (Fig. 6a and Fig. S5A). SHP2 inhibitor (NSC87877) treatment reduced the expression levels of Notch1 and NICD in SHED (Fig. 6a). Western blot analysis showed that ERK inhibitor (PD98059) treatment reduced the expression levels of Notch1 and NICD, while Notch inhibitor (DAPT) treatment had no significant effect on the ERK expression in SHED (Fig. 6b and Fig. S5B). These data suggest that Notch signaling may act as a downstream target of PD-1/SHP2/ERK pathway in SHED. To identify the role of Notch signaling in PD-1/SHP2/ERK-mediated SHED proliferation, we used Notch activator (DLL1) treatment to elevate the expression levels of Notch1 and NICD in SHED (Fig. S5C). BrdU labeling and continued culture assay showed that DLL1 treatment rescued the decreased proliferation rate and population doublings in PD98059-treated SHED (Fig. 6c, d). These data indicate that Notch signaling is downstream of the PD-1/SHP2/ERK cascade in controlling the proliferation of MSC-DP.

PD-1 maintains SHED proliferation via SHP2/ERK/Notch, and inhibits SHED differentiation via SHP2/ERK/β-catenin. a Western blot analysis showed that the expression levels of Notch1 and NICD were downregulated in SHED treated with PD-1 siRNA, SHP2 siRNA, or SHP2 inhibitor (NSC87877). b Western blot analysis showed that the expression levels of Notch1 and NICD were decreased in ERK inhibitor (PD98059)-treated SHED. c BrdU labeling assay showed that Notch activator (DLL1) treatment rescued the decreased proliferation rate seen in PD98059-treated SHED. d Continued culture assay showed that DLL1 treatment rescued the decreased number of population doublings seen in PD98059-treated SHED. e Western blot analysis showed that the expression level of active β-catenin, but not total β-catenin, was increased in SHED treated with PD-1 siRNA, SHP2 siRNA, or NSC87877. f Western blot analysis showed that the expression level of active β-catenin, but not total β-catenin, was elevated in PD98059-treated SHED. g Wnt/β-catenin inhibitor (XAV939) treatment rescued mineralized nodule formation in PD98059-treated SHED, as assessed by alizarin red staining. h Western blot analysis showed that XAV939 treatment rescued the decreased Runx2 and OCN expression levels seen in PD98059-treated SHED, n = 3 in each group; *P < 0.05; **P < 0.01; ***P < 0.005; error bars: mean ± SD. Vehicle: scrambled siRNA-treated SHED
Previous study showed that Wnt/β-catenin signaling plays a critical role in SHED differentiation [26]. Here we examined the expression levels of active and total β-catenin in PD-1 and SHP2 siRNA-treated SHED and found that the expression levels of active β-catenin were significantly increased in both siRNA treatment groups, but there were no significant changes in total β-catenin expression (Fig. 6e). We also found that the expression level of active β-catenin, but not total β-catenin, was markedly elevated in NSC87877-treated SHED (Fig. 6e). Next, we examined whether ERK signaling is responsible for PD-1-regulated Wnt/β-catenin expression. Western blot analysis showed that ERK inhibitor treatment increased the expression levels of active β-catenin, while Wnt/β-catenin inhibitor (XAV939) treatment had no significant effect on the ERK expression in SHED (Fig. 6f and Fig. S5B). These findings indicate that Wnt/β-catenin signaling may be downstream of PD-1/SHP2/ERK pathway in SHED. To identify the role of Wnt/β-catenin signaling in PD-1/SHP2/ERK-mediated SHED differentiation, we used XAV939 treatment to inhibit the expression level of active β-catenin in SHED (Fig. S5D) and found that this rescued the increased mineralized nodule formation and osteogenic marker expression in PD98059-treated SHED, as assessed by alizarin red staining and Western blotting (Fig. 6g, h). These data imply that PD-1 regulates the osteo-/odontogenic differentiation of SHED via SHP2/ERK/β-catenin signaling pathway.
To explore whether the interplay between Notch and β-catenin contributes to PD-1/SHP2-mediated proliferation and differentiation in SHED, we used PD-1 CRISPR/Cas9 knockout plasmid to generate PD-1−/− SHED. We found that inhibition of β-catenin, by XAV939 treatment, had no effects on the Notch1 and NICD expression in PD-1−/− SHED. However, activation of Notch signaling by DLL1 treatment suppressed active β-catenin expression in PD-1−/− SHED. Furthermore, we showed that the inhibition of Notch by DAPT treatment elevated active β-catenin expression in SHED (Fig. S6). These data indicate that PD-1/SHP2 may regulate Wnt/β-catenin through Notch pathway.
Discussion
PD-1 is a critical negative receptor that regulates central and peripheral T cell response through engagement with PD-L1 or PD-L2 to induce immune tolerance. It is well known that PD-1 is inducibly expressed in T cells, B cells, NK T cells, activated monocytes, and dendritic cells [15]. However, the function and expression of PD-1 in non-lymphoid organs are still largely unknown. Recently, it was reported that PD-1 expression was observed in the retinal ganglion cell (RGC) layer and the inner nuclear layer of naïve retina, and also in certain neurons of the cerebrum and cerebellum [27], suggesting that PD-1 may be expressed outside of the immune system. As an inhibitory signal, PD-1 may be involved in RGC apoptosis during retinal development through a caspase 3-dependent pathway [28]. PD-1 expression was shown to be significantly increased in RGCs after a crushing injury to the optic nerve [29]. These findings suggest that PD-1 might play a pivotal role in retinal neurodegeneration.
It is generally believed that MSCs express PD-1 ligand, but not PD-1 [19, 30]. In this study, we reveal constitutive expression of PD-1 in the cytomembrane of MSC-DP, but not in BMMSCs. We found that PD-1 ligand was expressed in the cytoplasm of MSC-DP and BMMSCs. This is the first study demonstrating PD-1 expression in stem cells and suggesting that PD-1 plays a critical role in maintaining stem cell properties in neural crest-derived SHED and DPSCs. Since MSCs, including MSC-DP, are heterogenic population stem cells, only 10–30% of MSCs expressed early MSC surface markers STRO-1, CD146, and CD105 [5, 26, 31, 32]. In this study, we showed that only around 11–17% SHED expressed PD-1, suggesting that PD-1 may represent an early stem cell marker. Moreover, we explored the role of PD-1 in regulating proliferation via SHP2/ERK/Notch pathway and differentiation via a SHP2/ERK/β-catenin cascade. When we knocked down PD-1 expression in SHED, their proliferation rate was dramatically decreased and they were easily differentiated into osteogenic cells. We isolated PD-1+ SHED and demonstrated that this population may represent a highly proliferative subset.
Upon TCR stimulation and ligation with PD-ligands, PD-1 is activated and recruits the src homology phosphotyrosyl phosphatase (SHP), which in turn results in dephosphorylation of downstream molecules and inhibition of downstream pathways including PI3K/AKT, Bcl-xL, and Ras [23]. SHP2, encoded by the PTPN11 gene, is ubiquitously expressed. It plays a critical role in regulating the migration, proliferation, survival, and differentiation of ESCs. SHP2 deletion suppresses ESC differentiation with increased self-renewal/proliferation [24]. Germline mutations in PTPN11 cause Noonan and LEOPARD syndromes, indicating that SHP2 is essential for neural crest cell function [33, 34]. Although SHP2 signaling may proceed in a cell type- and receptor-specific manner, it is required for the full activation of RAS/ERK pathway [35]. Our findings show that PD-1 is required for sustaining proliferation of MSC-DP via a SHP2/ERK cascade. Interestingly, we found that the role of SHP2 in supporting MSC-DP is the converse of its role in ESCs. PD-1 knockdown severely inhibited MSC-DP proliferation, accompanied by accelerated differentiation through downregulating SHP2. MAPK-ERK signaling is essential for lineage differentiation of osteogenic cells. However, the detailed role of ERK signaling in osteogenic cell differentiation is controversy [36]. In vitro studies show that the activation of MAP kinase, including ERK, JNK, and p38, is critical to support Runx2 activity and osteogenesis [37]. In contrast, ERK signaling is able to antagonize bone formation. Treatment with epidermal growth factor (EGF) or basic fibroblast growth factor (bFGF) can inhibit osteogenic differentiation of MSC via activation of ERK signaling [38, 39], implying that ERK-mediated MSC differentiation may depend on cell culture conditions and type of MSCs. Our studies further revealed that PD-1 maintains cell proliferation via a SHP2/ERK/Notch cascade and suppresses osteogenic differentiation via a SHP2/ERK/β-catenin cascade. Therefore, distinct intracellular signaling mechanisms regulate the self-renewal and maintenance of ESCs and MSC-DP.
In this study, we showed that the neural crest-derived MSC-DP express the transmembrane protein PD-1. PD-1 knockdown severely impairs MSC-DP proliferation and accelerates MSC-DP differentiation. PD-1 maintains cell proliferation via a SHP2/ERK/Notch cascade and suppresses osteogenic differentiation via a SHP2/ERK/β-catenin cascade. PD-1 can be used as a unique surface marker to isolate and characterize MSC-DP. This study identifies a previously unknown role of PD-1 in maintaining stem cell function.
Methods and materials
Animals
Beige nude/nude Xid (III) mice were purchased from Harlan (Indianapolis, IN, USA). All animal experiments were performed under institutionally approved protocol for the use of animal research (University of Pennsylvania IACUC #805478).
Antibodies and reagents
Anti-PD-1, PD-L1, OCN antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-runt related transcription factor 2 (Runx2), p-SHP2, SHP2, p-ERK, ERK, Notch1, NICD antibodies were purchased from Cell Signaling (Danvers, MA, USA). Active β-catenin and total β-catenin antibodies were purchased from Millipore (Billerica, MA, USA). Anti-β-actin antibody was purchased from Sigma-Aldrich Co (St. Louis, MO, USA). APC-conjugated anti-PD-1; PE-conjugated CD90, CD105, and CD73 were purchased from BD Biosciences (Franklin Lakes, NJ, USA). BrdU solution and BrdU imaging kit were purchased from Invitrogen (Carlsbad, CA, USA).
Isolation and culture of dental pulp MSCs
Human exfoliated deciduous incisors and third molars were obtained as discarded biological samples from the Dental Clinic of the University of Pennsylvania following the approved Institutional Review Board guidelines. SHED and DPSCs were cultured as reported previously [3, 5].
Isolation and culture of human BMMSCs
Human bone marrow aspirates from healthy human adult volunteers (20–35 years of age) were purchased from AllCells LLC. hBMMSCs were cultured as reported previously [40].
siRNA and chemical treatments
For siRNA transfection, SHED (0.5 × 106/well) were seeded in a 6-well culture plate under reduced serum medium (Opti-MEM, Gibco), and treated with PD-1 siRNA (Invitrogen), SHP2 siRNA (Santa Cruz Biotechnology), or control vehicle siRNA with lipofectamine reagent (Invitrogen) for 24 h, according to the manufacturer’s instructions. For chemical reagent treatments, serum-starved SHED were treated with 10 μM Wnt/β-catenin inhibitor (XAV939; Cayman Chemical), 5 μM ERK inhibitor (PD98059; Cayman Chemical), or 50 μM SHP2 inhibitor (NSC87877; Cayman Chemical) for 24 h. Treated cells were harvested and directly used for further experiments.
Cell proliferation assay
MSCs (10 × 103/well) were seeded on 2-well chamber slides (Nunc, Rochester, NY, USA) and cultured for 2–3 days. The cultures were incubated with BrdU solution (1:100) (Invitrogen) for 20 h, and stained with a BrdU staining kit (Invitrogen) according to the manufacturer’s instructions. The samples were then stained with hematoxylin. BrdU-positive and total cell numbers were counted in 10 images per subject. The number of BrdU-positive cells was indicated as a percentage of the total cell number. The BrdU assay was repeated on three independent samples for each experimental group.
Population doublings
MSCs were trypsinized and seeded at 2 × 105 cells in 35-mm dishes in complete growth medium at the first passage. Cells were harvested and seeded at the same number when they reached confluence. Population doublings (PD) were calculated by the following formula: PD = log2 (number of harvested cells/number of seeded cells). PD numbers were determined by the cumulative addition of total numbers generated from each passage until cells ceased dividing. The PD assay was repeated with 3 independent isolated cells for each group.
In vitro osteogenic differentiation assay
MSCs were cultured under osteogenic culture conditions in medium containing 1.8 mM monopotassium phosphate (Sigma-Aldrich) and 10 nM dexamethasone (Sigma-Aldrich). After 4 weeks of induction, the cultured cells were either stained with alizarin red to assess mineralized nodule formation or lysed for protein isolation to assess osteogenic gene expression.
Flow cytometry/cell sorting
SHED at passage 2 (P2) were trypsinized and washed in PBS supplemented with 2% heat inhibited FBS. Cells were then transferred to a 5 mL polystyrene tube (Falcon, Franklin Lakes, NJ, USA) and sorted using FACSAria II (BD Biosciences, San Jose, CA, USA). CD90+ SHED (PE-positive cells) were gated, and PD-1+ (APC-positive) SHED and PD-1− (APC-negative) SHED were collected for the following experiments.
Western blot analysis
Total protein was extracted using M-PER mammalian protein extraction reagent (Thermo, Rockford, IL). Twenty micrograms of protein were applied and separated on 4–12% NuPAGE gel (Invitrogen Co.), followed by transferring to nitrocellulose membranes (Millipore Inc.). Membranes were blocked with 5% non-fat dry milk and 0.1% Tween-20 for 1 h, followed by incubation with the primary antibodies at 4 °C overnight. HRP-conjugated secondary antibody (Santa Cruz Biotechnology; 1:10,000) was used to treat the membranes for 1 h. Immunoreactive proteins were detected using SuperSignal West Pico Chemiluminescent Substrate (Thermo) and BioMax film (Kodak, Rochester, NY, USA). Each membrane was also stripped using a stripping buffer (Thermo) and re-probed with anti-β-actin antibody to quantify the amount of loaded protein.
Implantation of SHED into immunocompromised mice
About 6.0 × 106 SHED were mixed with 40 mg of hydroxyapatite/tricalcium phosphate (HA/TCP) ceramic powder (Zimmer Inc.) and then implanted into the dorsal surface of 10-week-old immunocompromised mice as previously described [5]. These procedures were performed in accordance with specifications of an approved small animal protocol (University of Pennsylvania IACUC #805478). The implants were harvested 8 weeks post-implantation, fixed in 4% paraformaldehyde, and then decalcified with 10% EDTA (pH 8.0) for paraffin embedding. Paraffin sections were deparaffinized, rehydrated, and stained with hematoxylin and eosin (H&E). For quantification of new bone regeneration in vivo, we used 10 representative images from different regions of the SHED implants to calculate the area of bone formation using Image J software (NIH). This assay was repeated with 3 independent implants for each experimental group.
Statistics
SPSS 13.0 was used to perform statistical analysis. Comparisons between two groups were analyzed using independent two-tailed Student’s t-tests, and comparisons between more than two groups were analyzed using one-way ANOVA. P values <0.05 were considered statistically significant.
References
Chung IH, Yamaza T, Zhao H, Choung PH, Shi S, Chai Y. Stem cell property of postmigratory cranial neural crest cells and their utility in alveolar bone regeneration and tooth development. Stem Cells. 2009;4:866–77.
Zhao H, Chai Y. Stem cells in teeth and craniofacial bones. J Dent Res. 2015;94:1495–501.
Gronthos S, Mankani M, Brahim J, Robey PG, Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc Natl Acad Sci USA. 2000;97:13625–30.
Gronthos S, Brahim J, Li W, Fisher LW, Cherman N, Boyde A, et al. Stem cell properties of human dental pulp stem cells. J Dent Res. 2002;81:531–5.
Miura M, Gronthos S, Zhao M, Lu B, Fisher LW, Robey PG, et al. SHED: stem cells from human exfoliated deciduous teeth. Proc Natl Acad Sci USA. 2003;100:5807–12.
Sakai VT, Zhang Z, Dong Z, Neiva KG, Machado MA, Shi S, et al. SHED differentiate into functional odontoblasts and endothelium. J Dent Res. 2010;89:791–6.
Zhao Y, Wang L, Jin Y, Shi S. Fas ligand regulates the immunomodulatory properties of dental pulp stem cells. J Dent Res. 2012;91:948–54.
Li Z, Jiang CM, An S, Cheng Q, Huang YF, Wang YT, et al. Immunomodulatory properties of dental tissue-derived mesenchymal stem cells. Oral Dis. 2014;20:25–34.
Yamaza T, Kentaro A, Chen C, Liu Y, Shi Y, Gronthos S, et al. Immunomodulatory properties of stem cells from human exfoliated deciduous teeth. Stem Cell Res Ther. 2010;1:5.
Liu Y, Wang L, Liu SY, Liu DW, Chen C, Xu XT, et al. Transplantation of SHED prevents bone loss in the early phase of ovariectomy-induced osteoporosis. J Dent Res. 2014;93:1124–32.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–95.
Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–72.
Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34.
Latchman Y, Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8.
Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010;236:219–42.
Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–51.
Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22.
Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015;21:24–33.
English K, Barry FP, Field-Corbett CP, Mahon BPIFN-gamma. and TNF-alpha differentially regulate immunomodulation by murine mesenchymal stem cells. Immunol Lett. 2007;110:91–100.
Pedoeem A, Azoulay-Alfaguter I, Strazza M, Silverman GJ, Mor A. Programmed death-1 pathway in cancer and autoimmunity. Clin Immunol. 2014;153:145–52.
Jin HT, Ahmed R, Okazaki T. Role of PD-1 in regulating T-cell immunity. Curr Top Microbiol Immunol. 2011;350:17–37.
Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54.
Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209:1201–17.
Feng GS. Shp2-mediated molecular signaling in control of embryonic stem cell /proliferation and differentiation. Cell Res. 2007;17:37–41.
Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self-renewal/proliferation and differentiation. Curr Top Dev Biol. 2010;92:367–409.
Liu Y, Chen C, Liu SY, Liu DW, Xu XT, Chen X, Shi S. Acetylsalicylic acid treatment improves differentiation and immunomodulation of SHED. J Dent Res. 2015;94:209–18.
Chen L, Pai V, Levinson R, Sharpe AH, Freeman GJ, Braun J, et al. Constitutive neuronal expression of the immune regulator, programmed death 1 (PD-1), identified during experimental autoimmune uveitis. Ocul Immunol Inflamm. 2009a;17:47–55.
Chen L, Sham CW, Chan AM, Francisco LM, Wu Y, Mareninov S, et al. Role of the immune modulator programmed cell death-1 during development and apoptosis of mouse retinal ganglion cells. Invest Ophthalmol Vis Sci. 2009b;50:4941–8.
Wang W, Chan A, Qin Y, Kwong JM, Caprioli J, Levinson R, et al. Programmed cell death-1 is expressed in large retinal ganglion cells and is upregulated after optic nerve crush. Exp Eye Res. 2015;140:1–9.
Augello A, Tasso R, Negrini SM, Amateis A, Indiveri F, Cancedda R, et al. Bone marrow mesenchymal progenitor cells inhibit lymphocyte proliferation by activation of the programmed death 1 pathway. Eur J Immunol. 2005;35:1482–90.
Ma L, Makino Y, Yamaza H, Akiyama K, Hoshino Y, Song G, et al. Cryopreserved dental pulp tissues of exfoliated deciduous teeth is a feasible stem cell resource for regenerative medicine. PLoS ONE. 2012;7:e51777.
Jeon M, Song JS, Choi BJ, Choi HJ, Shin DM, Jung HS, et al. In vitro and in vivo characteristics of stem cells from human exfoliated deciduous teeth obtained by enzymatic disaggregation and outgrowth. Arch Oral Biol. 2014;59:1013–23.
Nakamura T, Gulick J, Colbert MC, Robbins J. Protein tyrosine phosphatase activity in the neural crest is essential for normal heart and skull development. Proc Natl Acad Sci USA. 2009;106:11270–5.
Stewart RA, Sanda T, Widlund HR, Zhu S, Swanson KD, Hurley AD, et al. Phosphatase dependent and independent functions of Shp2 in neural crest cells underlie LEOPARD syndrome pathogenesis. Dev Cell. 2010;18:750–62.
Neel BG, Gu H, Pao L. The shping news: SH2 domain-containing protein tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–93.
Schindeler A, Little DG. Ras-MAPK signaling in osteogenic differentiation: friend or foe? J Bone Miner Res. 2006;21:1331–8.
Jaiswal RK, Jaiswal N, Bruder SP, Mbalaviele G, Marshak DR, Pittenger MF. Adult human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by mitogen-activated protein kinase. J Biol Chem. 2000;275:9645–52.
Kretzschmar M, Doody J, Massagué J. Opposing BMP and EGF signalling pathways converge on the TGF-beta family mediator Smad1. Nature. 1997;389:618–22.
Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H, et al. Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. J Bone Miner Res. 2002;17:1785–94.
Shi S, Gronthos S, Chen S, Reddi A, Counter CM, Robey PG, et al. Bone formation by human postnatal bone marrow stromal stem cells is enhanced by telomerase expression. Nat Biotechnol. 2002;20:587–91.
Acknowledgements
This work was supported by Beijing Tason Biotech Co. LTD and National Natural Science Foundation of China (81600825 to Y.L.).
Author contributions
Y.L. designed and performed the experiments, analyzed and interpreted the data, and wrote the manuscript. H.J., X.X., C.C., and D.L. performed the experiments and analyzed and interpreted the data. Y.J., L.L., and S.S. designed the experiments, analyzed the data, wrote the manuscript, and supervised the laboratory studies.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Liu, Y., Jing, H., Kou, X. et al. PD-1 is required to maintain stem cell properties in human dental pulp stem cells. Cell Death Differ 25, 1350–1360 (2018). https://doi.org/10.1038/s41418-018-0077-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-018-0077-8
This article is cited by
-
TA-MSCs, TA-MSCs-EVs, MIF: their crosstalk in immunosuppressive tumor microenvironment
Journal of Translational Medicine (2022)
-
Blockade of PD-L1/PD-1 signaling promotes osteo-/odontogenic differentiation through Ras activation
International Journal of Oral Science (2022)
-
CD146 controls the quality of clinical grade mesenchymal stem cells from human dental pulp
Stem Cell Research & Therapy (2021)
-
Role of PD-L1 in licensing immunoregulatory function of dental pulp mesenchymal stem cells
Stem Cell Research & Therapy (2021)
-
Emerging understanding of apoptosis in mediating mesenchymal stem cell therapy
Cell Death & Disease (2021)
