
Abstract
Clinical efficacy of intravitreal anti-VEGF drugs has been widely demonstrated in several angiogenesis-driven eye diseases including diabetic macular edema and the neovascular form of age-related macular degeneration. Pegaptanib, ranibizumab, and aflibercept have been approved for use in the eye, whereas bevacizumab is widely used by ophthalmologists to treat patients “off-label”. These drugs are active in the nanomolar to picomolar range; however, caution is required when establishing the rank order of affinity and potency due to in vitro inter-experimental variation. Despite the small doses used for eye diseases and the intravitreal route of administration may limit systemic side effects, these drugs can penetrate into blood circulation and alter systemic VEGF with unknown clinical consequences, particularly in vulnerable groups of patients. Clinical pharmacokinetics of ocular anti-VEGF agents should therefore be taken into account when choosing the right drug for the individual patient. The gaps in current understanding that leave open important questions are as follows: (i) uncertainty about which drug should be given first, (ii) how long these drugs can be used safely, and (iii) the choice of the best pharmacological strategy after first-line treatment failure. The current review article, based on the information published in peer-reviewed published papers relevant to anti-VEGF treatments and available on the PubMed database, describes in detail the clinical pharmacology of this class of drugs to provide a sound pharmacological basis for their proper use in ophthalmology clinical practice.
Similar content being viewed by others

Introduction
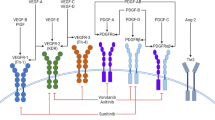
Angiogenesis plays an important role in tissue development and function and in the pathogenesis of many ocular diseases, including ocular ischemic syndrome, proliferative retinopathies, and neovascular glaucoma [1]. Angiogenesis was found to be regulated by a complex signaling network composed by vascular endothelial growth factors (VEGFs) and their cognate receptors (VEGFRs), placental growth factor (PlGF), angiopoietin and Tie receptors, platelet-derived growth factor-B (PDGF-B), stromal-derived factor-1 (SDF-1), hypoxia-inducible factor-1 (HIF-1), and signals from extracellular matrix [2]. The VEGF–VEGFR pathway has been shown to be important in regulating embryo vasculogenesis as well as adult angiogenesis [3]. In the eye, VEGF is mainly produced by vascular endothelial cells or pericytes and also by retinal neurons and astrocytes, Müller cells, retinal pigment epithelium, and non-pigmented ciliary epithelium [4]. Low-oxygen conditions cause upregulation of VEGF through the induction of HIF-1 and the consequent transcriptional activation of target genes [5]. Increased VEGF transcription and upregulation of angiogenesis serve to restore oxygen and nutrition supply for tissues affected by hypoxia [6]. VEGF may also contribute to the inflammatory process by inducing the expression of vascular cell adhesion molecule 1 (VCAM-1) enhancing leukocyte recruitment and endothelial cell adhesion and increasing blood–retinal blood barrier breakdown [5]. Beside angiogenesis, inflammation may also be involved in the development and progression of eye diseases such as retinal vein occlusion (RVO), diabetic retinopathy, neovascular age-related macular degeneration (AMD), or neovascular glaucoma [5]. For these reasons, anti-VEGF therapy represents as a potent and effective weapon against neovascular AMD, complications of diabetic retinopathy, and RVOs.
Structural features
Intravitreal anti-VEGF agents are the result of innovative biotechnology processes aimed at creating high-affinity-targeted drugs. Different structural features of these molecules represent the fundamental basis for the comprehension of their clinical pharmacology. Following are the approved drugs for human use in eye: pegaptanib, ranibizumab, and aflibercept. Pegaptanib was the first aptamer approved for use in humans. It is a 40-kDa RNA polyethylene glycol-linked molecule having a VEGF-binding sequence of 27 nucleotides plus an additional 3′-3′-terminal deoxythymidine [7]. Ranibizumab is a 48-kDa recombinant humanized immunoglobulin G1κ isotype monoclonal antibody fragment (Fab) devoid of the Fc portion [8] Aflibercept is a 115-kDa fusion protein obtained combining the Fc portion of a full monoclonal antibody and the two highest affinity domains of VEGF receptor type-1 (R1) and VEGFR2 [9]. Bevacizumab is a fully humanized IgG1 of 148 kDa administered by intravenous route in cancer patients [10]. This drug is also widely used intravitreally by ophthalmologists to treat patients “off-label” since there is no Federal Drug Administration (FDA) approval for it to be used as the treatment of wet AMD or diabetic macular edema (DME).
Pharmacodynamics
Intravitreal anti-VEGF drugs inhibit the functional activity of proangiogenic factors with different target selectivity, affinity, and potency. Pegaptanib selectively binds to VEGF165, whereas ranibizumab and bevacizumab bind to all the VEGF-A isoforms, while aflibercept is able to trap VEGF-A, VEGF-B, and PlGF (Table 1). Two major parameters that are used as pharmacodynamic biomarkers are as follows: the “drug affinity” and the “potency”. Drug affinity measures how strong a drug can bind to its receptor, while potency is the amount of drug needed to produce a pharmacological effect (the smaller the dosage required, the more potent the drug) (Fig. 1). All these drugs have affinity and potency in the nanomolar to picomolar range with a remarkable between-study variation [11,12,13,14,15]. For example, in some studies the dissociation constant (Kd) value is tenfold lower for ranibizumab than bevacizumab (<179 and 1800 pM, respectively) [11, 13], whereas others found that the two drugs had similar affinities (Kd values of 46 and 58 pM, respectively) [12]. This discrepancy could be due to the different methodologies used to evaluate the parameter (e.g., ELISA versus Biacore) and the assay used, particularly when binding kinetics and affinities were measured by Biacore [14]. Indeed, when measured over three assay formats, Kd values for ranibizumab ranged from <9.2 to 67 pM, those for bevacizumab from 75.4 to 4456 pM, and those for aflibercept from 1.8 to 9263 pM [14]. Interesting to note, Biacore analysis by Papadopoulos et al. showed a binding affinity for aflibercept about 100-fold higher than ranibizumab and bevacizumab with a Kd value of 0.49 pM [12]. On the other hand, there was no difference in terms of VEGF affinity between aflibercept and ranibizumab when sedimentation velocity analytical ultracentrifugation was used to support the binding affinities determined by Biacore [14] (Table 1).

Mechanism of action of intravitreal antiangiogenic drugs. Angiogenic factors (e.g., VEGF) stimulate their specific receptors (e.g., VEGFR) activating the signaling cascade. Binding of intravitreal antiangiogenic drugs to their ligands (e.g., VEGF) form the drug–ligand complex (DL) preventing ligand/receptor interaction and keeping (or turning) the angiogenic switch off. Affinity is defined as the degree of attraction between drug and the target, and it is expressed by the dissociation constant, Kd (that is, the ratio between dissociation (Koff) and association (Kon) rates). Kd is the inverse of the affinity to the binding site (i.e., the lower the Kd, the higher the affinity). Potency is the amount of drug needed to produce the pharmacological effect. For these drugs, potency is expressed by the half maximal inhibitory concentration (IC50), a measure of the effectiveness of a substance in inhibiting a specific biological function (the lower the IC50, the higher the potency)
Potency of anti-VEGF drugs was tested on VEGF-A- or PlGF-2-induced activation of VEGFR1 using a specific luciferase assay in human HEK293 cells [12]. In these experimental models, aflibercept showed a 45–92-fold greater blocking potency compared to either ranibizumab or bevacizumab, with an IC50 mean value of 16 pM for blocking VEGFR1 activation induced by 20 pM VEGF-A165 (Table 1). Furthermore, aflibercept also blocked luciferase activity induced by human PlGF-2 (40 pM) with an IC50 value of 2.9 nM [12]. These findings are in contrast with those obtained through bioassay analyses of VEGF-stimulated proliferation of bovine retinal microvascular endothelial cells, a well-established physiologically relevant cell type to investigate angiogenesis [14, 15]. In this in vitro model, ranibizumab and aflibercept were found to have very similar potencies (IC50 values of 88 and 90 pM, respectively), whereas bevacizumab was about fivefold less potent (IC50 value of 500 pM) than its competitors [15] (Table 1). Finally, it has been recently demonstrated by scratch test assay that ranibizumab was more effective than aflibercept in reducing the angiogenic potential of the human endothelial vascular cell line HECV [16]. As the authors stated, these findings appear to be noteworthy since choroidal neovascularization (i.e., a pathologic feature of several vascular diseases of the retina) can be referred to as wound healing or tissue repair [16]. Binding studies performed on human umbilical vein endothelial cells and on human dermal microvascular endothelial cells demonstrated that pegaptanib inhibited binding of 125I-labeled VEGF to its receptor with IC50 values in a concentration range of 0.75–1.4 nM [7] (Table 1).
Pharmacokinetics of anti-VEGF drugs
Pharmacokinetics of intravitreal anti-VEGF drugs has been studied in experimental models as well as in humans with remarkable interspecies variation. In a rabbit model, the vitreous half-life (t1/2) of VEGF-Trap (3.63 days) [17] has proven to be shorter than that of bevacizumab (6.99 days) [18] and longer than that of ranibizumab (2.51 days) [19]. In apparent contrast with these findings are those showing that t1/2 values in aqueous humor of macaque eyes were similar for ranibizumab and aflibercept (2.3 and 2.2 days, respectively) [20]. Vitreous elimination t1/2 for ranibizumab was calculated to be 9 days in patients with AMD [21] and 7.2 days in non-vitrectomized eyes of patients with both clinically significant cataract and macular edema secondary to diabetic retinopathy [22]. In patients with choroidal neovascularization, vitreous pharmacokinetics of bevacizumab followed a two-compartment model with a terminal t1/2 of 6.7 days [23]. Thus, notwithstanding these molecules have different molecular sizes and structures they display comparable mean vitreous t1/2 in humans.
Several evidences from preclinical and clinical studies clearly demonstrated that the presence of the neonatal Fc receptor (FcRn) in the blood retinal barrier might affect the ability of intravitreally injected Fc-carrying molecules to penetrate into the systemic circulation (Fig. 2). The FcRn-dependent penetration of intravitreal-administered bevacizumab through the blood retinal barrier into the blood system has been demonstrated in wild type, but not in FcRn knockout mice [24]. These findings are in line with those derived from a pharmacokinetic study investigating intravitreal ranibizumab (0.5 mg) and bevacizumab (1.25 mg) in a rabbit model [25]. In this experimental condition, small amounts of intravitreal bevacizumab have been detected in the serum as well as in the fellow uninjected eye, while no ranibizumab was detected [25].

Pharmacokinetic model for intravitreal antiangiogenic drugs. Intravitreally injected Fc-carrying molecules can penetrate into the systemic circulation by a FcRn-dependent transport. The presence of the Fc portion may also reduce systemic clearance thus prolonging drug exposure (see text for further details). Renal clearance may depend on the molecular size; in particular, the higher the molecular weight, the lower the drug clearance (e.g., pegylated molecules, full-length monoclonal antibodies). FcRn neonatal Fc receptor, BRB blood retinal barrier
Beyond distribution from the eye into the blood circulation, systemic clearance of intravitreal anti-VEGF drugs may depend on their molecular size and presence of the Fc function. For example, the pegylated-aptamer pegaptanib displays a mean plasma t1/2 of about 7–8 days for an intravitreal dose of 0.3 mg [26], with no plasma accumulation after eight doses repetition [26]. In particular, those with lower creatinine clearance and/or weight had higher pegaptanib plasma concentrations. Full-length mAbs are not excreted into urine because of their molecular size [27], while they are recycled from endothelial cells back into the bloodstream though a FcRn-mediated mechanism [27, 28]. In line with these data, systemic pharmacokinetics of intravenous bevacizumab follows a two-compartmental model, with an elimination t1/2 of about 19 days [29]. At variance with this, the systemic elimination t1/2 for ranibizumab following intravitreal administration was calculated to be ~2 h, although the apparent plasma t1/2 of ranibizumab would instead be 9 days, that is equivalent to its vitreous t1/2 because vitreous elimination is the rate-limiting step [21]. Aflibercept structure was specifically designed to extend in vivo t1/2 while maintaining the high affinity of the initial soluble decoy receptor; this aim was obtained by fusing the first three Ig domains of VEGFR1 to the Fc portion of human IgG1. The prolonged in vivo pharmacokinetics of aflibercept warrants an effective suppression of the growth and vascularization of tumors in vivo [30]. Although no data are available in humans for intravitreal aflibercept, terminal elimination t1/2 of free drug in plasma was estimated to be 5–6 days after intravenous injection [31]. Accordingly, while aflibercept is also currently used as an anticancer agent, ranibizumab is licensed only for intravitreal administration (Table 1).
These findings are in line with those recently obtained in wet AMD patients administered intravitreally with 0.5 mg ranibizumab, 1.25 mg bevacizumab, or 2 mg aflibercept, where systemic exposure levels, in terms of peak and trough concentrations and area under the curve, were bevacizumab > aflibercept > ranibizumab with no evidence of accumulation after repeated doses for ranibizumab only [32].
These findings clearly showed that Fab fragments are eliminated more rapidly than intact mAbs, which can be explained by the fact that these molecules lack an Fc part and hence protection by the FcRn (Fig. 2).
Pharmacokinetic–pharmacodynamic relationships
One important point that needs to be addressed is whether plasma concentrations reached after intravitreal administration of anti-VEGF drugs can produce measurable effects on target proteins. In patients with neovascular AMD, aflibercept substantially suppressed plasma-free VEGF to mean levels below lower limit of quantitation (10 pg/ml) starting from 3 h until ≥7 days post dose. Ranibizumab, instead, did not affect mean free VEGF levels with mean trough level of 14.4 pg/ml compared with baseline of 17 pg/ml [32]. This is in line with data showing that, after monthly and quarterly intravitreal administration, the plasma concentrations of ranibizumab at steady state (for both the 0.3 and 0.5 mg per eye dose levels) were estimated to be below the range needed to inhibit VEGF-A-induced endothelial cell proliferation in vitro by 50% (i.e., IC50) [21]. Plasma VEGF levels were also strongly reduced (about 80%) by bevacizumab from 89.7 pg/ml to 25.1 pg/ml after 7 days (p = 0.01), and to 22.8 pg/ml after 1 month (p = 0.008), in patients with exudative AMD [33]. Noteworthy, the same results were obtained in patients with DME, where systemic VEGF reduction by bevacizumab was observed with a significant decrease of baseline level from 72.2 pg/ml to 13.7 pg/ml after 7 days (p = 0.008) and 17.1 pg/ml at 4 weeks with (p = 0.012). No significant reductions of plasma VEGF levels were observed in patients receiving ranibizumab or pegaptanib during follow-up [33]. Furthermore, Wang et al. found that aflibercept decreased the baseline VEGF levels from 28.3 pg/ml to below the detectable limit at 1 week (p < 0.0001) in neovascular AMD patients, whereas no significant difference was observed in the ranibizumab group [34]. The systemic inhibitory effect on VEGF-A was more pronounced in neovascular AMD patients receiving intravitreal aflibercept compared to those administered with bevacizumab, while ranibizumab did not affect systemic VEGF [35]. Noteworthy, they also found a significant upregulation, a slight statistically non-significant increase and no detectable effects on systemic PlGF after intravitreal aflibercept, bevacizumab, and ranibizumab, respectively [35]. Since it has been widely recognized that PlGF acts as a synergistic amplifier of VEGF-driven angiogenesis [36], these findings demonstrate the presence of a host counter-regulatory response to systemic VEGF-A inhibition induced by intravitreal aflibercept, bevacizumab, and ranibizumab, respectively [35].
Clinical efficacy
Efficacy of anti-VEGF therapy has been proven by several independent phase III clinical trials. For instance, the CRUISE and BRAVO studies demonstrated the efficacy of monthly ranibizumab for macular edema after central RVO [37, 38]. Specifically, patients with macular edema due to RVO experienced clinically and statistically significant improvements in best-corrected visual acuity (BCVA), as compared with patients receiving sham injections. Alternative treatment schedules with less frequent than monthly ranibizumab injection are currently used in AMD patients and could also be applied to patients with RVO. The HORIZON trial assessing long-term safety and efficacy of intraocular ranibizumab injections in patients with macular edema after RVO demonstrated that reduced follow-up and fewer ranibizumab injections in the second year of treatment were associated with a decline in vision in central RVO patients, while vision in branch RVO patients remained stable. Therefore, central RVO patients may require more frequent follow-up than every 3 months [39].
DME represents another important field of application of anti-VEGF therapy. The RESOLVE, RESTORE, RISE, and RIDE studies for DME [40,41,42,43,44] demonstrated how single-agent ranibizumab gained the best BCVA when compared to laser treatment alone or in association with laser treatment itself. In 2014, aflibercept also obtained the FDA approval for DME based on two studies, VISTADME and VIVIDDME, aimed at comparing aflibercept with macular laser photocoagulation. Patients treated with aflibercept were able to read, on an average, two additional lines on an eye chart, while control patients showed no improvement [45]. When initial visual acuity loss was mild, there were no apparent differences among treatment groups in terms of efficacy, whereas patients with a severe loss of visual acuity at baseline respond better to aflibercept than bevacizumab or ranibizumab [46].
The largest application of anti-VEGF agents in ophthalmology is represented by neovascular AMD [47,48,49]. Pegaptanib, the first VEGF inhibitor to obtain US FDA approval for CNV in AMD in 2004, is administered as an intravitreal injection every 6 weeks. The VEGF Inhibition Study in Ocular Neovascularization (VISION) trial demonstrated that 70% of patients treated with pegaptanib lost less than three lines of vision compared with 55% of controls (p < 0.001). Unfortunately, a minority of patients gained vision with this therapy [50]. Since 2004 till 2006, prior to ranibizumab approval, anecdotal evidence led to the widespread off-label use of bevacizumab in wet AMD despite the lack of clinical evidence to support its safety or efficacy. In 2006, data from the Minimally Classic/Occult Trial of the anti-VEGF antibody ranibizumab in the treatment of neovascular AMD (MARINA) trial, demonstrated the efficacy of ranibizumab in patients with minimally classic or occult CNV secondary to AMD. Over 2 years, monthly injections of ranibizumab significantly reduced retinal thickness with ~90% of patients losing fewer than 15 lines of vision [51]. The anti-VEGF antibody for the treatment of predominantly classic choroidal neovascularization in AMD (ANCHOR) study demonstrated that ~40% of patients treated with monthly ranibizumab injections had gained 15 or more lines of vision compared with 6% of PDT-treated patients [52]. Many efforts have been made to understand the best anti-VEGF regimen to be used. Current guidelines for ranibizumab suggest starting with an initiation/induction phase followed by an individual maintenance phase that can be achieved by the following two different approaches: traditional PRN or “treat and extend” [53, 54]. The analysis of these reports highlights an important trend: the best visual acuity results derived from the study with the largest average number of treatments and the closest follow up, whereas the poorest outcomes were observed in the study with the lowest mean number of treatments and office visits. No difference in efficacy was observed between bevacizumab and ranibizumab, when the two drugs were administered with the same treatment schedule [55].
Aflibercept received the FDA approval for the treatment of neovascular AMD at the recommended dose of 2.0 mg every 8 weeks after an induction period of 3 monthly injections. Clinical efficacy of intravitreal aflibercept was supported by VIEW 1 and VIEW 2, two randomized, multicenter, double-masked, controlled clinical trials that demonstrated noninferiority of aflibercept compared to ranibizumab [53, 54]. Noteworthy, the use of a less frequent treatment regimen (i.e., ranibizumab every 2 months instead of monthly injections) may reduce treatment burden for patients and healthcare costs. With particular regard to neovascular AMD, it is also important to emphasize how the availability of several anti-VEGF molecules may be a great advantage for those patients who do not respond to or develop tachyphylaxis after first-line treatments.
There have also been attempts to apply anti-VEGF drugs in the management of other forms of diabetic retinopathy, retinopathy of prematurity, and choroidal neovascularisation caused by angioid streaks, pathologic myopia, traumatic choroidal rupture, or ocular histoplasmosis [56], and for the treatment of corneal neovascularisation [57]. Furthermore, anti-VEGF drugs might be used as potent adjuvant treatments in glaucoma filtering surgery and in the management of neovascular glaucoma [58]. Based on current knowledge, anti-VEGF therapy neither causes structural destructive changes in the retina (as lasers do), nor induces cataract formation (as steroids do) [56].
Local and systemic safety concerns
The LacZ-tagged allele is a knock-in strategy generated to assess the role of VEGF during embryonic development. It consists in introducing a LacZ reporter gene into the 3′-untranslated region of the endogenous VEGF locus by homologous recombination [59]. The VEGF-LacZ mice have also been used to demonstrate that VEGF is expressed in virtually every tissue, having a pivotal role in adults to stabilize mature vessels [60]. Therefore, it is important to take into account any possible local and systemic adverse events in patients receiving intravitreal anti-VEGF drugs. For example, it has been proposed that anti-VEGF injections may accelerate the onset and progression of geographic atrophy (GA) in eyes previously affected by choroidal neovascularization [61]. Preclinical models demonstrated indeed that endogenous VEGF provides critical trophic support necessary for retinal function. For example, conditionally knocking out VEGF-A in adult mouse retinal pigmented epithelial cells leads to vision loss and ablation of the choriocapillaris [62]. Furthermore, mice lacking of VEGF120 and 164 isoforms were shown to experience changes similar to GA including atrophy of the choriocapillaris, abnormalities of retinal pigment epithelium and Bruch’s membrane as well as increased photoreceptor apoptosis [63]. Although not yet tested in prospective studies, preclinical evidences and reviewed data from the IVAN, CATT, and HARBOR trials [64,65,66] seem to support such an hypothesis.
Systemically delivered anti-VEGF drugs are widely recognized to reduce vascular hyperpermeability, raise systemic arterial blood pressure and promote the development of thromboembolic events [67]. Furthermore, preclinical findings demonstrated that a decoy VEGFR promotes left ventricular dilatation and contractile dysfunction [68].
Bevacizumab treatment has been associated with an increased risk of significant heart failure in patients with breast cancer [69]. Furthermore, it has been reported that the lowest approved doses for intravenous bevacizumab (i.e., 2.5 mg/week) can be sufficient for reaching the saturation level to promote cardiac function impairment [69]. Therefore, on the basis of these considerations, it could be assumed that these same risks might occur after intravitreal injection, provided that anti-VEGF drugs are able to reach the circulation at biologically active concentrations.
In the present paper, we have already discussed about structural and pharmacological differences among ocular anti-VEGF drugs. Compared to bevacizumab and aflibercept, ranibizumab lacks the Fc portion and has a small molecular size, which account for its lower systemic exposure and reduced ability to suppress circulating plasma-free VEGF following intravitreal injection. Even though only small amounts of bevacizumab and aflibercept are released from the eye into the systemic circulation, compared with the amount released after the high dosages used in oncology, mean serum concentrations of these drugs after intravitreal administration remained above their IC50 values for VEGF-A ≥ 7 days post dose. As previously mentioned, these exposure levels were sufficient to suppress circulating VEGF in vivo [32]. Overall, these findings underline the importance to consider clinical pharmacokinetic and pharmacodynamics parameters of ocular anti-VEGF drugs as clinically meaningful risk indicators for systemic cardiovascular adverse events.
The clinical question is whether extensive systemic VEGF inhibition actually increases the risk of cardiovascular side effects in patients with AMD and/or DME. This is currently unknown since registration trials were not statistically powered to answer this specific question. A study designed with this aim would have to enroll tens of thousands of patients to detect a meaningful difference for these rare events, especially in a target population with comorbidities that include the study end points. Furthermore, it is worthy of mention that absence of evidence is not evidence of absence and, based on compelling clinical trial subgroup analysis and pharmacokinetic–pharmacodynamic considerations, the risk of cardiovascular adverse events after intravitreal administration of anti-VEGF drugs cannot be excluded a priori, particularly in patients with comorbidities. Finally, even if we do not have a direct demonstration on such a relationship, we do have some indirect evidences of it. For example, it has been reported that, in patients not treated with intravitreal anti-VEGF drugs who experienced a myocardial infarction (n = 293), low plasma VEGF levels (<61.0 pg/ml) 7 days after the onset of the acute episode were associated with a significantly increased risk for further major adverse cardiovascular and cerebrovascular events (i.e., cardiac death, recurrent acute coronary syndrome, hospital readmission for heart failure, or stroke) compared to those with middle (61–176 pg/ml; n = 294) and high (≥176 pg/ml; n = 292) levels [70]. Interestingly, 7 days after intravitreal bevacizumab, baseline plasma VEGF levels were 25.1 pg/ml (p = 0.01) and 13.7 pg/ml (p = 0.008) in patients with ARMD and DME, respectively [33]. After intravitreal injection of aflibercept, plasma VEGF-A levels at baseline were also significantly low (p < 0.001): values were below the minimum detectable dose in 17 of 19 patients, resulting in a median and an interquartile range of <9.0 pg/ml, 7 days post treatment [35].
This information should be taken into careful account in vulnerable groups, including patients of 85 years and older, those with prior strokes, diabetics with significant comorbidities, pregnant and lactating women, presence of retinopathy of prematurity, and previous experience of adverse events likely related to VEGF suppression during intravitreal anti-VEGF therapy. Strategy for treatment optimization of intravitreal anti-VEGF treatment in these conditions could comprise drugs with less systemic exposure (e.g., ranibizumab instead of bevacizumab or aflibercept) administered at the minimum effective dose (e.g., 0.3 versus 0.5 mg for ranibizumab).
The role of PlGF deficiency in ischemic cardiovascular disease has been recently demonstrated [71]. It opens another intriguing question regarding the clinical consequences associated to a prolonged systemic PlGF inhibition in patients with eye diseases treated with aflibercept. PlGF-deficient mice display reduced angiogenesis in the border zone of the infarcted myocardium, whereas PlGF gene or protein transfer in infarcted mice stimulates angiogenesis improving cardiac recovery [71]. It has also been reported that elevated serum levels of the PlGF trap, sFLT1, may be an independent predictor of adverse outcome in patients with suspected acute myocardial infarction [72, 73]. Nonetheless, it is worth mentioning that systemic upregulation of PlGF induced by intravitreal aflibercept (as a counter-regulatory response to VEGF-A inhibition) cannot be prevented by its PlGF antagonism [35], suggesting that systemic aflibercept concentrations could be insufficient to alter PlGF homeostatis. Aflibercept has indeed an IC50 for PlGF in the nanomolar range (i.e., 2.89 nM) [12], and systemic concentrations reached after intravitreal injections are well under this value [32].
Predictive factors of drug response
There is uncertainty about which anti-VEGF drug should be given first, how long these drugs can be used safely and the appropriate pharmacological strategy after the first-line treatment failure (e.g., drug dosage and drug-free intervals, combination treatments with drugs from a different class, and treatment with different members within the same class).
A trade-off analysis aimed at comparing efficacy and safety for all currently used anti-VEGF formulations in patients with AMD found no substantial difference between aflibercept 2 mg and ranibizumab 0.5 mg. Furthermore, these treatments demonstrated only a modest superiority over ranibizumab 0.3 mg, aflibercept 0.5 mg, and bevacizumab 1.25 mg [74]. Therefore, the equivalence in terms of efficacy among the anti-VEGF drugs used in these studies point to VEGF-A as a major determinant of drug response in wet AMD, whereas VEGF-B and PlGF appeared to have a secondary role. An interesting clinical question is whether these latter angiogenic factors may be considered as predictors of drug response in angiogenesis-based eye diseases other than AMD. Some clinical difference among anti-VEGF drugs has emerged with aflibercept leading to a greater visual acuity improvement over bevacizumab or ranibizumab in the DME patients at worse levels of initial visual acuity [46, 75]. At variance with other anti-VEGF agents, aflibercept can also block PlGF with high affinity and preclinical investigations demonstrated that PlGF is a key angiogenic and proinflammatory factor that may play a role in ocular angiogenesis [76,77,78]. Although these findings suggest that patients with worse baseline visual acuity may benefit better from aflibercet than ranibizumab or bevacizumab, caution is required before translating these results into clinical recommendations. Indeed, it has been recently demonstrated that, while aflibercept maintained superior 2-year visual acuity outcomes compared with bevacizumab, superiority of aflibercept over ranibizumab noted at 1 year was no longer observed at year 2 with respect to the worse seeing group [79]. According to preclinical data showing a substantial equivalence between ranibizumab and aflibercept in terms of VEGF-A affinity and potency [14, 15], the long-term comparable benefit produced by these two drugs suggests that VEGF-A remains the key determinant of drug response also in DME patients and that the ability to block the function of VEGF-A is the major mechanism of action responsible for aflibercept efficacy. To further reinforce this notion, compelling evidences were provided that VEGF-A, but not PlGF, impairs the barrier function of immortalized bovine retinal endothelial cells, a well-recognized model able to predict the altered permeability of retinal endothelial cells observed in diabetic retinopathy [80].
Another interesting clinical question is whether the raise in other angiogenic signaling pathways aimed to compensate the blocked activity of VEGF might be responsible for the attenuated response after repeated administration of anti-VEGF drugs observed in some clinical trials. Several lines of evidence suggest a role of PlGF in coordination with VEGF-A during ocular angiogenesis. For example, it has been clearly demonstrated in transgenic mice that endothelial cells can amplify their responsiveness to VEGF during the angiogenic switch by upregulating PlGF [81]. These finding are in agreement with those showing that co-inhibition of VEGF-A and PlGF significantly reduces the vessel density in a laser burn-induced experimental choroideal neovascularization mouse model [78]. Finally, intravitreal aflibercept has been demonstrated to simultaneously upregulate and downregulate systemic PlGF and VEGF-A, respectively, in AMD patients [35].
Although these evidences seem to support the role of PlGF as a possible mechanism of drug resistance, a lot of factors other than PlGF could be associated to poor or non-responsive phenotype. These include: suboptimal dosing, prolonged dosing intervals, delayed administration (i.e., when disease is already at an advanced stage), lesion type, genetic variation and tachyphylaxis/tolerance [82,83,84,85,86,87]. It is also worth mentioning that poor responders are a minority compared to patients who benefit from treatment with anti-VEGF drugs. In particular, it has been found that only 15% of treated patients did not sufficiently respond to ranibizumab or bevacizumab (loss of three lines of distance acuity, increase of retinal thickness or lesion size) [88]. Furthermore, based on their molecular mechanism of action it is expected that clinical drug resistance to anti-VEGF drugs due to tachyphylaxis is uncommon. As a matter of fact, of 976 patients treated with ranibizumab, only 2% of them developed tachyphylaxis [89] and tachyphylaxis was observed in only 8% of patients treated with intravitreal bevacizumab after 100 weeks and eight intravitreal injections. Noteworthy in this study, non-response could not be rescue with increasing dosage [90]. Tolerance is a slow loss of efficacy over time that could occur as a consequence of an increased expression of VEGF or its receptors, changes in signal transduction pathways, a switch towards angiogenic factors other than VEGF-A (pharmacodynamic tolerance), and the development of neutralizing antibodies (pharmacokinetic tolerance). In line with this notion, significant benefit for non-responders were commonly observed in tolerant patients when treatment was changed to a different anti-VEGF drug [91,92,93,94,95].
Therefore, the application of the best treatment strategy in these specific patients’ categories would require the knowledge of the precise mechanism underlying the clinical resistance to anti-VEGF drugs. For example, in the presence of circulating neutralizing antibodies against ranibizumab [52] and bevacizumab [90], the use of a nonimmunogenic drug (e.g., aflibercept), could be a valuable treatment option. Furthermore, in the presence of pharmacodynamic tolerance, increasing the dosage or shortening treatment intervals, while maintaining the same drug or switching to a similar drug with different properties, could overcome tolerance. This strategy seemed to work in patients with residual center-involved DME following intravitreal bevacizumab who responded to 0.5 mg ranibizumab (i.e., improved visual and anatomic outcomes) [96]. Noteworthy, increased ranibizumab dosage to 2.0 mg provided additional benefit in those patients who did not respond to 0.5 mg ranibizumab [96].
The identification of factors or clinical conditions able to predict drug response may be helpful for treatment individualization. For example, the difference between BCVA under optimal luminance and baseline low-luminance visual acuity (LLVA) was a predictor of ranibizumab response in wet AMD patients; specifically, a smaller baseline BCVA-LLVA gap predicted higher BCVA gains over 24 months [97]. Furthermore, activated forms of VEGF receptors, PDGF receptors, and c-KIT shed into the vitreous of patients with wet AMD have been proposed as possible biomarkers for predicting drug response [98]. Moreover, in this pilot study, the authors found that vitreous levels of VEGFR Y1175, VEGFR Y996, and PDGFRβ Y751 were significantly higher in AMD patients who respond to intravitreal bevacizumab [98].
Math methods are commonly used in pharmacokinetic modeling and such an approach has also been applied to intravitreal anti-VEGF drugs [99]. In particular, this model predicted the in vivo activity of aflibercept at 10–12 weeks as comparable to that of an equimolar amount of ranibizumab at 30 days [99]. However, it is worth mentioning that the Authors’ conclusions were only based on a published article showing that aflibercept had 100-fold higher binding affinity for VEGF than ranibizumab [12], while other studies demonstrate no difference between aflibercept and ranibizumab in terms of affinity [14] and potency [15]. Such apparent discrepancies are most probably related to the very high affinity of the anti-VEGF agents tested and a number of experimental variables (e.g., the type and the temperature of assays employed), which make difficult obtaining accurate values. In line with this data, neovascular AMD patients studied in real-life clinical practice and treated with the same number of injections of ranibizumab and aflibercept had equivalent functional and morphologic outcomes [100].
Most recently, it has been proposed that a reduction in central retinal thickness by ≤25%, 1 month after one anti-VEGF injection, is predictive of poor response to anti-VEGF treatment in patients with macular edema secondary to RVOs. These patients may benefit from earlier switching to intravitreal dexamethasone implant [83].
Summary
Intravitreal anti-VEGF drugs are the mainstay of important angiogenesis-driven eye diseases. They have affinity and potency in the nanomolar to picomolar range of concentrations; however, inter-experimental variation makes data interpretation difficult and caution is required when establishing the rank order of affinity and potency for this class of drugs. Data on clinical efficacy obtained in a number of randomized clinical trials reinforce the importance of VEGF-A as the major targetable determinant in DME and wet AMD. The pathologic switch towards other angiogenic factors than VEGF-A (e.g., PlGF) might occur in severe disease states, during disease progression and/or development of anti-VEGF tolerance. Intravitreal anti-VEGF drugs can penetrate into the systemic circulation and alter systemic VEGF. Drug safety profile and low incidence of the most important side effects have been demonstrated in several randomized clinical trials; however, these studies lack the power to adequately assess small differences in these uncommon events and possible concerns, particularly in patients with important co-morbidity and in special populations, cannot be excluded a priori. The gaps in current understanding that leave open important questions in drug management are: (i) uncertainty about which drug should be given first, (ii) how long these drugs can be used safely, and (iii) the choice of the best pharmacological strategy after first-line treatment failure. Finally, the absence of a unanimous consensus on to how to classify the optimal response to intravitreal anti-VEGF drugs makes difficult to clearly define the poor or non-response phenotype.
References
Fogli S, Mogavero S, Egan CG, Del Re M, Danesi R. Pathophysiology and pharmacological targets of VEGF in diabetic macular edema. Pharmacol Res. 2016;103:149–57.
Campochiaro PA. Molecular pathogenesis of retinal and choroidal vascular diseases. Prog Retin Eye Res. 2015;49:67–81.
Senger DR, Davis GE. Angiogenesis. Cold Spring Harb Perspect Biol. 2011;3(8):a005090–a005090.
Chalam KV, Brar VS, Murthy RK. Human ciliary epithelium as a source of synthesis and secretion of vascular endothelial growth factor in neovascular glaucoma. JAMA Ophthalmol. 2014;132(11):1350–4.
Kaur C, Foulds WS, Ling E-A. Hypoxia-ischemia and retinal ganglion cell damage. Clin Ophthalmol. 2008;2(4):879–89.
Kim M, Lee C, Payne R, Yue BYJT, Chang J-H, Ying H. Angiogenesis in glaucoma filtration surgery and neovascular glaucoma: a review. Surv Ophthalmol. 2015;60(6):524–35.
Ng EWM, Shima DT, Calias P, Cunningham ET, Guyer DR, Adamis AP. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat Rev Drug Discov. 2006;5(2):123–32.
Narayanan R, Kuppermann BD, Jones C, Kirkpatrick P. Ranibizumab. Nat Rev Drug Discov. 2006;5(10):815–6.
Stewart MW, Grippon S, Kirkpatrick P. Aflibercept. Nat Rev Drug Discov. 2012;11(4):269–70.
Ellis LM. Bevacizumab. Nat Rev Drug Discov. 2005; Suppl: S8–9.
Lowe J, Araujo J, Yang J, Reich M, Oldendorp A, Shiu V, et al. Ranibizumab inhibits multiple forms of biologically active vascular endothelial growth factor in vitro and in vivo. Exp Eye Res. 2007;85(4):425–30.
Papadopoulos N, Martin J, Ruan Q, Rafique A, Rosconi MP, Shi E, et al. Binding and neutralization of vascular endothelial growth factor (VEGF) and related ligands by VEGF trap, ranibizumab and bevacizumab. Angiogenesis. 2012;15(2):171–85.
Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57(20):4593–9.
Yang J, Wang X, Fuh G, Yu L, Wakshull E, Khosraviani M, et al. Comparison of binding characteristics and in vitro activities of three inhibitors of vascular endothelial growth factor A. Mol Pharm. 2014;11(10):3421–30.
Yu L, Liang XH, Ferrara N. Comparing protein VEGF inhibitors: In vitro biological studies. Biochem Biophys Res Commun. 2011;408(2):276–81.
Puddu A, Sanguineti R, Traverso CE, Viviani GL, Nicolò M. Response to anti-VEGF-A treatment of endothelial cells in vitro. Exp Eye Res. 2016;146:128–36.
Park SJ, Oh J, Kim Y-K, Park JH, Park JY, Hong HK, et al. Intraocular pharmacokinetics of intravitreal vascular endothelial growth factor-Trap in a rabbit model. Eye. 2015;29(4):561–8.
Nomoto H, Shiraga F, Kuno N, Kimura E, Fujii S, Shinomiya K, et al. Pharmacokinetics of bevacizumab after topical, subconjunctival, and intravitreal administration in rabbits. Invest Ophthalmol Vis Sci. 2009;50(10):4807–13.
Ahn SJ, Ahn J, Park S, Kim H, Hwang DJ, Park JH, et al. Intraocular pharmacokinetics of ranibizumab in vitrectomized versus nonvitrectomized eyes. Invest Ophthalmol Vis Sci. 2014;55(1):567–73.
Niwa Y, Kakinoki M, Sawada T, Wang X, Ohji M. Ranibizumab and aflibercept: intraocular pharmacokinetics and their effects on aqueous VEGF level in vitrectomized and nonvitrectomized macaque eyes. Invest Ophthalmol Vis Sci. 2015;56(11):6501–5.
Xu L, Lu T, Tuomi L, Jumbe N, Lu J, Eppler S, et al. Pharmacokinetics of ranibizumab in patients with neovascular age-related macular degeneration: a population approach. Invest Ophthalmol Vis Sci. 2013;54(3):1616–24.
Krohne TU, Liu Z, Holz FG, Meyer CH. Intraocular pharmacokinetics of ranibizumab following a single intravitreal injection in humans. Am J Ophthalmol. 2012;154(4):682–686.e2.
Zhu Q, Ziemssen F, Henke-Fahle S, Tatar O, Szurman P, Aisenbrey S, et al. Vitreous levels of bevacizumab and vascular endothelial growth factor-A in patients with choroidal neovascularization. Ophthalmology. 2008;115(10):1750–5–1755.e1.
Kim H, Robinson SB, Csaky KG. FcRn receptor-mediated pharmacokinetics of therapeutic IgG in the eye. Mol Vis. 2009;15:2803–12.
Bakri SJ, Snyder MR, Reid JM, Pulido JS, Ezzat MK, Singh RJ. Pharmacokinetics of intravitreal ranibizumab (Lucentis). Ophthalmology. 2007;114(12):2179–82.
Basile AS, Hutmacher M, Nickens D, Nielsen J, Kowalski K, Whitfield L, et al. Population pharmacokinetics of pegaptanib in patients with neovascular, age-related macular degeneration. J Clin Pharmacol. 2012;52(8):1186–99.
Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507.
Sarav M, Wang Y, Hack BK, Chang A, Jensen M, Bao L, et al. Renal FcRn reclaims albumin but facilitates elimination of IgG. J Am Soc Nephrol. 2009;20(9):1941–52.
Li J, Gupta M, Jin D, Xin Y, Visich J, Allison DE. Characterization of the long-term pharmacokinetics of bevacizumab following last dose in patients with resected stage II and III carcinoma of the colon. Cancer Chemother Pharmacol. 2013;71(3):575–80.
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99(17):11393–8.
Syed YY, McKeage K. Aflibercept: a review in metastatic colorectal cancer. Drugs. 2015;75(12):1435–45.
Avery RL, Castellarin AA, Steinle NC, Dhoot DS, Pieramici DJ, See R, et al. Systemic pharmacokinetics following intravitreal injections of ranibizumab, bevacizumab or aflibercept in patients with neovascular AMD. Br J Ophthalmol. 2014;98(12):1636–41.
Zehetner C, Kirchmair R, Huber S, Kralinger MT, Kieselbach GF. Plasma levels of vascular endothelial growth factor before and after intravitreal injection of bevacizumab, ranibizumab and pegaptanib in patients with age-related macular degeneration, and in patients with diabetic macular oedema. Br J Ophthalmol. 2013;97(4):454–9.
Wang X, Sawada T, Sawada O, Saishin Y, Liu P, Ohji M. Serum and plasma vascular endothelial growth factor concentrations before and after intravitreal injection of aflibercept or ranibizumab for age-related macular degeneration. Am J Ophthalmol. 2014;158(4):738–744.e1.
Zehetner C, Bechrakis NE, Stattin M, Kirchmair R, Ulmer H, Kralinger MT, et al. Systemic counterregulatory response of placental growth factor levels to intravitreal aflibercept therapy. Invest Ophthalmol Vis Sci. 2015;56(5):3279–86.
Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lambrechts D, et al. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med. 2003;9(7):936–43.
Campochiaro PA, Brown DM, Awh CC, Lee SY, Gray S, Saroj N, et al. Sustained benefits from ranibizumab for macular edema following central retinal vein occlusion: twelve-month outcomes of a phase III study. Ophthalmology. 2011;118(10):2041–9.
Brown DM, Campochiaro PA, Bhisitkul RB, Ho AC, Gray S, Saroj N, et al. Sustained benefits from ranibizumab for macular edema following branch retinal vein occlusion: 12-month outcomes of a phase III study. Ophthalmology. 2011;118(8):1594–602.
Heier JS, Campochiaro PA, Yau L, Li Z, Saroj N, Rubio RG, et al. Ranibizumab for macular edema due to retinal vein occlusions: long-term follow-up in the HORIZON trial. Ophthalmology. 2012;119(4):802–9.
Massin P, Bandello F, Garweg JG, Hansen LL, Harding SP, Larsen M, et al. Safety and efficacy of ranibizumab in diabetic macular edema (RESOLVE Study): a 12-month, randomized, controlled, double-masked, multicenter phase II study. Diabetes Care. 2010;33(11):2399–405.
Nguyen QD, Shah SM, Heier JS, Do DV, Lim J, Boyer D, et al. Primary end point (six months) results of the ranibizumab for edema of the macula in diabetes (READ-2) study. Ophthalmology. 2009;116(11):2175–81.e1.
Kimoto K, Kubota T. Anti-VEGF agents for ocular angiogenesis and vascular permeability. J Ophthalmol. 2012;2012(14):852183–11.
Simha A, Braganza A, Abraham L, Samuel P, Lindsley K. Anti-vascular endothelial growth factor for neovascular glaucoma. Cochrane Database Syst Rev. 2013;66(10):CD007920.
Nguyen QD, Brown DM, Marcus DM, Boyer DS, Patel S, Feiner L, et al. Ranibizumab for diabetic macular edema: results from 2 phase III randomized trials: RISE and RIDE. Ophthalmology. 2012;119(4):789–801.
Korobelnik J-F, Do DV, Schmidt-Erfurth U, Boyer DS, Holz FG, Heier JS, et al. Intravitreal aflibercept for diabetic macular edema. Ophthalmology. 2014;121(11):2247–54.
Diabetic Retinopathy Clinical Research Network, Wells JA, Glassman AR, Ayala AR, Jampol LM, Aiello LP, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N Engl J Med. 2015;372(13):1193–203.
Akpek EK, Smith RA. Current treatment strategies for age-related ocular conditions. Am J Manag Care. 2013;19(5 Suppl):S76–84.
Singh RSJ, Kim JE. Ocular hypertension following intravitreal anti-vascular endothelial growth factor agents. Drugs Aging. 2012;29(12):949–56.
Chang J-H, Garg NK, Lunde E, Han K-Y, Jain S, Azar DT. Corneal neovascularization: an anti-VEGF therapy review. Surv Ophthalmol. 2012;57(5):415–29.
Gragoudas ES, Adamis AP, Cunningham ET, Feinsod M, Guyer DR, VEGF Inhibition Study in Ocular Neovascularization Clinical Trial Group. Pegaptanib for neovascular age-related macular degeneration. N Engl J Med. 2004;351(27):2805–16.
Brown DM, Kaiser PK, Michels M, Soubrane G, Heier JS, Kim RY, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–44.
Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–31.
Spaide R. Ranibizumab according to need: a treatment for age-related macular degeneration. Am J Ophthalmol. 2007;143(4):679–80.
Brown DM, Regillo CD. Anti-VEGF agents in the treatment of neovascular age-related macular degeneration: applying clinical trial results to the treatment of everyday patients. Am J Ophthalmol. 2007;144(4):627–37.
Rosenfeld PJ. Bevacizumab versus ranibizumab for AMD. N Engl J Med. 2011;364(20):1966–7.
Al-Latayfeh M, Silva PS, Sun JK, Aiello LP. Antiangiogenic therapy for ischemic retinopathies. Cold Spring Harb Perspect Med. 2012;2(6):a006411–a006411.
Bahar I, Yeung SN, Sella R, Slomovic A. Anterior segment uses of bevacizumab. Curr Opin Ophthalmol. 2012;23(4):303–16.
Xiong Q, Li Z, Li Z, Zhu Y, Abdulhalim S, Wang P, et al. Anti-VEGF agents with or without antimetabolites in trabeculectomy for glaucoma: a meta-analysis. PLoS ONE. 2014;9(2):e88403.
Miquerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol. 1999;212(2):307–22.
Maharaj ASR, Saint-Geniez M, Maldonado AE, D’Amore PA. Vascular endothelial growth factor localization in the adult. Am J Pathol. 2006;168(2):639–48.
Gemenetzi M, Lotery AJ, Patel PJ. Risk of geographic atrophy in age-related macular degeneration patients treated with intravitreal anti-VEGF agents. Eye. 2016; 31(1):1–9.
Kurihara T, Westenskow PD, Bravo S, Aguilar E, Friedlander M. Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest. 2012;122(11):4213–7.
Saint-Geniez M, Kurihara T, Sekiyama E, Maldonado AE, D’Amore PA. An essential role for RPE-derived soluble VEGF in the maintenance of the choriocapillaris. Proc Natl Acad Sci USA. 2009;106(44):18751–6.
Comparison of Age-related Macular Degeneration Treatments Trials (CATT) Research Group, Martin DF, Maguire MG, Fine SL, Ying G-S, Jaffe GJ, et al. Ranibizumab and bevacizumab for treatment of neovascular age-related macular degeneration: two-year results. Ophthalmology. 2012;119(7):1388–98.
Chakravarthy U, Harding SP, Rogers CA, Downes SM, Lotery AJ, Culliford LA, et al. Alternative treatments to inhibit VEGF in age-related choroidal neovascularisation: 2-year findings of the IVAN randomised controlled trial. Lancet. 2013;382(9900):1258–67.
Ho AC, Busbee BG, Regillo CD, Wieland MR, Van Everen SA, Li Z, et al. Twenty-four-month efficacy and safety of 0.5 mg or 2.0 mg ranibizumab in patients with subfoveal neovascular age-related macular degeneration. Ophthalmology. 2014;121(11):2181–92.
Chen ZI, Ai DI. Cardiotoxicity associated with targeted cancer therapies. Mol Clin Oncol. 2016;4(5):675–81.
Izumiya Y, Shiojima I, Sato K, Sawyer DB, Colucci WS, Walsh K. Vascular endothelial growth factor blockade promotes the transition from compensatory cardiac hypertrophy to failure in response to pressure overload. Hypertension. 2006;47(5):887–93.
Choueiri TK, Mayer EL, Je Y, Rosenberg JE, Nguyen PL, Azzi GR, et al. Congestive heart failure risk in patients with breast cancer treated with bevacizumab. J Clin Oncol. 2011;29(6):632–8.
Matsudaira K, Maeda K, Okumura N, Yoshikawa D, Morita Y, Mitsuhashi H, et al. Impact of low levels of vascular endothelial growth factor after myocardial infarction on 6-month clinical outcome. Results from the Nagoya Acute Myocardial Infarction Study. Circ J. 2012;76(6):1509–16.
Dewerchin M, Carmeliet P. PlGF: a multitasking cytokine with disease-restricted activity. Cold Spring Harb Perspect Med. 2012;2(8):a011056–a011056.
Hochholzer W, Reichlin T, Stelzig C, Hochholzer K, Meissner J, Breidthardt T, et al. Impact of soluble fms-like tyrosine kinase-1 and placental growth factor serum levels for risk stratification and early diagnosis in patients with suspected acute myocardial infarction. Eur Heart J. 2011;32(3):326–35.
Heeschen C, Dimmeler S, Fichtlscherer S, Hamm CW, Berger J, Simoons ML, et al. Prognostic value of placental growth factor in patients with acute chest pain. JAMA. 2004;291(4):435–41.
Schmid MK, Bachmann LM, Fäs L, Kessels AG, Job OM, Thiel MA. Efficacy and adverse events of aflibercept, ranibizumab and bevacizumab in age-related macular degeneration: a trade-off analysis. Br J Ophthalmol. 2015;99(2):141–6.
Heier JS, Bressler NM, Avery RL, Bakri SJ, Boyer DS, Brown DM, et al. Comparison of aflibercept, bevacizumab, and ranibizumab for treatment of diabetic macular edema: extrapolation of data to clinical practice. JAMA Ophthalmol. 2016;134(1):95–99.
Mammadzada P, Gudmundsson J, Kvanta A, André H. Differential hypoxic response of human choroidal and retinal endothelial cells proposes tissue heterogeneity of ocular angiogenesis. Acta Ophthalmol 2016.
Kovacs K, Marra KV, Yu G, Wagley S, Ma J, Teague GC, et al. Angiogenic and inflammatory vitreous biomarkers associated with increasing levels of retinal ischemia. Invest Ophthalmol Vis Sci. 2015;56(11):6523–30.
Huo X, Li Y, Jiang Y, Sun X, Gu L, Guo W, et al. Inhibition of ocular neovascularization by co-inhibition of VEGF-A and PLGF. Cell Physiol Biochem. 2015;35(5):1787–96.
Wells JA, Glassman AR, Ayala AR, Jampol LM, Bressler NM, Bressler SB, et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema: two-year results from a comparative effectiveness randomized clinical trial. Ophthalmology. 2016;123(6):1351–9.
Deissler HL, Deissler H, Lang GK, Lang GE. VEGF but not PlGF disturbs the barrier of retinal endothelial cells. Exp Eye Res. 2013;115:162–71.
Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De Mol M, et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med. 2001;7(5):575–83.
Kim H, Lee SC, Kwon KY, Lee JH, Koh HJ, Byeon SH, et al. Subfoveal choroidal thickness as a predictor of treatment response to anti-vascular endothelial growth factor therapy for polypoidal choroidal vasculopathy. Graefes Arch Clin Exp Ophthalmol 2015: 1–7.
Wolfe JD, Shah AR, Yonekawa Y, Faran Al A, Franklin MS, Abbey AM, et al. Receiver operating characteristic curve to predict anti-VEGF resistance in retinal vein occlusions and efficacy of Ozurdex. Eur J Ophthalmol. 2016;26(2):168–73.
Amoaku WM, Chakravarthy U, Gale R, Gavin M, Ghanchi F, Gibson J, et al. Defining response to anti-VEGF therapies in neovascular AMD. Eye (Lond). 2015;29(6):721–31.
Zhao L, Grob S, Avery R, Kimura A, Pieramici D, Lee J, et al. Common variant in VEGFA and response to anti-VEGF therapy for neovascular age-related macular degeneration. Curr Mol Med. 2013;13(6):929–34.
Wang VM, Rosen RB, Meyerle CB, Kurup SK, Ardeljan D, Agron E, et al. Suggestive association between PLA2G12A single nucleotide polymorphism rs2285714 and response to anti-vascular endothelial growth factor therapy in patients with exudative age-related macular degeneration. Mol Vis. 2012;18:2578–85.
Kloeckener-Gruissem B, Barthelmes D, Labs S, Schindler C, Kurz-Levin M, Michels S, et al. Genetic association with response to intravitreal ranibizumab in patients with neovascular AMD. Invest Ophthalmol Vis Sci. 2011;52(7):4694–702.
Krebs I, Glittenberg C, Ansari-Shahrezaei S, Hagen S, Steiner I, Binder S. Non-responders to treatment with antagonists of vascular endothelial growth factor in age-related macular degeneration. Br J Ophthalmol. 2013;97(11):1443–6.
Eghøj MS, Sørensen TL. Tachyphylaxis during treatment of exudative age-related macular degeneration with ranibizumab. Br J Ophthalmol. 2012;96(1):21–23.
Forooghian F, Cukras C, Meyerle CB, Chew EY, Wong WT. Tachyphylaxis after intravitreal bevacizumab for exudative age-related macular degeneration. Retina. 2009;29(6):723–31.
Stepien KE, Rosenfeld PJ, Puliafito CA, Feuer W, Shi W, Al-Attar L, et al. Comparison of intravitreal bevacizumab followed by ranibizumab for the treatment of neovascular age-related macular degeneration. Retina. 2009;29(8):1067–73.
Ehlken C, Jungmann S, Böhringer D, Agostini HT, Junker B, Pielen A. Switch of anti-VEGF agents is an option for nonresponders in the treatment of AMD. Eye (Lond). 2014;28(5):538–45.
Almony A, Mansouri A, Shah GK, Blinder KJ. Efficacy of intravitreal bevacizumab after unresponsive treatment with intravitreal ranibizumab. Can J Ophthalmol. 2011;46(2):182–5.
Kumar N, Marsiglia M, Mrejen S, Fung AT-C, Slakter J, Sorenson J, et al. Visual and anatomical outcomes of intravitreal aflibercept in eyes with persistent subfoveal fluid despite previous treatments with ranibizumab in patients with neovascular age-related macular degeneration. Retina. 2013;33(8):1605–12.
Shiragami C, Ono A, Kobayashi M, Manabe S, Yamashita A, Shiraga F. Effect of switching therapy to pegaptanib in eyes with the persistent cases of exudative age-related macular degeneration. Med (Baltim). 2014;93(18):e116.
Dhoot DS, Pieramici DJ, Nasir M, Castellarin AA, Couvillion S, See RF, et al. Residual edema evaluation with ranibizumab 0.5 mg and 2.0 mg formulations for diabetic macular edema (REEF study). Eye (Lond). 2015;29(4):534–41.
Frenkel REP, Shapiro H, Stoilov I. Predicting vision gains with anti-VEGF therapy in neovascular age-related macular degeneration patients by using low-luminance vision. Br J Ophthalmol 2015: bjophthalmol–2015–307575.
Davuluri G, Espina V, Petricoin EF, Ross M, Deng J, Liotta LA, et al. Activated VEGF receptor shed into the vitreous in eyes with wet AMD: a new class of biomarkers in the vitreous with potential for predicting the treatment timing and monitoring response. Arch Ophthalmol. 2009;127(5):613–21.
Stewart MW, Rosenfeld PJ, Penha FM, Wang F, Yehoshua Z, Bueno-Lopez E, et al. Pharmacokinetic rationale for dosing every 2 weeks versus 4 weeks with intravitreal ranibizumab, bevacizumab, and aflibercept (vascular endothelial growth factor Trap-eye). Retina. 2012;32(3):434–57.
Böhni SC, Bittner M, Howell JP, Bachmann LM, Faes L, Schmid MK. Comparison of Eylea® with Lucentis® as first-line therapy in patients with treatment-naïve neovascular age-related macular degeneration in real-life clinical practice: retrospective case-series analysis. BMC Ophthalmol. 2015;15(1):109.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Rights and permissions
About this article

Cite this article
Fogli, S., Del Re, M., Rofi, E. et al. Clinical pharmacology of intravitreal anti-VEGF drugs. Eye 32, 1010–1020 (2018). https://doi.org/10.1038/s41433-018-0021-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-018-0021-7
This article is cited by
-
PFKFB3 in neovascular eye disease: unraveling mechanisms and exploring therapeutic strategies
Cell & Bioscience (2024)
-
Ocular and systemic vascular endothelial growth factor ligand inhibitor use and nephrotoxicity: an update
International Urology and Nephrology (2024)
-
Cardiovascular Outcomes with Intravitreal Anti-Vascular Endothelial Growth Factor Therapy in Patients with Diabetes: A Real-World Data Analysis
Diabetes Therapy (2024)
-
VIVEX: A Formula for Calculating Individual Vitreous Volume: A New Approach Towards Tailored Patient Dosing Regime in Intravitreal Therapy
Ophthalmology and Therapy (2024)
-
Progress in the Application of Microneedles in Eye Disorders and the Proposal of the Upgraded Microneedle with Spinule
Pharmaceutical Research (2024)
