
Abstract
Biliary atresia (BA) is a severe pediatric liver disease resulting in necroinflammatory obliteration of the extrahepatic biliary tree. BA presents within the first few months of life as either an isolated finding or with additional syndromic features. The etiology of isolated BA is unknown, with evidence for infectious, environmental, and genetic risk factors described. However, to date, there are no definitive causal genes identified for isolated BA in humans, and the question of whether single gene defects play a major role remains open. We performed exome-sequencing in 101 North American patients of European descent with isolated BA (including 30 parent-child trios) and considered several experimental designs to identify potentially deleterious protein-altering variants that may be involved in the disease. In a case-only analysis, we did not identify genes with variants shared among more than two probands, and burden tests of rare variants using a case-case control design did not yield significant results. In the trio analysis of 30 simplex families (patient and parent trios), we identified 66 de novo variants in 66 genes including potentially deleterious variants in STIP1 and REV1. STIP1 is a co-chaperone for the heat-shock protein, HSP90, and has been shown to have diverse functions in yeast, flies and mammals, including stress-responses. REV1 is known to be a key player in DNA repair pathway and to interact with HSP90. In conclusion, our results do not support the hypothesis that a simple genetic model is responsible for the majority of cases of isolated BA. Our finding of de novo variants in genes linked to evolutionarily conserved stress responses (STIP1 and REV1) suggests that exploration of how genetic susceptibility and environmental exposure may interact to cause BA is warranted.
Similar content being viewed by others

Introduction
Biliary atresia (BA) is an obstructive cholangiopathy with initial symptoms arising during the first days to weeks of life. BA occurs as an isolated finding in 85% of affected individuals, and with additional syndromic features (heterotaxy and/or other congenital birth defects) in 15%1. The incidence of BA varies across different populations, with estimates ranging from 1 in 5,000 to 1 in 14,000 live births2. BA presents clinically with neonatal cholestasis, elevated bilirubin and liver enzymes, although the differential diagnosis suggested by these findings is extensive. The diagnosis of BA is suggested by features of biliary obstruction on liver histology, including bile duct proliferation, portal tract expansion and bile plugs. The diagnosis is confirmed upon intraoperative cholangiogram showing lack of patency of the extrahepatic biliary tree.
Although the etiology of BA is not clear, a genetic component is supported by multiple lines of evidence. These include the presence of familial cases3,4,5, case reports of individuals with syndromic BA with likely causal genes identified (including FOXA26, CFC17,8, ZEB29, ZIC310, HNF1B11 and PKD1L112), animal models of BA involving the genes Sox1713, Foxm114, Invs15, Onecut116, and candidate susceptibility genes including ADD317, GPC118, EFEMP119 and ARF620 identified by genome-wide association studies (GWAS). A follow-up study of GPC1 using a zebrafish model confirmed that suppressing GPC1 expression gives rise to biliary defects21. A GWAS identified SNPs associated with BA upstream of XPNPEP1 and ADD3 in a Chinese cohort of isolated BA patients17, and this association has been confirmed in both a Thai22 and a European-American cohort23. In a study of 499 isolated and syndromic BA patients and 1928 controls, our group identified a GWAS signal within 2p16.1 implicating common variants in the gene EFEMP119. Another genome-wide association study of 63 BA patients and 1907 controls identified two SNPs downstream of the gene ARF6 associated with isolated BA20, although we could not replicate this finding in our study. However, to date, no genes have been definitively identified as a cause of isolated BA. Environmental exposure to viruses or toxins has long been proposed as a hypothesis to explain the etiology of the perinatal form of BA2, but the genetic susceptibility factors linking such exposures and BA are yet to be identified.
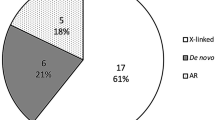
In this work, we hypothesized that rare and novel, deleterious, protein-altering variants transmitted in a Mendelian fashion could be involved in the etiology of isolated BA, and patients with isolated BA would be enriched with such variants. To test this hypothesis, we considered three experimental designs (Table 1). First, we conducted a case-only analysis where we looked for potentially deleterious novel variants. In the second design, we conducted a case-control analysis comparing the cumulative frequency of rare variants in the two groups. Finally, we performed trio analysis on a subset of 30 probands to identify genes with rare, protein-altering de novo and compound heterozygous or homozygous variants.
Patients and Methods
Patients with isolated BA and controls
We studied a cohort of 101 self-reported white, non-Hispanic patients with isolated BA. The patients were ascertained from a larger cohort of 1063 BA cases based on their self-reported race and ethnicity, availability of parental DNA samples, and absence of any other anomaly. Ninety-nine participants were enrolled in the NIDDK-funded Childhood Liver Disease Research Network (ChiLDReN) at one of the 16 participating institutions under Institutional Review Board (IRB) approved protocols (IRB 04–003655 and 06–004814). A set of affected dizygotic twins were consented into the approved IRB study at the Children’s Memorial Hospital, Chicago, IL. One of them was randomly selected for inclusion in the cohort of unrelated individuals (for over-representation and burden tests). Informed consent was obtained from parents or legal guardians for each patient enrolled in this study. The diagnosis of BA was made by clinical presentation, liver histology, and an intraoperative cholangiogram. Most patients also had BA confirmed by examination of the biliary remnant from a Roux-en-Y hepatic portoenterostomy (also known as a Kasai operation). The average age at Kasai was 0.16 years (range 0–1.16 years). There were no data available on BA subtype and gestational age for the majority of individuals included in the study. DNA samples were extracted from whole blood (n = 16) or lymphoblastoid cell lines (LCLs) (n = 85). For a subset of patients (n = 32), DNA samples were also available from both parents (LCL = 56; whole blood = 6).
Controls were individuals recruited from two different studies at the Children’s Hospital of Philadelphia, PediSeq (the CHOP cohort of the Clinical Sequencing and Exploratory Research (CSER) consortium)24, and Very Early Onset Inflammatory Bowel Disease (VEO-IBD)25, and enrolled in institution-approved IRB protocols (IRB 12–009169; 14–010826). Controls from the PediSeq dataset (n = 158) fall into four different disease cohorts (cardiovascular, hearing loss, mitochondrial and intellectual disability) but have no known liver disease. Controls from the VEO-IBD cohort (n = 145) were the unaffected parents of individuals with VEO-IBD. The patient recruitment, methods, and experimental protocols used in these studies were performed in accordance with relevant guidelines and regulations set by the aforementioned institutions. This study was approved by the Ethics Committee of the Children’s Hospital of Philadelphia.
Exome sequencing
Genomic DNA (3–5 ug) was supplied to Perkin Elmer, Inc. (Perkin-Elmer, Branford, CT) for exome sequencing of the BA patients and their parents. The DNA was amplified and enriched using the Agilent SureSelect V4 + UTR 71 Mb All Exon Capture Kit (Agilent Technologies, Santa Clara, CA). Sequencing was performed on the Illumina HiSeq 2000 (Illumina, San Diego, CA) at the Perkin-Elmer sequencing service center. Control samples were sequenced using Agilent SureSelect V4 51 Mb Exon Capture Kit (Agilent Technologies, Santa Clara, CA) on the Illumina HiSeq 2000 (Illumina, San Diego, CA) at the internal sequencing core at CHOP (BGI@CHOP, Philadelphia, PA). All of the exome sequencing data was produced with paired-end reads of 100 base-pairs in length (2 × 100 bp). The unaligned FASTQ reads of all samples were returned to us for read analysis and downstream processing as detailed below.
Variant calling and annotation
Raw sequencing reads were mapped to the GRCh37.69 reference genome using the Burrows-Wheeler Aligner26 to produce an aligned BAM file. BAM files were realigned around known indels using IndelRealigner in the GenomeAnalysisToolKit software (GATK) and base quality scores were recalibrated using the BaseQualityScoreRecalibrator in GATK27. Variant calling was performed using HaplotypeCaller and jointly genotyped using the GenotypeGVCFs command in GATK. The initial variant quality filtering was done using the VariantQualityScoreRecalibrator (VQSR) in GATK. SNPEff28 was used to annotate the variants with their expected functional consequence. Other annotations such as allele frequencies in non-Finnish Europeans (NFE) from the gnomAD variant database version 2.129, and deleteriousness scores such as CADD (Combined Annotation Dependent Depletion version 1.4)30 were added using the Gemini software31. We manually reviewed all variants found in the de novo analysis, and variant clusters with overlapping genomic positions that could be indicative of technical artifacts32. Individual genes were further annotated using the pLI, or probability of loss-of-function intolerance, a gene-level measure estimated by comparing the expected number of loss-of-function variants against the number observed in a population. The pLI score ranges from 0 to 1 with the highest value being the most intolerant33.
Quality control
The Peddy34 software tool was used to infer genetic ancestry from the exome sequencing data by comparing our samples against the samples from the 1000 Genomes Project Phase 335. All samples in the case and control cohorts clustered with the European populations in the 1000 Genomes Project confirming their self-reported race/ethnicity (Supp. Fig. 1). We also confirmed the sex of the participants based on heterozygosity of X chromosome variants. Pairwise relatedness (estimated by the kinship coefficient) between samples was investigated using the KING36 software, and confirmed that there were no related individuals in the entire cohort beside the known parent-child pairs used in the trio analysis and the twins. Distribution of the variant allele fraction (allele balance) for all heterozygous variants identified in the cases (Supp. Fig. 2), principal component analysis of pairwise genetic distances based on genotype data (Supp. Fig. 3), and the distribution of transition/transversions ratio (Supp. Fig. 4) stratified by source of DNA were used to compare results obtained from samples sequenced from whole-blood and LCLs, and did not reveal any systematic differences between the two sets of samples.
Variant prioritization
We filtered the variants identified from exome sequencing for stop gain, stop loss, frameshift and in-frame insertions/deletions (indels), missense and splicing variants. We excluded variants overlapping with segmental duplications and variants with call rates less than 90% from all analyses. In the case-only analysis, we included novel and potentially deleterious variants, defined as variants that were not present in the NFE population of the gnomAD v2.1 genomes and exomes and had a phred-scaled CADD score of at least 30, representing the top 0.1% of the potentially deterious variants. For the burden tests in the case-control experimental design, all protein-altering variants with a cohort (cases and controls combined) minor allele frequency (MAF) of <5% were included. We did not apply a CADD score threshold for this analysis. For the de novo analysis, we considered all variants irrespective of their frequency or the CADD score. For the analysis under the recessive model, we retained variants with an allele frequency in gnomAD v2.1 NFE less than or equal to 0.1% and a CADD score of at least 30. Details pertaining to variant prioritization are provided in Table 1.
Autosomal copy-number variant (CNV) analysis
Mean depth of coverage for each individual exonic interval in the autosomes was computed using the DepthOfCoverage routine in the GATK package and copy number variants were called using XHMM following the protocol described elsewhere37. We excluded CNVs with quality score (Q_SOME) less than 60 to retain only high quality CNVs. To identify novel CNVs, we excluded any variant found in the Database of Genomic Variants (DGV)38 and the ExAC version 1.039. We did not look for CNVs in the sex chromosomes as the ExAC dataset only had calls from autosomes.
Gene, pathway and gene-set definitions
For all the analyses, we used refFlat_hg19 gene definitions from the UCSC genome browser40. We used pathway definitions including KEGG (n = 293), Panther (n = 112), Reactome (n = 1530), WikiPathways (n = 437) and BioCarta (n = 237), and annotation gene-sets from Gene Ontology (GO) Biological Process (n = 5192), Molecular Functions (n = 1136) and Cellular Components (n = 641) downloaded from the Enrichr website41.
Case-only and trio analyses: Over representation tests
To determine if our lists of candidate genes from the case-only and trio analyses were enriched for specific gene-sets or pathways, we performed over-representation analysis using the R package enrichR41. This software essentially counts the number of genes with a given annotation that are present in our lists of candidate genes and genome-wide, and compares it to the number of genes that are not associated with the same annotation in our lists and genome-wide, using a Fisher exact test. Over representation tests were carried out on gene-sets and pathways.
Case-control analysis: burden tests
To test if a gene, gene-set or pathway contained a significantly different number of low frequency or rare variant (cohort MAF < 5%) in cases compared to controls, we performed two-sided Fisher’s exact tests using the Combined and Multivariate Collapsing (CMC) approach42 implemented in the exactCMC procedure from the software package rvtests43. Burden tests were carried out at different levels including (1) genes; (2) gene-sets (e.g. collection of genes with similar function or structure or cellular localization, such as sodium channel genes or membrane bound receptors); and (3) pathways, which include genes that function in a specific biological pathway (e.g. WNT or NOTCH signaling). The number of cases and controls with and without at least one rare variant in a given gene, gene-set, or pathway were compared using a two-sided Fisher exact test.
Trio and quad analysis
Segregation analysis was performed using Gemini software31 in a subset of 30 probands and a dizygotic twin pair for whom DNA from both parents was available. De novo variants were identified using the command ‘de_novo’ which detects SNVs and indels occuring in the proband and not in the parents. Compound heterozygous variants were identified using the command ‘comp_het’ which detects two heterozygous variants in the same gene inherited from different parents. Homozygous variants were identified using the ‘autosomal_recessive’ command which detects variants present in homozygosity in the proband and in heterozygosity in both parents.
Statistical significance
For gene level burden tests, the raw p-values were corrected for multiple testing using a Bonferroni correction for the total number of genes tested (n = 16,393). For the pathway based analyses, the raw P-values were corrected for multiple testing using a Bonferroni correction for the total number of pathway definitions (n = 2609). For the gene-set based analyses, we corrected for the total number of gene-sets used in the analysis (n = 6969). We considered a test to be statistically significant if the Bonferroni-adjusted p-value was less than or equal to 0.05. Post-hoc power analysis for the case-control burden analysis was performed using the software tool G*Power44. We used the maximum observed effect size from the gene-level burden test to compute the minimum sample size required to observe the same difference at a Bonferroni-corrected significance level based on the number of genes tested (p < 3 × 10−6).
Results
Case-only analysis
We identified a total of 496,654 variants that passed the quality filters in the 100 probands. Out of 63,473 single nucleotide variants (SNV) or small indels found to be protein-altering changes (stop gain, stop loss, frameshift and in-frame indels, missense and splicing variants), 332 variants in 326 genes were novel (not in the NFE population in gnomAD exomes and genomes) and had a phred-scaled CADD score of 30 or more. This formed our variant dataset for the case-only analyses. Almost all of the variants (331/332) were present in heterozygosity in a single individual, and 96 out of the 100 individuals in the cohort had at least one potentially deleterious novel variant. Six genes had two different variants in two different individuals (MAST2, WDR35, DNAH5, FBN2, SCUBE2 and PITPNM3). Over-representation analysis did not yield any significant gene, pathway or gene-set after correcting for multiple testing. Copy-number variant analysis did not reveal any novel variant in the probands after filtering for variants present in the population databases ExAC and DGV. Details of the SNVs and indels with annotations are provided in the Supp. Table 1.
Case-control analysis
In the second experimental design, we conducted burden tests for low frequency and rare variants (MAF < 5%) by comparing their cumulative number in a gene, gene-set or pathway in the 100 cases against the 303 controls. After correcting for multiple testing, we did not identify any gene, gene-set or a pathway with a statistically significant difference in burden of low frequency and rare variants in the cases compared to the controls. Analysis of QQ-plot from the gene-based burden tests of genes with three or more variants did not show a deviation of the observed p-values compared to the expected distribution (Supp. Fig. 5). Post-hoc power analysis using the most significant result from the gene-based burden analysis (proportion of individuals with a rare variant in cases = 2% and controls = 15%; p-value = 0.0001) suggested that 260 cases and controls (total n = 520) are required to achieve 80% power for the same effect size to reach statistical significance after correction for the number of genes tested (p < 3 × 10−6).
Trio and quad analysis
In the final experimental design, we sequenced the parents of a subset of the patients (30 singletons and one affected dizygotic twin pair) to identify de novo variants in the probands under the hypothesis that they are more likely to be involved in the etiology of the disease, and variants transmitted from the parents to the probands according to a recessive model (homozygous or compound heterozygous). We identified a total of 66 de novo, protein-altering and splice variants in 66 different genes in 25 out of 30 individuals, with a mean of 2.2 variants per individual (range: 0–7; median 3). We did not identify any de novo variants in five individuals. All the de novo variants were manually verified in the BAM file and a small subset of them were validated using Sanger sequencing (n = 4). There were 58 missense, 3 frameshift indels, 3 nonsense, and 2 splice-site variants. A prioritized list of 14 de novo variants (all loss-of-function variants, and missense variants with CADD greater than or equal to 30) is provided in Table 2, and all de novo variants are listed in the Supp. Table 2. Of the eight de novo loss-of-function variants, a premature stop-gain in the gene STIP1 (NM_001282652: p.(Cys30Ter)) and a splice-acceptor variant in the gene REV1 (NM_001037872: c.1674-2 A > G) were the most interesting as these genes are known to be intolerant to loss-of-function, with a pLI (probability of loss-of-function intolerance) score of 1. Sanger traces and a graphical representation of the sequence reads from the BAM files supporting their presence (read pile-up), are provided for these two variants in Supp. Figs. 6 and 7. Over-representation analysis of the 66 genes did not yield significant results after correcting for multiple testing.
By segregation analysis, we did not identify any compound heterozygous or homozygous variants at a maximum alternate allele frequency threshold of 0.1% and a phred-scaled CADD score of 30 in the 30 trios. After applying the same filters, we did not identify de novo, compound heterozygous or homozygous variants shared by both affected individuals in the twin quad.
Discussion
In this study, exome sequencing was used to identify genomic changes in a cohort of patients with isolated biliary atresia, and in spite of the utilization of several distinct strategies, we were unable to demonstrate an associated genetic change. We did identify new candidate genes by analysis of 30 trios, finding potentially deleterious de novo variants in STIP1 and REV1, which we hypothesize may result in susceptibility to toxin induced biliary disease as discussed below.
BA presents as an isolated finding in 85% of cases, and in a syndromic form with laterality and/or other congenital malformations in 15%. Mouse models of BA (Sox17, Onecut1) and gene mutations in patients with syndromic forms of BA (CFC1, ZIC3, FOXA2, PKD1L1) suggest an etiology involving genetic factors that influence left-right symmetry, and bile duct development and morphology. Genome-wide association studies in patients with isolated BA have suggested candidate genes and genomic regions (ADD3 and XPNPEP1 in 10q25.1, EFEMP1 in 2p16.1, GPC1 in 2q37.3, ARF6 in 14q21.3) but consistent proof of the involvement of each of these regions has been elusive. BA as a consequence of exposure to environmental toxins is also proposed as a model for the perinatal form of the disease based on research into the natural occurance of BA in Australian sheep, which led to the discovery of an isoflavonoid (biliatresone) found in specific plant species, later found to cause biliary defects in zebrafish45. RNA-seq experiments on biliatresone treated and control fish identified several differentially expressed stress signaling pathways including the glutathione antioxidant pathway, cellular redox response, and heat shock response46. However, the genetic susceptibility factors that might link exposure to these agents and BA are yet to be identified. In summary, from previous literature, there are genes that are known to cause syndromic forms of BA in humans and mice, genes that are associated with isolated BA identified via GWAS, and biological pathways that are perturbed in zebrafish treated with the toxin biliatresone, which has been associated with BA in sheep. However, in spite of these clues, there has not been a causal gene identified for isolated BA in humans.
In this work, we performed exome sequencing in 101 North American patients of European ancestry with isolated BA and looked for novel or low-frequency and rare protein-altering variants that may explain the BA phenotype (strategies presented in Table 1). Such study designs have been successful in finding novel genes responsible for rare Mendelian diseases47. Fewer exome sequencing studies with sufficiently large sample sizes have been successfully employed in identifying associations with complex phenotypes48,49. This study was primarily designed with the goal of finding genes with rare and highly deleterious variants that may explain the occurrence of isolated BA. We considered three experimental designs including a case-only analysis to look for novel, potentially deleterious and protein-altering variants, a case-control analysis to look for an excessive burden of low frequency and rare variants in cases compared to controls, and a segregation analysis, to look for de novo and homozygous or compound heterozygous variants.
In the case-only design, we identified 332 novel and potentially deleterious protein-altering variants in our cohort of 100 unrelated individuals with isolated BA. The majority of the variants were found in heterozygosity in a single individual. Six genes had variants in two individuals, and these variants were different from each other. This suggests that BA is unlikely to result from a single genetic change, although we cannot rule out extreme locus heterogeneity, with a small percentage of cases having a genetic etiology, perhaps in combination with an environmental insult. Case-control analysis did not yield any statistically significant result, and this may suggest that there are no variants or genes with large enough effect sizes present in the cases as compared to the controls or our sample size is too small to identify such a difference between cases and controls.
Trio-based segregation analysis identified 66 de novo potentially protein-altering variants in 66 genes in 25 out of 30 unrelated patients. All putative loss-of-function de novo variants and a prioritized list of de novo missense variants with a CADD score greater than or equal to 30 are included in Table 2. Of all the genes with putative loss-of-function de novo variants, a splice-acceptor variant in the gene REV1 and a premature stop in one of the transcripts in the gene STIP1 were the most interesting for several reasons. These genes are known to be intolerant to loss-of-function variants with a perfect pLI score of 1. STIP1 is a chaperone that assists in the transfer of proteins from HSP70 to HSP90, which are an integral part of the heat shock response pathway. Mass spectrometry experiments to identify differentially expressed proteins in 20 BA liver biopsies compared to 12 non-BA, neonatal cholestasis livers found that HSP90 was significantly down-regulated in BA livers, and was the most significantly altered protein50. REV1 encodes a protein similar to the S. cerevisiae mutagenesis protein Rev1 and is known to be involved in DNA repair. REV1 is shown to be regulated by the heat-shock protein HSP90 in the DNA repair pathway51. Based on the de novo variant found in the human exome data, along with evidence that the heatshock response pathway is differentially expressed in zebrafish treated with the plant toxin biliatresone, our collaborators used CRISPR/Cas9 methods to introduce a frameshift mutation in the zebrafish stip1 gene. When treated with biliatresone toxin, stip1 heterozygous fish were highly sensitive to a low dose that is normally inactive in wild-type fish52. Similar sensitization was detected in human cholangiocytes following siRNA knockdown of the STIP1 isoform targeted by the BA variant52. Heterozygous rev1 mutation in zebrafish and siRNA knockdown of REV1 in human cholangiocytes were sensitized to low dose biliatresone, similar to mutation and knockdown of stip1/STIP152.
Our study has several limitations including a small sample size (which is a function of the disease frequency) and the failure of exome sequencing to identify all possible disease causing genomic changes. The sample size was relatively small for statistical analyses such as rare variant burden tests and for the trio analysis to identify genes with recurring de novo variants. Exome sequencing is a targeted capture experiment which focuses only on the exonic regions53, and coverage might not be uniform through the coding parts of the genome. This limits our ability to study intronic regions and/or poorly targeted parts of the genome. Copy-number detection from exome sequencing is challenging and the XHMM software tool used in this work is tuned for specificity rather than sensitivity. In addition, exome sequencing does not allow us to look for structural variation in these samples. Finally, the majority (85/101) of the DNA samples used in this study came from LCLs, including those containing the de novo variants in STIP1 and REV1 discussed above, and we do not have access to a primary source of DNA to confirm their presence. It is known that LCLs are susceptible to mutational changes over passages (cell line artifacts)53. However, in our study, we did not see any evidence of systematic differences in the results obtained from DNA samples extracted from whole blood or LCLs.
Together, our analyses do not support a simple genetic model in which a small number of genes are responsible for the majority of cases with isolated BA. However, the identification of de novo variants in the genes STIP1 and REV1 and the evidence for their sensitivity to the biliatresone toxin in zebrafish open new opportunities for further investigating BA as a consequence of environmental exposure in genetically susceptible individuals. Other genes in the heat shock response pathway or other pathways identified in the zebrafish experiments could also be considered as BA susceptibility candidate genes.
References
Schwarz, K. B. et al. Extrahepatic anomalies in infants with biliary atresia: results of a large prospective North American multicenter study. Hepatology 58, 1724–1731, https://doi.org/10.1002/hep.26512 (2013).
Lakshminarayanan, B. & Davenport, M. Biliary atresia: A comprehensive review. J. Autoimmun. 73, 1–9, https://doi.org/10.1016/j.jaut.2016.06.005 (2016).
Gunasekaran, T. S., Hassall, E. G., Steinbrecher, U. P. & Yong, S. L. Recurrence of extrahepatic biliary atresia in two half sibs. Am. J. Med. Genet. 43, 592–594, https://doi.org/10.1002/ajmg.1320430317 (1992).
Strickland, A. D., Shannon, K. & Coln, C. D. Biliary atresia in two sets of twins. J. pediatrics 107, 418–420 (1985).
Lachaux, A. et al. Familial extrahepatic biliary atresia. J. pediatric gastroenterology Nutr. 7, 280–283 (1988).
Tsai, E. A. et al. Heterozygous deletion of FOXA2 segregates with disease in a family with heterotaxy, panhypopituitarism, and biliary atresia. Hum. Mutat. 36, 631–637, https://doi.org/10.1002/humu.22786 (2015).
Davit-Spraul, A., Baussan, C., Hermeziu, B., Bernard, O. & Jacquemin, E. CFC1 gene involvement in biliary atresia with polysplenia syndrome. J. pediatric gastroenterology Nutr. 46, 111–112, https://doi.org/10.1097/01.mpg.0000304465.60788.f4 (2008).
Jacquemin, E., Cresteil, D., Raynaud, N. & Hadchouel, M. CFCI gene mutation and biliary atresia with polysplenia syndrome. J. pediatric gastroenterology Nutr. 34, 326–327 (2002).
Cui, S., Erlichman, J., Russo, P., Haber, B. A. & Matthews, R. P. Intrahepatic biliary anomalies in a patient with Mowat-Wilson syndrome uncover a role for the zinc finger homeobox gene zfhx1b in vertebrate biliary development. J. pediatric gastroenterology Nutr. 52, 339–344, https://doi.org/10.1097/MPG.0b013e3181ff2e5b (2011).
Ware, S. M. et al. Identification and functional analysis of ZIC3 mutations in heterotaxy and related congenital heart defects. Am. J. Hum. Genet. 74, 93–105, https://doi.org/10.1086/380998 (2004).
Kotalova, R. et al. Hepatic phenotypes of HNF1B gene mutations: a case of neonatal cholestasis requiring portoenterostomy and literature review. World J. Gastroenterol. 21, 2550–2557, https://doi.org/10.3748/wjg.v21.i8.2550 (2015).
Berauer, J. P. et al. Identification of PKD1L1 Gene Variants in Biliary Atresia Patients with Structural Malformations (BASM). AASLD 2017 (2017).
Uemura, M. et al. Sox17 haploinsufficiency results in perinatal biliary atresia and hepatitis in C57BL/6 background mice. Dev. 140, 639–648, https://doi.org/10.1242/dev.086702 (2013).
Krupczak-Hollis, K. et al. The mouse Forkhead Box m1 transcription factor is essential for hepatoblast mitosis and development of intrahepatic bile ducts and vessels during liver morphogenesis. Dev. Biol. 276, 74–88, https://doi.org/10.1016/j.ydbio.2004.08.022 (2004).
Shimadera, S. et al. The inv mouse as an experimental model of biliary atresia. J. pediatric Surg. 42, 1555–1560, https://doi.org/10.1016/j.jpedsurg.2007.04.018 (2007).
Clotman, F. et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Dev. 129, 1819–1828 (2002).
Garcia-Barcelo, M. M. et al. Genome-wide association study identifies a susceptibility locus for biliary atresia on 10q24.2. Hum. Mol. Genet. 19, 2917–2925, https://doi.org/10.1093/hmg/ddq196 (2010).
Leyva-Vega, M. et al. Genomic alterations in biliary atresia suggest region of potential disease susceptibility in 2q37.3. Am. J. Med. Genet. A 152A, 886–895, https://doi.org/10.1002/ajmg.a.33332 (2010).
Chen, Y. et al. A genome-wide association study identifies a susceptibility locus for biliary atresia on 2p16.1 within the gene EFEMP1. Plos Genet. 14, e1007532, https://doi.org/10.1371/journal.pgen.1007532 (2018).
Ningappa, M. et al. The Role of ARF6 in Biliary Atresia. PLoS one 10, e0138381, https://doi.org/10.1371/journal.pone.0138381 (2015).
Cui, S. et al. Evidence from human and zebrafish that GPC1 is a biliary atresia susceptibility gene. Gastroenterology 144, 1107–1115.e1103, https://doi.org/10.1053/j.gastro.2013.01.022 (2013).
Kaewkiattiyot, S., Honsawek, S., Vejchapipat, P., Chongsrisawat, V. & Poovorawan, Y. Association of X-prolyl aminopeptidase 1 rs17095355 polymorphism with biliary atresia in Thai children. Hepatol. Res. 41, 1249–1252, https://doi.org/10.1111/j.1872-034X.2011.00870.x (2011).
Tsai, E. A. et al. Replication of a GWAS signal in a Caucasian population implicates ADD3 in susceptibility to biliary atresia. Hum. Genet. 133, 235–243, https://doi.org/10.1007/s00439-013-1368-2 (2014).
Green, R. C. et al. Clinical Sequencing Exploratory Research Consortium: Accelerating Evidence-Based Practice of Genomic Medicine. Am. J. Hum. Genet. 99, 246, https://doi.org/10.1016/j.ajhg.2016.06.002 (2016).
Kelsen, J. R. et al. Exome sequencing analysis reveals variants in primary immunodeficiency genes in patients with very early onset inflammatory bowel disease. Gastroenterology 149, 1415–1424, https://doi.org/10.1053/j.gastro.2015.07.006 (2015).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinforma. 26, 589–595, https://doi.org/10.1093/bioinformatics/btp698 (2010).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinforma. 43, 11.10.11–33, https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Cingolani, P. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 6, 80–92, https://doi.org/10.4161/fly.19695 (2012).
Karczewski, K. J. et. al. The Genome Aggregation Database Consortium Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Preprint at, https://doi.org/10.1101/531210 (2019).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46, 310–315, https://doi.org/10.1038/ng.2892 (2014).
Paila, U., Chapman, B. A., Kirchner, R. & Quinlan, A. R. GEMINI: integrative exploration of genetic variation and genome annotations. Plos computational biology 9, e1003153, https://doi.org/10.1371/journal.pcbi.1003153 (2013).
Li, H. Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinforma. 30, 2843–2851, https://doi.org/10.1093/bioinformatics/btu356 (2014).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nat. 536, 285–291, https://doi.org/10.1038/nature19057 (2016).
Pedersen, B. S. & Quinlan, A. R. Who’s Who? Detecting and Resolving Sample Anomalies in Human DNA Sequencing Studies with Peddy. Am. J. Hum. Genet. 100, 406–413, https://doi.org/10.1016/j.ajhg.2017.01.017 (2017).
Genomes Project, C. et al. A global reference for human genetic variation. Nat. 526, 68–74, https://doi.org/10.1038/nature15393 (2015).
Manichaikul, A. et al. Robust relationship inference in genome-wide association studies. Bioinforma. 26, 2867–2873, https://doi.org/10.1093/bioinformatics/btq559 (2010).
Fromer, M. & Purcell, S. M. Using XHMM Software to Detect Copy Number Variation in Whole-Exome Sequencing Data. Curr. Protoc. Hum. Genet. 81, 7 23 21–21, https://doi.org/10.1002/0471142905.hg0723s81 (2014).
MacDonald, J. R., Ziman, R., Yuen, R. K., Feuk, L. & Scherer, S. W. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic acids Res. 42, D986–992, https://doi.org/10.1093/nar/gkt958 (2014).
Ruderfer, D. M. et al. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat. Genet. 48, 1107–1111, https://doi.org/10.1038/ng.3638 (2016).
Casper, J. et al. The UCSC Genome Browser database: 2018 update. Nucleic acids Res. 46, D762–D769, https://doi.org/10.1093/nar/gkx1020 (2018).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic acids Res. 44, W90–97, https://doi.org/10.1093/nar/gkw377 (2016).
Li, B. & Leal, S. M. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am. J. Hum. Genet. 83, 311–321, https://doi.org/10.1016/j.ajhg.2008.06.024 (2008).
Zhan, X., Hu, Y., Li, B., Abecasis, G. R. & Liu, D. J. RVTESTS: an efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinforma. 32, 1423–1426, https://doi.org/10.1093/bioinformatics/btw079 (2016).
Faul, F., Erdfelder, E., Lang, A. G. & Buchner, A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav. Res. Methods 39, 175–191 (2007).
Lorent, K. et al. Identification of a plant isoflavonoid that causes biliary atresia. Sci. Transl. Med. 7, 286RA267, https://doi.org/10.1126/scitranslmed.aaa1652 (2015).
Zhao, X. et al. Glutathione antioxidant pathway activity and reserve determine toxicity and specificity of the biliary toxin biliatresone in zebrafish. Hepatology 64, 894–907, https://doi.org/10.1002/hep.28603 (2016).
Bamshad, M. J. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 12, 745–755, https://doi.org/10.1038/nrg3031 (2011).
Do, R. et al. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nat. 518, 102–106, https://doi.org/10.1038/nature13917 (2015).
Lange, L. A. et al. Whole-exome sequencing identifies rare and low-frequency coding variants associated with LDL cholesterol. Am. J. Hum. Genet. 94, 233–245, https://doi.org/10.1016/j.ajhg.2014.01.010 (2014).
Dong, R. et al. Identification of HSP90 as potential biomarker of biliary atresia using two-dimensional electrophoresis and mass spectrometry. PLoS one 8, e68602, https://doi.org/10.1371/journal.pone.0068602 (2013).
Pozo, F. M. et al. Molecular chaperone Hsp90 regulates REV1-mediated mutagenesis. Mol. Cell Biol. 31, 3396–3409, https://doi.org/10.1128/MCB.05117-11 (2011).
Xiao, Z et. al. Protein Quality Control is a Risk Factor and Therapeutic Target in Toxin-Induced Biliary Atresia. Preprint at https://doi.org/10.1101/821967 (2019).
Conrad, D. F. et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 43, 712–714, https://doi.org/10.1038/ng.862 (2011).
Acknowledgements
We would like to thank all the patients and their families who participated in this study; the ChiLDReN investigators, research coordinators and the Data Coordinating Center for the ChiLDReN network. We would like to acknowledge Dr. Judith Kelsen and Dr. Ian Krantz for providing exome sequencing data for the control samples. We also would like to acknowledge Dr. David Piccoli and Dr. Barbara Haber for their support. We would like to acknowledge Dr. Michael Pack and Dr. Xiao Zhao at the University of Pennsylvania for the collaborative work on zebrafish models. This work is funded by the NIH/NIDDK grant R01-DK090045 and the Fred and Suzanne Biesecker Pediatric Liver Center at Children’s Hospital of Philadelphia. The Childhood Liver Disease Research Network is supported by U01 grants from the National Institute of Diabetes, Digestive and Kidney Disease (NIDDK): DK062481, DK062456, DK062497, DK084536, DK062500, DK062503, DK062466, DK062453, DK062452, DK062436, DK103149, DK103135, DK084575, DK084538, DK062470, DK103140 and DK062455. In addition, the project was supported by UL1 grants from the National Institutes of Health Clinical and Translational Sciences Award (CTSA) program through the National Center for Advancing Translational Science (NCATS): TR000130, TR001872, TR001857, TR001108, TR002535, TR000454, TR000423, RR025014, TR000077 and TR000003. The control samples used in this study were generated using funds from NIH/NIDDK grant R01-DK111843 and NIH/NHGRI grant U01-HG006546.
Author information
Authors and Affiliations
Contributions
M.D., K.L. and N.B.S. designed the experiments. R.R. and E.T. performed the computational and bioinformatics analysis. C.G. performed validation studies. K.L. and S.K. recruited patients. R.R., E.T., K.L., N.B.S. and M.D. were involved in the interpretation of the results. R.R. wrote the manuscript and all authors contributed in editing the manuscript
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rajagopalan, R., Tsai, E.A., Grochowski, C.M. et al. Exome Sequencing in Individuals with Isolated Biliary Atresia. Sci Rep 10, 2709 (2020). https://doi.org/10.1038/s41598-020-59379-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-59379-4
This article is cited by
-
Biliatresone: progress in biliary atresia study
World Journal of Pediatrics (2023)
-
Novel findings from family-based exome sequencing for children with biliary atresia
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
