
Abstract
Surveillance of human immunodeficiency virus (HIV) molecular diversity and drug resistance-associated mutations (DRMs) among treatment-naïve blood donors is critical for monitoring viral evolution and blood safety. From 2016-2017, 199 plasma samples were collected from 24 blood centers and confirmed as HIV viral load positive or serologically reactive in National Centers for Clinical Laboratories (NCCL), of which 179 were sequenced and subtyped in the gag, protease (PR)-reverse transcriptase (RT), integrase (IN) and/or envelope (env) regions. DRMs in PR-RT and IN regions were analyzed in Stanford HIVdb Program. The majority of subtypes were circulating recombinant form (CRF) 07_BC (34.6%) and CRF01_AE (32.4%); many unique recombinant forms (URFs) (39, 21.8%) and other rare CRFs were observed in the study. Notably, CRF02_AG and CRF06_cpx strains typically found in Africa were firstly identified amongst Chinese blood donors. DRMs were common, with 28 of 179 (15.6%) specimens carrying DRMs, including the PR N88S and RT K103N mutations, which have been implicated in elevated resistance to antiretroviral drugs. Furthermore, 4 HIV-1 isolates (2.4%, 4/168) had surveillance drug-resistance mutation (SDRM), including 3 nonnucleosidereverse transcriptase inhibitors (NNRTI) SDRMs (1 K101E, 2 K103N) and 1 protease inhibitor (PI) SDRM (M46I). The HIV viral diversity among blood donors observed in this study suggest that ongoing HIV-1 recombination is becoming progressively complex in China, and lots of DRMs found in the study exacerbate the primary drug resistance landscape, which highlight the necessity of timely genotypic drug resistance monitoring and molecular surveillance of HIV-1 among blood donors.
Similar content being viewed by others
Introduction
According to a recent report from the National Center for Acquired Immunodeficiency Syndrome/Sexually Transmitted Disease (AIDS/STD) Control and Prevention (China CDC), there were approximately 849,602 people living with HIV and 262,442 reported HIV-associated deaths in China as of September, 20181. Epidemiological evidence indicates that the HIV epidemic has shifted from high risk populations into general populations in China, including blood donors2,3. In 2003, the “Four Frees and One Care” policy was implemented to reduce AIDS-related mortality in China by providing free antiretroviral drugs, including highly active antiretroviral therapy4. However, the effectiveness of antiretroviral therapy (ART) may be limited by the transmission of HIV drug resistant strains to ART-naïve patients5, which is a major obstacle to viral suppression6,7. Therefore, the World Health Organization strongly recommends surveillance of transmitted drug resistance (TDR) amongst HIV infected populations8. Characterization of HIV genetic diversity and TDR among volunteer blood donors (treatment-naïve populations) is essential for monitoring viral evolution and optimal ART selection, both have important roles in blood safety9. Moreover, viral diversity has the potential to impact the sensitivity and accuracy of HIV blood screening tests, potentially putting the blood supply at risk as new strains emerge10,11,12. Molecular epidemiological analyses have been powerful tools to investigate the origin and evolution of HIV-1 variants around the world13,14. In China, CRF07_BC, CRF01_AE, CRF08_BC and subtype B are the four most prevalent HIV-1 strains, according to a nationwide molecular epidemiologic survey by China CDC between 2006–200815. In 2012, the US National Heart, Lung and Blood Institute (NHLBI) initiated a surveillance study of the molecular epidemiology of HIV among Chinese blood donor population as an important part of the Recipient Epidemiology and Donor Evaluation Study9. Since this study focused on only 5 blood centers, limited HIV diversity data are currently available. Furthermore, the prevalence of TDR amongst Chinese blood donors is unknown.
In this study, HIV diversity and DRM prevalence were examined among Chinese blood donors from 24 blood screening laboratories between January 2016 and December 2017, covering 17 provinces or municipalities, including all geographic regions (North China, South China, Northwestern District of China and Qinghai-Tibet region). This large-scale study provides the most recent and comprehensive data on HIV-1 molecular epidemiology and TDR among Chinese blood donors, which may inform optimal delivery of ART, improve HIV screening strategies, and serve as a resource for blood centers in China.
Results
Demographic characteristics of blood donors
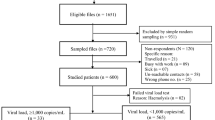
A total of 199 blood donors confirmed as HIV-1 seropositive or viral load positive were enrolled in this study, and 179 donations were successfully sequenced. From the 179 plasma samples, 168 gag, 170 IN, 168 PR-RT and 166 env sequences were generated by Sanger methods. Demographic information from all participants is summarized in Table 1. Notably, the majority of study participants were male (92.2%, 165/179), first-time donors (62.0%, 111/179), Han (95.0%, 170/179) and aged 18–35 years old (73.2%,131/179). Most of the HIV-1 infected blood donors had a lower educated level (Associate degree and secondary school or below: 80.4%, 144/179).
HIV-1 subtype classification
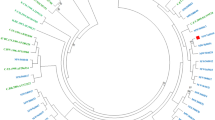
After initial HIV-1 genotyping by the HIV BLAST tool, REGA HIV-1 Subtyping Tool-Version 3.0 and jpHMM program, the specimen classifications were performed using phylogenetic inference of the gag, PR-RT, IN and env regions (Fig. 1). HIV-1 subtype was confirmed by the consistent results from the subtyping tools above, between different gene regions. All potential unique recombinant sequences (the sequences with inconsistent subtyping results from the tools above) were further analyzed by SimPlot 3.5.1 software to determine recombination breakpoints (Fig. S1) and subtypes. Recombinant composition of URFs were displayed in Table 2. It is noted that a limitation of the recombinant HIV-1 drawing tool used to generate Fig. S1 does not allow CRF labels other than CRF01_AE or CRF02_AG. Therefore, regions that were classified as a CRF with a strong bootstrap value and branching pattern are labeled as the parental strains for that CRF, including regions where no recombinant breakpoints are present. In addition to subtype B and C sequences, a diverse set of CRFs were identified amongst the sequenced regions, including CRF01_AE, CRF02_AG, CRF06_cpx, CRF07_BC, CRF08_BC, CRF15_01B, CRF52_01B, CRF55_01B, CRF59_01B, CRF65_cpx, CRF67_01B, CRF77_cpx, CRF78_cpx, CRF79_0107, CRF83_cpx, and CRF85_BC. The majority of specimens were classified as CRF07_BC (34.6%, 62/179) or CRF01_AE (32.4%, 58/179), with URFs being nearly as common as these CRFs (21.8%, 39/179). Although the relative prevalence of each classification varied between geographic regions in China (Number of samples ≥10), URFs were present in all locations (Fig. 2).

Neighbor-joining phylogenetic tree analysis of HIV-1 isolates in blood donors. Sequences from HIV-1 infected blood donors and references are respectively in red and black in the trees and boxes indicate relevant nodes with >70 bootstrap. (a) Phylogenetic tree analysis of gag sequences. (b) Phylogenetic tree analysis of PR-RT sequences. (c) Phylogenetic tree analysis of IN sequences. (d) Phylogenetic tree analysis of env sequences.

The geographic distribution of HIV- I subtype among blood donors. (a) The Northwestern District of China was excluded on account of the small sample sizes. Others represent the rare CRFs including CRF02_AG, CRF55_01B, CRF59_01B, CRF65_cpx, CRF67_01B, CRF79_0107 and CRF85_BC.
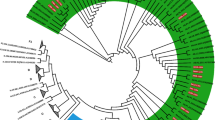
Notably, several sequences (Shaanxi-001, Shaanxi-015, Shaanxi-017) were identified with rare classifications, CRF02_AG and CRF06_cpx, which are uncommon in China (Figs. 3 and 4). Of these, CRF06_cpx regions were present within two different URF strains, whereas the CRF02_AG sequences did not display evidence of recombination (Figs. 3 and 4). The three rare recombinant partial-genome maps were shown in Fig. 5. Furthermore, the details of bootscan and similarity analyses among the other 7 rare CRF strains including CRF55_01B, CRF59_01B, CRF65_cpx, CRF67_01B, CRF79_0107 and CRF85_BC subtypes were described in Figs. S2 and S3 respectively.

Bootscan plots of the three rare recombinant partial-genome sequences from the HIV-1 infected blood donors. Each bootscan plot was performed with Kimura-2 model of nucleotide substitution with a window size of 200 and a step size of 20. The color-coded key represents the different subtypes, sub-subtypes and CRFs of HIV-1. (a) Shaanxi-001 env sequence. (b) Shaanxi-001 IN sequence. (c) Shaanxi-015 env sequence. (d) Shaanxi-015 IN sequence. (e) Shaanxi-017 env sequence.

Similarity plots of the three rare recombinant partial-genome sequences from the HIV-1 infected blood donors. Each similarity plot was performed with Kimura-2 model of nucleotide substitution with a window size of 200 and a step size of 20. The color-coded key represents the different subtypes, sub-subtypes and CRFs of HIV-1. (a) Shaanxi-001 env sequence. (b) Shaanxi-001 IN sequence. (c) Shaanxi-015 env sequence. (d) Shaanxi-015 IN sequence. (e) Shaanxi-017 env sequence.

Three rare recombinant partial-genome maps. (a) Shaanxi-001. (b) Shaanxi-015. (c) Shaanxi-017.
ARV drug resistance–associated mutation analysis
The overall prevalence of DRMs was 15.6% (28/179) in this study population (Table 3). There were 4 (14.3%, 4/28) protease inhibitor (PI) accessory DRMs, 3 PI major DRMs and 22 (78.6%, 22/28) nonnucleoside reverse transcriptase inhibitors (NNRTI) DRMs. No accessory or major NRTI DRMs and Integrase Inhibitors (INSTIs) DRMs were found in these samples. The majority of blood donors with DRMs were infected with CRF07_BC or URF strains (60.7%, 17/28). Most of the PI accessory DRMs were Q58E (3 out of 4) and all the HIV-1 isolates with Q58E mutations in our study were CRF07_BC strains. The PI major DRMs included M46L, M46I and N88S. Most of the NNRTI DRMs (V179D/E [72.7%, 16/22]) were observed in HIV-1 infected blood donors and a combination of V179D and K103R were found in two samples may synergistically reduce ARV drug susceptibility. Furthermore, two blood donors with K103N mutation in the reverse transcriptase gene would be anticipated to have high-level resistance (HLR) to HIV-1 drug. Overall, the prevalence of primary DRMs among each geographic region was as follows, excluding the Northwestern District of China for small sample sizes (Table S1): North China: 14.8% (18/122), South China: 21.7% (5/23), Qinghai-Tibet region 16.1% (5/31).
TDR analysis in PR and PR by Calibrated Population Resistance tool showed that 4 HIV-1 isolates (2.4%, 4/168) had surveillance drug-resistance mutation (SDRM), including 3 NNRTI SDRMs (1 K101E, 2 K103N) and 1 PI SDRM (M46I) (Table 3). Of these 3 were collected from Beijing Tongzhou district blood bank, 1 was from Heilongjiang blood center.
Discussion
During the past two decades, the HIV-1 epidemic has expanded from high risk groups (injection drug users, men who have sex with men, female sex workers etc.) to the general population, including blood donors16. Surveillance of the molecular epidemiology and diversity of HIV amongst blood donors is critical to determining the origin and evolution of HIV-1 variants in China and to prevention of transfusion-transmitted HIV-1 infections. Our study is the most geographically comprehensive epidemiological investigation of HIV among blood donors to date, encompassing 17 provinces and municipalities. The majority of the HIV-1 positive blood donors in the study were males with low educated level aged between 18–35 years. Prevention and screening strategies targeted towards these populations may have a greater impact towards ending the HIV pandemic.
Molecular characterization of the HIV-1 strains circulating within the blood donor population revealed unique patterns in comparison to other groups. Notably, subtype B and CRF08_BC accounted for 5.0% and 1.7% of blood donor infections, respectively, which was lower than the national prevalence reported in the 2006 survey15. Furthermore, the majority of subtype B strains (6/9) identified in this study were from Henan province, and 28.6% (6/21) of the HIV-1 strains from Henan blood centers were subtype B. During the mid-1990s, commercial plasma collection in central China (Henan and Shanxi provinces) led to an outbreak of subtype B among blood donors17. Since the prohibition of commercial blood collection, the prevalence of subtype B transmitted via blood transfusion has significantly decreased, which is reflected by the low proportion of subtype B among enrolled samples in this study. The absence of donor samples from Yunnan, Guizhou and Sichuan provinces in our study may have contributed to the observed low percentage of CRF08_BC, since CRF08_BC predominates in these provinces17. The geographic subtype distribution in Fig. 2 (excluding Qinghai-Tibet region: number of samples ≥10) indicated that CRF01_AE and CRF07_BC were the two main genotypes in each geographic region, which was consistent with previous studies amongst high-risk populations15,18,19,20,21,22,23. The presence of these same strains amongst the local blood donor population was evidence of the expansion of HIV-1 from high-risk groups to blood donor groups. This was consistent with the NHLBI Retrovirus Epidemiology Donor Study-II9, which also found many URFs in the Chinese blood donor population, indicating that ongoing HIV-1 recombination is becoming progressively complex in China. Due to the limited sampling of some blood centers and lack of samples from Qinghai-Tibet region in this study, we cannot make conclusions on the genotype distribution among blood donors throughout China. Future research must focus on expanded geographical coverage to get a more comprehensive dataset to improve blood safety, ART strategy and HIV control and prevention in China.
Our study was also consistent with previous work demonstrating that most Chinese URFs consist of CRF01_AE, B, and C regions (Table 2)24,25. However, the identification of two URFs containing CRF06_cpx regions and an ostensibly pure CRF02_AG infection are novel observations in a Chinese blood donor population. CRF02_AG is a subtype A/G recombinant form endemic to Africa26. Since the first identification of CRF02_AG in neighboring Taiwan in 199827, several CRF02_AG variants have been reported in China28,29, but CRF02_AG has not previously been found amongst volunteer blood donors in China. Likewise, CRF06_cpx was first reported in Burkina Faso in 1998 and had circulated widely in West African countries30. Although a few CRF06_cpx isolates have been found in Beijing, Shenzhen and Hong Kong, this strain has not been reported in volunteer blood donors in China31,32,33. It was noteworthy that the CRF06_cpx regions found in Shaanxi blood donors in our study had recombined with CRF01_AE, A3, B and C strains. Therefore, it was likely that the recombination events that gave rise to these CRF06_cpx-containing URFs occurred in China. Complete genome sequencing of these strains and the CRF02_AG isolate will be required to identify all recombination breakpoints and estimate when these strains may have entered the Chinese blood donor population.
Generally, all HIV-1 positive blood donors are presumed to be treatment-naïve and the presence of ART resistant strains in this group is a reflection of the rate of transmitted drug resistance in a population34. DRM determinations were analyzed by Stanford HIVdb Program, which was based on subtype B and had biased the results for non-B strains35. In the present study, 15.6% (28/179) of HIV-1 infected blood donors had accessory or major DRMs. Notably, Q58E and V179D/E were the most common DRMs to PI and NNRTI respectively in the study, which is consistent with a previous study focused on five blood centers in China9. Q58E was identified as the potential low-level resistance (PLLR)-related muation to Nelfinavir (NFV) and low-level resistance (LLR)-related mutation to Tipranavir (TPV)36.
In particular, the Q58E DRM may be more common in CRF07_BC strains, which was also consistent with a recent study9. The PI major DRMs included M46L, M46I and N88S. M46I/L caused PLLR to many INSTIs among HIV-1 positive individuals, while N88S could result in HLR to Atazanavir (ATV) and NFV, LLR to Indinavir (IDV) and Saquinavir (SQV)37,38,39. Q58E and other DRMs (M46L/I, N88S) that confer resistance to PIs were present in our study. About PIs, only Lopinavir (LPV) was included in the Free AIDS Antiretroviral Therapy Manual, and not included in the first-line ART in China40, suggesting that either these DRMs were imported or they did not arise from selective pressure during treatment. In contrast, the most common NNRTI DRMs in our study, V179D/E mutations, were observed within a variety of strains, consistent with selective pressure from use of NNRTI in China40,41. A combination of RT V179D and K103R found in two samples with HIV-1 infection may synergistically reduce EFV and NVP susceptibility about 10-fold42, RT mutations with combination of V179D and K103R were also observed in treatment-naïve individuals in China43. The RT K103N mutation found in two strains can reduce EFV and NVP susceptibility by about 20- and 50-fold, respectively44.
Drug resistance analysis demonstrated that 2.4% of HIV-1 isolates contained at least one NNRTI (K101E, K103N) or PI (M46I) SDRMs, the overall prevalence of TDR was lower than previous reports in Zhejiang (11.1%) and Shijiazhuang (6.1%) among treatment-naïve HIV-infected individuals45,46, but similar to a nationwide cross-sectional survey about prevalence of TDR (3.6%) in 2015 in China47. Although the rate of TDR remained relatively low in Chinese blood donors in this study, the detection of 3 major NNRTI mutations and 1 PI mutation underlined the importance of a continuous surveillance of resistance mutations.
Overall, the prevalence of DRMs in South China was higher than other regions. The distribution of HIV-1 with DRMs in this study (Table S1) suggests that the HIV-1 strains isolated from positive blood donors in urban centers such as Beijing, Zhengzhou (Henan provincial capital) and Shenzhen had higher rates of DRMs. Moreover, 75% (3/4) HIV-1 isolates with SDRMs in the study were from Beijing Tongzhou district blood bank. It is possible that increased international travel and immigration in these populations may have contributed to the observed higher rates of DRMs.
In summary, our study characterized increasing HIV-1 diversity and high rates of drug resistance in the Chinese blood donor population, with unique province-level trends observed therein. The main HIV-1 subtypes of blood donors in most provinces were consistent with the local high-risk populations, suggesting that the HIV-1 epidemic has expanded from high risk groups to the general population. Most importantly, the integration of imported CRF02_AG and CRF06_cpx strains into the Chinese blood donor population is further evidence of the newly emerging migration patterns of the global HIV-1 pandemic. Furthermore, lots of DRMs and several TDR were found in treatment-naïve blood donors, underscoring the need for continued molecular surveillance to monitor and appropriately respond to expanding local HIV-1 diversity with diagnostic tests and therapeutics that are effective for circulating strains.
Limitations
Since the prevalence of HIV-1 among Chinese blood donors remains low and not all the HIV-1 positive donations in blood screening laboratories were enrolled in our study, the limited sample sizes from several blood screening laboratories may bias the molecular epidemiological results. Furthermore, the socio-demographic data of the HIV-1 infected blood donor lack possible mode of transmission which would be used for analysis of the risks of HIV-1 transmission. For HIV-1 subtype analysis, HIV-1 genome sequencing including env, pol and gag genes is most reliable for subtype classification, but it’s hard to get HIV-1 genome sequence, due to the long length of genome sequence, low viral load in several samples.
Future research must focus on expanded geographical coverage and HIV-1 genome sequences to get a more comprehensive dataset to improve blood safety, ART strategy and HIV control and prevention in China.
Materials and Methods
Study samples
From January 2016 to December 2017, a total of 199 blood donations collected from 24 blood screening laboratories were confirmed as HIV viral load positive by the Abbott RealTime HIV-1 (Abbott Molecular Diagnostics, Des Plaines, IL, USA) test or serologically reactive by the Abbott ARCHITECT HIV Ag/Ab Combo test (Abbott Diagnostics, Weisbaden, Germany). These samples were tested non-reactive for Hepatitis B surface antigen (HBsAg), antibody to Hepatitis C Virus (anti-HCV) and antibody to treponema pallidum (anti-TP) in blood screening laboratories. Of these, at least two HIV regions were successfully sequenced for 179 plasma samples (Supplementary materials Table S1). Geographical localizationof blood screening laboratories in the study and the number of blood donations in these laboratories from January 2016 to December 2017 were shown in Fig. S4.
RNA extraction, amplification and sequencing
HIV-1 RNA was extracted from 140 µL of HIV-1 positive plasma using QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s protocol. Amplifications of the HIV-1 gag p24, env gp41 and polymerase genes (pol) (encoding PR, RT and IN), were performed by QIAGEN OneStep RT-PCR Kit (Qiagen, Hilden, Germany) and nested PCR using AmpliTaq DNA Polymerase (Applied Biosystems, Foster City, USA). HIV-1 gag (HXB2: 1074–2047), PR-RT sequence (HXB2: 2068–3521), IN (HXB2: 4175–5214) and env gp41 (HXB2: 7648–8365) regions were amplified with outer primers respectively in the first round. The amplification of the env fragment was performed at 50 °C for 30 min for reverse transcription and then 95 °C for 15 min, followed by 50 cycles at 94 °C for 15 s, 50 °C for 30 s, and 72 °C for 1 min, with a final extension at 72 °C for 7 min. The other three regions were amplified following the same PCR conditions, except with an annealing temperature of 55 °C. The fist-round PCR products for four regions (5 µL) along with their respective inner primers were used in the nested PCR48. Nested PCR for pol-PR-RT was conducted with one cycle at 94 °C for 2 min, followed by 40 cycles at 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, and finally an extension of 10 min at 72 °C. Nested PCR for gag, IN and env genes followed the same procedure but with an annealing temperature of 50 °C. The nested PCR products were purified and sequenced by Sangon Biotech (Shanghai) Co., Ltd using Sanger methods. Details of the primers used in the study are described in Supplementary materials Table S2.
HIV-1 genotype and phylogenetic analysis
The sample sequences were edited and aligned by Geneious 9.1.2 (https://www.geneious.com/products/prime/resources/download/previous-versions). Gag and two pol gene sequences covering PR and part of RT and the entire IN were submitted to the Los Alamos HIV BLAST tool for initial HIV-1 subtyping (https://www.hiv.lanl.gov/content/sequence/BASIC_BLAST/basic_blast.html)49 and then analyzed by REGA HIV-1 Subtyping Tool-Version 3.0 (http://dbpartners.stanford.edu:8080/RegaSubtyping/stanford-hiv/typingtool)50 and jpHMM program (http://jphmm.gobics.de/submission_hiv.html)51. Previous reports support utilizing the env, gag and pol regions for reliable subtype assignment52,53,54. The sequences were aligned with HIV-1 reference sequences (Accession numbers: Table S3) obtained from the Los Alamos database (https://www.hiv.lanl.gov) and then the nucleotide alignments were used to build phylogenetic tree for further HIV-1 subtyping by MEGA 7.0.2 (https://www.megasoftware.net) using the neighbor-joining algorithm based on Kimura 2-parameter model in 1000 bootstrap replicates55. The final subtype of HIV-1 isolate was confirmed by the consistent results from all the subtyping tools above. Boot-scanning and intra-genomic breakpoints analyses were conducted on sequences with possibly unidentified recombinant strains (the sequences with inconsistent results from all the subtyping tools above) through SimPlot 3.5.1 (https://www.softpedia.com/get/Science-CAD/SimPlot.shtml)56. Sequences with recombinant patterns that did not match established CRFs were classified as URFs6,57. The genome maps of URFs were generated through Recombinant HIV-1 Drawing Tool (https://www.hiv.lanl.gov/content/sequence/DRAW_CRF/recom_mapper.html). For finalization of phylogenetic classifications, neighbor-joining phylogenetic trees were prepared using PHYLIP 3.5 as previously described58 and simplified trees were visualized using FigTree v1.4.2 (University of Edinburgh, UK) to prepare figures.
Drug resistance mutation analysis
DRM analyses in PR-RT and IN regions were performed in the Stanford HIV Drug Resistance Database (https://hivdb.stanford.edu)59 using Stanford HIVdb Program Genotypic Resistance Interpretation Algorithm (https://hivdb.stanford.edu/hivdb/by-sequences)60. Inferred levels of resistance of HIV-1 (PR, RT, IN) to 24 US Food and Drug Administration (FDA)-approved ARV drugs can be analyzed by the HIVdb program using total drug score derived by scores of each DRM related with the antiviral drug, and program reports are shown as the 5 levels of inferred drug resistance: HLR, intermediate resistance (IR), LLR,PLLR and susceptible61. Furthermore, populations of PR and RT sequences were submitted to CRP tool (http://cpr.stanford.edu/cpr.cgi) to perform standardized genotypic estimation of TDR62.
Statistical analysis
Demographic data were obtained from the donor/donation database from each blood center and bank. SPSS 21.0 software was utilized for statistical analysis.
Sequence data
The sequences described in this article have been deposited in the GenBank Nucleotide Sequence Database under accession numbers MK771158-MK771325 (pol reverse transcriptase sequences), MK771326-MK771493 (gag sequences), MK771495-MK771664 (pol protease sequences) and MK771494, MK771665-MK771829 (env sequences).
Ethics approval and consent to participate
The study was approved by the Ethics Committee in Beijing Hospital (Ethics board approval number: 2016BJYYEC-118–01). Ethics statement in Chinese shown in supplementary material. The methods in the study were in accordance with the guidelines of the Declaration of Helsinki. Written informed consent was obtained from all subjects before blood donation.
Data availability
The data for this study is available from the corresponding author on reasonable request.
References
National Center for AIDS/STD Control and Prevention. National AIDS and STD epidemic in the third quarter of 2018. Chinese Journal of AIDS & STD, 24 (2018).
Wang, J. et al. An analysis of risk factors for human immunodeficiency virus infection among Chinese blood donors. Transfusion 53, 2431–2440, https://doi.org/10.1111/trf.12062 (2013).
Wang, J. et al. Prevalence, incidence, and residual risks for transfusion-transmitted human immunodeficiency virus Types 1 and 2 infection among Chinese blood donors. Transfusion 53, 1240–1249, https://doi.org/10.1111/j.1537-2995.2012.03940.x (2013).
He, N. & Detels, R. The HIV epidemic in China: history, response, and challenge. Cell Res. 15, 825–832, https://doi.org/10.1038/sj.cr.7290354 (2005).
Han, X. et al. Genotypic resistance mutations to antiretroviral drugs in treatment-naive HIV/AIDS patients living in Liaoning Province, China: baseline prevalence and subtype-specific difference. AIDS Res. Hum. Retroviruses 23, 357–364, https://doi.org/10.1089/aid.2006.0094 (2007).
Nii-Trebi, N. I. et al. HIV-1 drug-resistance surveillance among treatment-experienced and -naive patients after the implementation of antiretroviral therapy in Ghana. PLoS one 8, e71972, https://doi.org/10.1371/journal.pone.0071972 (2013).
Gupta, R. K. et al. Global trends in antiretroviral resistance in treatment-naive individuals with HIV after rollout of antiretroviral treatment in resource-limited settings: a global collaborative study and meta-regression analysis. Lancet 380, 1250–1258, https://doi.org/10.1016/s0140-6736(12)61038-1 (2012).
Bennett, D. E., Bertagnolio, S., Sutherland, D. & Gilks, C. F. The World Health Organization’s global strategy for prevention and assessment of HIV drug resistance. Antivir. Ther. 13(Suppl 2), 1–13 (2008).
Zeng, P. et al. The infection staging and profile of genotypic distribution and drug resistance mutation among the human immunodeficiency virus-1 infected blood donors from five Chinese blood centers, 2012-2014. PLoS one 12, e0179328 (2017).
Qiu, X. et al. Comparative evaluation of three FDA-approved HIV Ag/Ab combination tests using a genetically diverse HIV panel and diagnostic specimens. J. Clin. Virol. 92, 62–68, https://doi.org/10.1016/j.jcv.2017.05.005 (2017).
Brennan, C. A. et al. HIV global surveillance: foundation for retroviral discovery and assay development. J. Med. Virol. 78(Suppl 1), S24–29, https://doi.org/10.1002/jmv.20603 (2006).
Ly, T. D. et al. The variable sensitivity of HIV Ag/Ab combination assays in the detection of p24Ag according to genotype could compromise the diagnosis of early HIV infection. J. Clin. Virol. 55, 121–127, https://doi.org/10.1016/j.jcv.2012.06.012 (2012).
Travers, S. A. et al. Timing and reconstruction of the most recent common ancestor of the subtype C clade of human immunodeficiency virus type 1. J. Virol. 78, 10501–10506, https://doi.org/10.1128/jvi.78.19.10501-10506.2004 (2004).
Ou, C. Y. et al. Molecular epidemiology of HIV transmission in a dental practice. Science 256, 1165–1171, https://doi.org/10.1126/science.256.5060.1165 (1992).
He, X. et al. A comprehensive mapping of HIV-1 genotypes in various risk groups and regions across China based on a nationwide molecular epidemiologic survey. PLoS one 7, e47289, https://doi.org/10.1371/journal.pone.0047289 (2012).
Zeng, P. et al. The human immunodeficiency virus-1 genotype diversity and drug resistance mutations profile of volunteer blood donors from Chinese blood centers. Transfusion 52, 1041–1049, https://doi.org/10.1111/j.1537-2995.2011.03415.x (2012).
Su, Y., Liu, H., Wu, J., Zhu, L. & Wang, N. Distribution of HIV-1 genotypes in China: a systematic review. Chinese Journal of Epidemiology 35, 1164–1168 (2014).
Li, Q. H. et al. Molecular Genotyping of HIV-1 Strains from Newly Infected MSM in Harbin, China. Aids Res. Hum. Retroviruses 32, 595–600, https://doi.org/10.1089/AID.2016.0028 (2016).
Bin, Z. et al. New trends of primary drug resistance among HIV type 1-infected men who have sex with men in Liaoning Province, China. Aids Res. Hum. Retroviruses 27, 1047–1053, https://doi.org/10.1089/AID.2010.0119 (2011).
Xing, A. H., Xiang, L. I. & Zhang, M. Y. Molecular epidemiological study on HIV type 1 among MSM in Shaanxi Province. Chinese Journal of Aids & Std 18, 741–745 (2012).
Zhao, J. et al. The dynamics of the HIV epidemic among men who have sex with men (MSM) from 2005 to 2012 in Shenzhen, China. Sci. Rep. 6, 28703, https://doi.org/10.1038/srep28703 (2016).
Yan, M. et al. HIV-1 diversity and drug-resistant mutations in infected individuals in Changchun, China. PLoS one 9, e100540, https://doi.org/10.1371/journal.pone.0100540 (2014).
Jing-Rong, Y. et al. Genetic diversity of HIV type 1 isolated from newly diagnosed subjects (2006-2007) in Beijing, China. Aids Res. Hum. Retroviruses 28, 119–123, https://doi.org/10.1089/AID.2011.0012 (2012).
Lee, N. Y. et al. Characterization of the Near Full-Length Genome of a Novel HIV-1 CRF01_AE/CRF07_BC Recombinant in an Injection Drug User from Southern Taiwan. AIDS Res. Hum. Retroviruses 32, 588–594, https://doi.org/10.1089/aid.2015.0246 (2016).
Wang, N. et al. Near full-length genome characterization of a new CRF01_AE/CRF08_BC recombinant transmitted between a heterosexual couple in Guangxi, China. Aids Res. Hum. Retroviruses 30, 484–488, https://doi.org/10.1089/aid.2013.0230 (2014).
Howard, T. M. & Rasheed, S. Genomic structure and nucleotide sequence analysis of a new HIV type 1 subtype A strain from Nigeria. AIDS Res. Hum. Retroviruses 12, 1413–1425, https://doi.org/10.1089/aid.1996.12.1413 (1996).
Lee, C. N. et al. HIV type 1 env subtype A variants in Taiwan. AIDS Res. Hum. Retroviruses 14, 807–809, https://doi.org/10.1089/aid.1998.14.807 (1998).
Li, Q. H. et al. Molecular Genotyping of HIV-1 Strains from Newly Infected Men Who Have Sex with Men in Harbin, China. AIDS Res. Hum. Retroviruses 32, 595–600, https://doi.org/10.1089/aid.2016.0028 (2016).
Li, X. et al. Molecular epidemiology of HIV-1 in Jilin province, northeastern China: emergence of a new CRF07_BC transmission cluster and intersubtype recombinants. PLoS one 9, e110738, https://doi.org/10.1371/journal.pone.0110738 (2014).
Montavon, C. et al. The identification of a complex A/G/I/J recombinant HIV type 1 virus in various West African countries. AIDS Res. Hum. Retroviruses 15, 1707–1712, https://doi.org/10.1089/088922299309757 (1999).
Ye, J. R. et al. The prevalence of drug resistance mutations among treatment-naive HIV-infected individuals in Beijing, China. AIDS Res. Hum. Retroviruses 28, 418–423, https://doi.org/10.1089/aid.2011.0097 (2012).
Chen, J. H. et al. Evaluation of an in-house genotyping resistance test for HIV-1 drug resistance interpretation and genotyping. J. Clin. Virol. 39, 125–131, https://doi.org/10.1016/j.jcv.2007.03.008 (2007).
Zhao, G. L. et al. Study on the molecular-epidemiological characteristics of HIV-1 in Shenzhen, 1992-2008. Chinese Journal of Epidemiology 33, 82–87 (2012).
Yang et al. The emergence of HIV-1 primary drug resistance genotypes among treatment-naive men who have sex with men in high-prevalence areas in China. Arch. virology 158, 839–844, https://doi.org/10.1007/s00705-012-1557-7 (2013).
Snoeck, J. et al. Discordances between interpretation algorithms for genotypic resistance to protease and reverse transcriptase inhibitors of human immunodeficiency virus are subtype dependent. Antimicrob. Agents Chemother. 50, 694–701, https://doi.org/10.1128/aac.50.2.694-701.2006 (2006).
Baxter, J. D. et al. Genotypic changes in human immunodeficiency virus type 1 protease associated with reduced susceptibility and virologic response to the protease inhibitor tipranavir. J. Virol. 80, 10794–10801, https://doi.org/10.1128/jvi.00712-06 (2006).
Dolling, D. I. et al. Low frequency of genotypic resistance in HIV-1-infected patients failing an atazanavir-containing regimen: a clinical cohort study. J. Antimicrob. Chemother. 68, 2339–2343, https://doi.org/10.1093/jac/dkt199 (2013).
Dandache, S. et al. In vitro antiviral activity and cross-resistance profile of PL-100, a novel protease inhibitor of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 51, 4036–4043, https://doi.org/10.1128/aac.00149-07 (2007).
PI Resistance Notes, https://hivdb.stanford.edu/dr-summary/resistance-notes/PI/ (2019).
National Center for AIDS/STD Control and Prevention, China CDC. Free AIDS Antiretroviral Therapy Manual in China, People’s Medical Publishing House (2016).
Li, J. et al. Drug resistance evolution in patients with human immunodeficiency virus-1 under long-term antiretroviral treatment-failure in Yunnan Province, China. Virol. J. 16, 5, https://doi.org/10.1186/s12985-018-1112-6 (2019).
Parkin, N. T., Gupta, S., Chappey, C. & Petropoulos, C. J. The K101P and K103R/V179D mutations in human immunodeficiency virus type 1 reverse transcriptase confer resistance to nonnucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 50, 351–354, https://doi.org/10.1128/aac.50.1.351-354.2006 (2006).
Qiang, R. et al. Investigation on Drug Resistance Threshold of Newly Found HIV Infected Persons in Shaanxi. J. Mod. Laboratory Med. 32, 136–138 (2017).
Bacheler, L. et al. Genotypic correlates of phenotypic resistance to efavirenz in virus isolates from patients failing nonnucleoside reverse transcriptase inhibitor therapy. J. Virol. 75, 4999–5008, https://doi.org/10.1128/jvi.75.11.4999-5008.2001 (2001).
Xu, Y. et al. Characterization of HIV-1 subtypes and transmitted drug resistance among treatment-naive HIV-infected individuals in Zhejiang, China, 2014-2017. Arch. virology 163, 2233–2237, https://doi.org/10.1007/s00705-018-3839-1 (2018).
Wang, X. et al. Epidemiological surveillance of HIV-1 transmitted drug resistance among newly diagnosed individuals in Shijiazhuang, northern China, 2014-2015. PLoS one 13, e0198005, https://doi.org/10.1371/journal.pone.0198005 (2018).
Zhao, S. et al. Prevalence of Transmitted HIV drug resistance in antiretroviral treatment naive newly diagnosed individuals in China. Sci. Rep. 8, 12273, https://doi.org/10.1038/s41598-018-29202-2 (2018).
Winters, M. A. et al. A 6-basepair insert in the reverse transcriptase gene of human immunodeficiency virus type 1 confers resistance to multiple nucleoside inhibitors. J. Clin. Invest. 102, 1769–1775, https://doi.org/10.1172/jci4948 (1998).
Kuiken, C., Korber, B. & Shafer, R. W. HIV sequence databases. Aids Rev. 5, 52–61 (2003).
Pineda-Peña, A.-C. et al. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 19, 337–348, https://doi.org/10.1016/j.meegid.2013.04.032 (2013).
Schultz, A. K. et al. jpHMM: improving the reliability of recombination prediction in HIV-1. Nucleic acids Res. 37, W647–651, https://doi.org/10.1093/nar/gkp371 (2009).
Njouom, R. et al. Assessment of HIV-1 subtyping for Cameroon strains using phylogenetic analysis of pol gene sequences. J. Virological Methods 110, 1–8, https://doi.org/10.1016/s0166-0934(03)00080-6 (2003).
Yang, C. et al. Detection of phylogenetically diverse human immunodeficiency virus type 1 groups M and O from plasma by using highly sensitive and specific generic primers. J. Clin. Microbiol. 37, 2581–2586, https://doi.org/10.1016/j.fertnstert.2007.10.071 (1999).
Swanson, P., Devare, S. G. & Hackett, J. J. Molecular Characterization of 39 HIV Isolates Representing Group M (Subtypes A-G) and Group O: Sequence Analysis of Gag p24, Pol Integrase, and Env gp41. Aids Res. Hum. Retroviruses 19, 625–629, https://doi.org/10.1089/088922203322231003 (2003).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evolution 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Tagliamonte, M. et al. Genetic and phylogenetic characterization of structural genes from non-B HIV-1 subtypes in Italy. AIDS Res. Hum. Retroviruses 22, 1045–1051, https://doi.org/10.1089/aid.2006.22.1045 (2006).
Lu, X. et al. HIV-1 Genetic Diversity and Transmitted Drug Resistance Among Recently Infected Individuals at Men Who Have Sex with Men Sentinel Surveillance Points in Hebei Province, China. Aids Res. Hum. Retroviruses 31, 1038–1045, https://doi.org/10.1089/AID.2015.0160 (2015).
Rodgers, M. A. et al. Identification of rare HIV-1 Group N, HBV AE, and HTLV-3 strains in rural South Cameroon. Virology 504, 141–151, https://doi.org/10.1016/j.virol.2017.01.008 (2017).
Rhee, S. Y. et al. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic acids Res. 31, 298–303, https://doi.org/10.1093/nar/gkg100 (2003).
Liu, T. F. & Shafer, R. W. Web resources for HIV type 1 genotypic-resistance test interpretation. Clin. Infect. diseases: an. Off. Publ. Infect. Dis. Soc. Am. 42, 1608–1618, https://doi.org/10.1086/503914 (2006).
RELEASE NOTES For HIVdb, HIVseq, and HIValg https://hivdb.stanford.edu/page/release-notes (2019).
Gifford, R. J. et al. The calibrated population resistance tool: standardized genotypic estimation of transmitted HIV-1 drug resistance. Bioinformatics 25, 1197–1198, https://doi.org/10.1093/bioinformatics/btp134 (2009).
Acknowledgements
We thank clinical laboratory directors from 24 blood centers respectively for participating in sample collection and testing. This work was supported by grants from National Science and Technology Major Project of the Ministry of Science and Technology of China (2018ZX10306412-004 and 2018ZX10732101-003-002).
Author information
Authors and Affiliations
Contributions
J.Z., L.W. and X.L. designed the study. J.Z., X.L., F.G. conducted the laboratory tests. J.Z. collected and analyzed data and prepared the manuscript. L.W., M.R. and L.C. edited and reviewed the manuscript. All Authors critically reviewed and revised the manuscript drafts, approved the final version of the manuscript and take responsibility for the integrity of the data and accuracy of data analysis.
Corresponding author
Ethics declarations
Competing interests
X.L., B.H., P.Y., J.H., G.C. and M.R. are employees and shareholders in Abbott Laboratories.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, J., Lv, X., Chang, L. et al. HIV-1 molecular epidemiology and drug resistance-associated mutations among treatment-naïve blood donors in China. Sci Rep 10, 7571 (2020). https://doi.org/10.1038/s41598-020-64463-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-64463-w
This article is cited by
-
Characterization of an imported HIV-1 A1/A7/G recombinant in China
Virology Journal (2024)
-
Transmitted HIV drug resistance and subtype patterns among blood donors in Poland
Scientific Reports (2021)
-
Genetic characteristics of HIV-1 CRF06_cpx and CRF56_cpx strains isolated in Guangzhou, China
Archives of Virology (2021)
-
HTLV screening of blood donors using chemiluminescence immunoassay in three major provincial blood centers of China
BMC Infectious Diseases (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
