
Summary:
I-cell disease or mucolipidosis type II, a rare inherited storage disorder of lysosomal enzyme localization, is characterized by dysostosis multiplex, progressive severe psychomotor retardation and death by 5–8 years from congestive heart failure and recurrent pulmonary infections. A 19-month old girl with I-cell disease received a bone marrow transplant (BMT) from an HLA-identical carrier brother. At the age of 7 years, 5 years after BMT, she has no history of respiratory infections. Her cardiac function remains normal with a shortening fraction of 47%, and she continues to gain neurodevelopmental milestones, albeit at a very slow rate. Musculoskeletal deformities have worsened despite BMT. This is the first report describing neurodevelopmental gains and prevention of cardiopulmonary complications in I-cell disease after BMT.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others
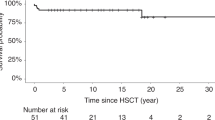

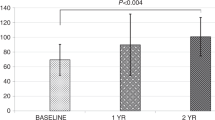
References
Leroy JG, Spranger JW, Feingold M et al. I-cell disease: a clinical picture. J Pediatr 1971; 79: 360–365.
Kornfeld S, Sly WS . I-cell disease and pseudo-Hurler polydystrophy: disorders of lysosomal enzyme phosphorylation and localization. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds). The Metabolic and Molecular Bases of Inherited Disease, 8th edn. McGraw-Hill: New York, 2001, pp 3469–3482.
Patriquin HB, Kaplan P, Kind HP et al. Neonatal mucolipidosis II (I-cell disease): clinical and radiologic features in three cases. AJR Am J Roentgenol 1977; 129: 37–43.
Hille-Rehfeld A . Mannose 6-phosphate receptors in sorting and transport of lysosomal enzymes. Biochim Biophys Acta 1995; 1241: 177–194.
Hickman S, Neufeld EF . A hypothesis for I-cell disease: defective hydrolases that do not enter lysosomes. Biochem Biophys Res Commun 1972; 49: 992–999.
Burgess J, Traycoff C, Hardwick A et al. Separation of clinical quantities of CD34+ cells from human marrow using immunomagnetic procedures. Prog Clin Biol Res 1994; 389: 293–302.
Kurobane I, Inoue S, Gotoh Y et al. Biochemical improvement after treatment by bone marrow transplantation in I-cell disease. Tohoku J Exp Med 1986; 150: 63–68.
Imaizumi M, Gushi K, Kurobane I et al. Long-term effects of bone marrow transplantation for inborn errors of metabolism: a study of four patients with lysosomal storage diseases. Acta Paediatr Jpn 1994; 36: 30–36.
Peters C, Shapiro EG, Anderson J et al. Hurler syndrome: II. Outcome of HLA-genotypically identical sibling and HLA-haploidentical related donor bone marrow transplantation in fifty-four children. The storage disease collaborative study group. Blood 1998; 91: 2601–2608.
Peters C, Balthazor M, Shapiro EG et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996; 87: 4894–4902.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Grewal, S., Shapiro, E., Braunlin, E. et al. Continued neurocognitive development and prevention of cardiopulmonary complications after successful BMT for I-cell disease: a long-term follow-up report. Bone Marrow Transplant 32, 957–960 (2003). https://doi.org/10.1038/sj.bmt.1704249
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.bmt.1704249
Keywords
This article is cited by
-
Mucolipidosis II and III alpha/beta: mutation analysis of 40 Japanese patients showed genotype–phenotype correlation
Journal of Human Genetics (2009)
-
Mukolipidosen
Monatsschrift Kinderheilkunde (2006)
