
Abstract
Ursodeoxycholic acid (UDCA), a relatively nontoxic bile acid, enhanced the apoptotic response of tumor cells to both photosensitizers that cause photodamage to Bcl-2 and to the nonpeptidic Bcl-2/Bcl-xL antagonist HA14-1. The latter agent binds to the surface pocket formed by the BH1, BH2 and BH3 domains of Bcl-2 and Bcl-xL. Fluorescence polarization studies indicated that affinity of HA14-1 for Bcl-2 was enhanced in the presence of UDCA. Moreover, Bcl-2 photodamage was promoted by UDCA using a photosensitizing agent with affinity for the endoplasmic reticulum, a site of Bcl-2 localization. Fluorescence resonance energy transfer (FRET) studies revealed that the proximity of Bcl-2 to a hydrophobic photosensitizing agent embedded in liposomes was enhanced by UDCA. Since photodamage will occur only if a protein is in close contact with a photosensitizing agent, we propose that these findings support the hypothesis that UDCA causes a conformational change in Bcl-2, promoting HA14-1 binding and enhancing affinity for certain membrane-bound photosensitizers.
Similar content being viewed by others


Introduction
Photodynamic therapy (PDT) is a procedure for cancer control using drugs that sensitize neoplastic tissues and their vasculature to light.1 The basis of this therapy is the ability of the photosensitizing agents to catalyze the light-mediated conversion of dissolved oxygen to a highly toxic product, singlet molecular oxygen. The latter can catalyze oxidative damage to proteins and lipids that are in very close proximity to the sensitizer.1 The subcellular targets for photodamage vary among the different photosensitizing agents. In 1999, we identified the target of an important class of photosensitizing agents: the antiapoptotic protein Bcl-2.2 Agents in this class include the tin etiopurpurin SnET2, the phthalocyanine Pc 4, the chlorin m-THPC and a porphycene termed 9-capronyloxy-tetrakis(methoxyethyl) porphycene (CPO).2, 3, 4, 5, 6 It is likely that any photosensitizing agent that localizes in the vicinity of Bcl-2 will be in this drug class. We and Oleinick's group have examined the consequences of Bcl-2 photodamage and subsequent inactivation. These include release of cytochrome c from mitochondria, activation of caspase-3 and initiation of the apoptotic program.4, 5, 6, 7
Ursodeoxycholic acid (UDCA) (Figure 1) is a nontoxic bile acid that has been reported to protect hepatocytes, hepatoma cells, osteogenic sarcomas and HeLa cells from apoptosis induced by okadaic acid, hydrogen peroxide, ethanol, and more hydrophobic bile acids, for example, DCA.8, 9, 10, 11, 12, 13, 14, 15 Other investigators found that UDCA promoted apoptosis in some systems.16, 17 These diverse reports prompted an examination of the effects of UDCA on PDT-induced apoptosis in cell culture. We found a significant promotion of the apoptotic cell death initiated by the photosensitizer SnET2.6, 18 The latter agent shows affinity for both lysosomes and subcellular membranes including the endoplasmic reticulum (ER), and can therefore mediate photodamage at a variety of sites.3 To better delineate the mechanism of action of UDCA, we focused on two sensitizers known to selectively localize in either subcellular membranes including the ER (CPO) or in lysosomes (NPe6). A mode of action of UDCA was suggested by our finding that UDCA also enhanced the proapoptotic effects of the nonpeptidic Bcl-2 antagonist HA14-1.5 The latter agent binds to the surface cleft on Bcl-2 and Bcl-xL.19, 20 Preliminary results suggested that the proapoptotic effects of UDCA might involve interactions with antiapoptotic members of the Bcl-2 family, a hypothesis that we have pursued in the present work.

Structures of DCA and UDCA. Labeled compounds contain 14C in the terminal COOH residue
Results
Intracellular accumulation and toxicity of bile acids
To characterize bile acid transport, we initially examined lysyl-fluorescein derivatives of UDCA and DCA,21 but found that these fluorescent analogs were excluded from L1210 cells. We therefore prepared radioactive UDCA and DCA to assess bile acid transport, stability of intracellular pools, and competition between these two compounds for cellular binding sites.
A steady state was rapidly reached with 14COOH-labeled DCA or UDCA (Table 1), with the equilibrium distribution ratio higher when the more hydrophobic DCA was tested. There was no significant inhibition of 14C-DCA or -UDCA accumulation by an equimolar level of either nonradioactive bile acid, indicating a high capacity for accumulation. Once a steady state was reached, washing cells for 10 min at 37°C in fresh medium resulted in a loss of ∼90% of intracellular UDCA or DCA. This was unaffected by prolonging the washing interval to 30 min or by the presence of unlabeled bile acids in the medium (not shown).
A ‘dose–response’ study was carried out over a range of UDCA concentrations from 3–100 μM. This experiment revealed that neither the distribution ratio nor the percentage of UDCA retained after washing was altered over this concentration range (Table 2).
Exposure of L1210 cells to 100 μM DCA for 60 min resulted in a substantial loss of viability. This was not reversed by addition of 100 μM UDCA, nor was viability significantly affected by 100 μM UDCA alone (Table 3).
Photosensitizer localization and PDT effects
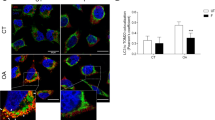
Fluorescence microscopy demonstrated that CPO showed an affinity for a variety of subcellular membranes. NPe6 was localized in lysosomes, and SnET2 was detected at both loci (Figure 2, top). Irradiation of cells sensitized with CPO or SnET2, using LD90 PDT conditions, resulted in a substantial loss of Bcl-2, but not of Bcl-xL or Bax, as detected by Western blots (Figure 2, bottom). NPe6 was inactive in this regard. In the presence of 100 μM UDCA, the phototoxicities of SnET2 and CPO were substantially promoted, while NPe6 phototoxicity was unaffected (Table 4). Studies shown in Table 4 were carried out with LD50 PDT conditions so that the effect of UDCA could more readily be detected. These results demonstrate that PDT efficacy is promoted by the bile acid UDCA when Bcl-2 is a direct target for photodamage.

(Top) Localization of photosensitizers in L1210 cells: (a) CPO, (b) NPe6, (c) SnET2. (Bottom) Western blots showing the extent of PDT-induced photodamage to Bax, Bcl-2, Bax or Bcl-xL after LD90 PDT doses of CPO, NPe6 and SnET2
As the PDT light dose was increased (using CPO as the sensitizer) a progressively greater loss of Bcl-2 was observed (Figure 3). Bcl-2 photodamage, at each light dose tested, was promoted by an extracellular UDCA concentration of 100 μM. When cells were incubated with 2 μM CPO, irradiated (180 mJ/cm2) and incubated for an additional 60 min at 37°C, 32±5% of the cell population exhibited an apoptotic morphology, detected by HO342 labeling (Figure 4b). In the presence of 100 μM UDCA, this value increased to 66±4% (Figure 4c). This result was not altered when cells were loaded with UDCA (100 μM), and then washed for 10 min at 37°C before PDT (Figure 4d). The effect of UDCA on PDT efficacy is therefore not readily reversible. We also measured DEVDase activation 10 min after irradiation (Table 5). It is noteworthy that loss of 90% of the initial UDCA pool did not affect the ability of the bile acid to promote DEVDase activation after PDT. Complementary results were obtained in colony-survival assays (Table 2). A dose–response study was carried out using an LD50 PDT dose of CPO and graded UDCA levels (Table 2). UDCA-enhanced photokilling was unaffected by a subsequent 10 min wash at 37°C, regardless of the initial bile acid concentration.

Photodamage to Bcl-2 as a function of the light dose and presence of UDCA. Cells were treated with 2 μM CPO and irradiated with specified light doses (mJ/cm2). UDCA (100 μM) was present where specified. Under these conditions, a 135 mJ/cm2 light dose results in a 50–60% loss of viability, 180 mJ/cm2 in a 90–95% loss of viability

Apoptotic effects of UDCA and PDT. L1210 cells were photosensitized with CPO and irradiated (180 mJ/cm2), then incubated at 37° for 60 min and labeled with Höchst dye HO33342: (a) control cells; (b) cells treated with an LD90 PDT dose; (c) effect of 100 μM UDCA treatment before PDT as specified in the text; (d) cells treated with UDCA, then washed for 10 min at 37°C before irradiation. White bar=10 μm
Effects of UDCA on Bax insertion into the mitochondrial membrane
We previously reported that PDT conditions sufficient to cause Bcl-2 photodamage markedly promoted the migration of Bax to mitochondria. These latter studies were carried out with murine L1210 leukemia cells and the porphycene CPO.6 In the study summarized in Figure 5, we examined the effect of UDCA alone, and the ability of UDCA to alter the effects of CPO-catalyzed photodamage on the association of Bax with mitochondria. The results indicate a substantial promotion of this association when UDCA is added to a PDT protocol.

Promotion by UDCA of Bax binding to mitochondria. (a) Control, (b) UDCA control, (c) CPO+90 mJ/cm2, (d) CPO+180 mJ/cm2, (e) CPO+100 μM UDCA+90 mJ/cm2, (f) CPO+100 μM UDCA+180 mJ/cm2
Fluorescence polarization
HA14-1 was initially identified on the basis of its ability to compete with a fluorescent Bak-BH3 peptide (flu-Bak) for binding to the surface cleft on Bcl-2, as monitored by fluorescence polarization.19 A Bcl-2 preparation lacking the C-terminal membrane domain (Bcl-2Δ21) was used in the latter study. Consistent with data described in Wang et al.19, we found a significant increase in fluorescence polarization (P) of the flu-Bak peptide upon binding to Bcl-2 (Figure 6). The maximum P-value obtained was 0.2. This increase reflects the decreased mobility of the fluorescent peptide upon binding to a large molecule. Addition of HA14-1 caused a decrease in fluorescence polarization, indicating competition between HA14-1 and flu-Bak for the binding site on Bcl-2. The effect of UDCA on the competition between HA14-1 and flu-Bak for Bcl-2 binding was then examined. In the absence of UDCA, a 20 μM concentration of HA14-1 decreased flu-Bak : Bcl-2 fluorescence polarization binding by half. The corresponding HA14-1 concentration was decreased 10-fold in the presence of 50 μM UDCA (Figure 6), reflecting a selective promotion of HA14-1 binding to Bcl-2 by the bile acid.

Promotion of binding of HA14-1 to Bcl-2 by UDCA as detected by fluorescence polarization. Bcl-2Δ21 was incubated with flu-Bak along with graded levels of HA14-1, in the presence or absence of 50 μM UDCA, prior to the determination of fluorescence polarization. Data represent mean±S.D. of five determinations. ○=UDCA absent; •=UDCA present
In the absence of HA14-1, the P-value for the Bcl-2 : flu-Bak complex was unaffected by addition of 50 or 100 μM UDCA (not shown). When Bcl-2 was omitted from the system, the P-value for flu-Bak was <0.01, indicating that the freedom of rotation of the molecule is unrestricted in the absence of a protein. This value was unaffected by addition of UDCA.
Fluorescence resonance energy transfer (FRET) analysis
FRET occurs when the fluorescence emission spectrum of one fluorophore overlaps the excitation spectrum of the other, and the fluorophores are sufficiently close.22 Bcl-2 contains a sufficient number of aromatic amino acids so that it will fluoresce upon excitation at 280 nm. The resulting fluorescence emission spectrum overlaps the excitation spectrum of CPO (Figure 7a). The latter was therefore chosen as the second fluorophore in a FRET study. The hydrophobicity of CPO23 insures that there will be no CPO fluorescence unless the porphycene is bound to a hydrophobic site. In order to examine the possibility that UDCA could promote an interaction between Bcl-2 and a sensitizer in a model membrane system, we embedded CPO and full-length recombinant Bcl-2 in dioleyl phosphatidylcholine liposomes.

(a) FRET spectral overlap between CPO and Bcl-2. The Bcl-2 excitation spectrum (I) was obtained using 360 nm emission, and the emission spectrum (II) was acquired with 280 nm excitation. For CPO, excitation spectrum (III) was obtained using 670 nm emission, and the emission spectrum (IV) was obtained using 400 nm excitation. (b) Fluorescence emission spectrum of CPO embedded in dioleyl phosphatldylcholine liposomes (trace 1), after addition of 100 μM UDCA (trace 2), after addition of 900 ng/ml Bcl-2 (trace 3), after addition of UDCA+Bcl-2 (trace 4). Excitation wavelength=400 nm. (c) Analysis of FRET between CPO and Bcl-2 embedded in dioleyl phosphatidylcholine liposomes. Excitation wavelength=280 nm. Trace A solid line=CPO alone; trace A dashed line=CPO+100 μM UDCA; Trace B=CPO+900 ng/ml Bcl-2; Trace C=CPO, Bcl-2+100 μM UDCA
The effects of Bcl-2 and/or UDCA on the fluorescence emission spectrum of CPO embedded in the liposomes (excitation=400 nm) are shown in Figure 7b. The fluorescence emission intensity of CPO (trace 1) was only slightly promoted by UDCA (trace 2), Bcl-2 (trace 3) or UDCA+Bcl-2 (trace 4). When liposomes contained CPO alone, excitation at 280 nm resulted in a very slight fluorescence emission at 670 nm, attributed to the long tail of the CPO excitation spectrum into the UV (Figure 7c, trace A, solid line). The fluorescence intensity was not enhanced by addition of an equal volume of 10 mM Triton X-100 detergent, indicating that no CPO remained in an aggregated state outside of the liposomes (data not shown). The fluorescence of CPO in a liposomal environment was also unaffected by addition of 100 μM UDCA (Figure 7c, trace A, dashed line).
Addition of Bcl-2 to liposomes containing CPO resulted in a significant promotion of 670 nm fluorescence (Figure 7c, trace B) upon excitation at 280 nm. We attribute this effect to FRET involving both fluorophores. The FRET signal was further enhanced by addition of 100 μM of UDCA (Figure 7c, trace C). The latter result indicates that UDCA has altered the system so as to promote a greater interaction between liposome-bound CPO and Bcl-2.
Bcl-2 homodimerization analysis
An examination of the effect of 100 μM UDCA on Bcl-2 homodimer formation was carried out as described by Conus et al.24 Gel exclusion chromatography on Superose 12 yielded a single Bcl-2 peak that corresponded to the monomer, molecular weight=26 kDa, whether or not UDCA was present (Figure 8).

Elution pattern of marker proteins and Bcl-2 from a Superose 12 column. Proteins were detected by measurement of 280 nm absorbance except for Bcl-2, which was detected using the fluorescence probe NanoOrange, as described in the text
Discussion
Several earlier reports indicated that UDCA could protect cells in culture from the proapoptotic effects of a variety of reagents including more hydrophobic bile acids, for example, DCA.11, 12, 13, 14, 15 This protective mechanism was attributed to stabilization of the mitochondrial structure, thereby preventing loss of cytochrome c.15 Translocation of cytochrome c to the cytosol is known to trigger an apoptotic response via the Apaf-1/caspase-9 pathway.25 In contrast, our initial findings identified a different property of UDCA: promotion of apoptotic cell death after photodynamic therapy.18 A clue concerning the nature of the UDCA effect was provided by the finding that UDCA also enhanced the apoptotic response to the nonpeptidic Bcl-2 antagonist HA14-1.5 The implication is that UDCA can magnify effects of Bcl-2 inactivating procedures.
Competition studies (Table 1) showed no effect of an equimolar concentration of nonradioactive UDCA or DCA on accumulation or stability of 14C-DCA or -UDCA pools. These results indicate that L1210 cells have a high capacity for bile acid accumulation. Washout experiments revealed that 90% of the initial UDCA or DCA was rapidly lost during a subsequent wash. Such a loss did not affect the ability of UDCA to promote the apoptotic response to PDT (Figure 4, Table 2), indicating that the proapoptotic effect of UDCA is at least temporarily irreversible. Moreover, UDCA did not protect L1210 cells from the toxic effects of DCA (Table 3). This result is consistent with other reports indicating that UDCA does not always offer protection from toxic effects of more hydrophobic bile acids.16, 17
Our initial photodynamic studies with UDCA utilized SnET2, a photosensitizing agent that has a broad localization pattern (Figure 2, top). In this study, we examined two photosensitizing agents, NPe6 and CPO, with more selective targets. UDCA did not promote the phototoxic effect of NPe6 (Table 4), a sensitizer that localizes to lysosomes (Figure 2, top). Upon irradiation of NPe6-sensitized cells, the ensuing lysosomal photodamage ultimately leads to Bid cleavage, activation of procaspases-9 and -3, and an apoptotic outcome.26, 27 This pathway does not directly involve Bcl-2, and no Bcl-2 photodamage was observed after NPe6 photodamage (Figure 2, bottom). In contrast, the photosensitizer CPO, an agent that localizes in subcellular membranes (Figure 2), catalyzed substantial Bcl-2 photodamage (Figure 2, bottom). Both the phototoxicity of CPO and its ability to cause Bcl-2 photodamage were enhanced by UDCA (Figure 3, Table 4). These results indicate that the ability of UDCA to enhance the apoptotic response to SnET2 also derives from Bcl-2 photodamage.
In the presence of UDCA, Bcl-2 photodamage was enhanced (Figure 3). Even at an LD90 PDT dose, the Western blot indicated that some Bcl-2 remained undamaged. In this regard, Usuda et al.28 have shown that a minor degree of photodamage to Bcl-2 can interfere with its anti-apoptotic function.
Results shown in Figure 6 suggest an explanation for the ability of UDCA to promote HA14-1-induced apoptosis described in Kessel et al.5 Fluorescence polarization studies show that UDCA increases the affinity of HA14-1 for the surface cleft in the Bcl-2 protein, as indicated by an enhanced ability to displace flu-Bak from this site, resulting in a decrease in P. This effect can be explained if we postulate that UDCA alters the conformation of the Bcl-2 molecule so as to promote HA14-1 binding. Furthermore, this result demonstrates that UDCA preferentially promotes HA14-1 binding to Bcl-2. If binding of flu-Bak were similarly enhanced, we would observe no difference in flu-Bak fluorescence polarization in the presence of UDCA. A conformational change in Bcl-2, upon exposure to UDCA, can also explain enhanced Bcl-2 photodamage (see below).
We previously reported the effects of graded levels of UDCA on HA14-1 dose-dependent cytotoxicity and DEVDase activation.5 In the latter study, only extracellular levels of UDCA were reported. Data obtained in the current study using radioactive UDCA show that the pool of UDCA stable to washing is ∼10–15% of the initial intracellular level (Table 1). Nevertheless, this pool was sufficient for enhanced DEVDase activation (Table 5) and photokilling (Table 2). It is not clear whether this represents an irreversible consequence of an initially high concentration of intracellular UDCA, or the effect of a persisting stable pool of the bile acid.
Data shown in Figure 2 indicate that Bcl-xL photodamage does not occur with either CPO or SnET2. Oleinick had initially reported a similar result4 using a different photosensitizer, but later found that with a different antibody Bcl-xL photodamage was observed.29 It is possible that Bcl-xL is located in L1210 cells at sites where binding of CPO and SnET2 is minimal. A study involving the photosensitizing agent protoporphyrin, derived from 5-aminolevulinic acid administration, indicated that Bcl-2 was more sensitive to photodamage than Bcl-xL.30
With regard to the determinants of Bcl-2 photodamage, it is clear from the work of Usuda et al.7 that only proteins in very close proximity to sites of photosensitizer localization will be targets for photodamage. Studies with the photosensitizer Pc 4 demonstrated that a mutant Bcl-2 lacking the transmembrane domain, and therefore confined to the cytosol, was protected from photodamage.7 We therefore propose that the enhanced Bcl-2 photodamage that occurs in the presence of UDCA reflects a conformational change in Bcl-2 that enhances its proximity to membrane-bound photosensitizer. We sought proof for this hypothesis by designing a liposomal system to examine fluorescence resonance energy transfer between embedded Bcl-2 and an appropriate fluorophore.
In the current studies the ideal acceptor fluorophore for FRET studies should have an excitation spectrum that overlaps the fluorescence emission of Bcl-2. The emission spectrum should also show a significant Stokes shift, so that the probe emission will be well separated from the excitation wavelength. The spectral properties of CPO fit these requirements (Figure 7a). While CPO is a photosensitizing agent, the light intensity used in FRET studies is several orders of magnitude below the level needed to cause photodamage.
When liposomes containing CPO alone were excited at 280 nm, there was a low level of fluorescence emission at 670 nm, reflecting the tail of the excitation curve of CPO (Figure 7c). Addition of Triton X-100 or UDCA did not alter the intensity of fluorescence indicating that there was no free CPO in the system, and that UDCA did not affect affinity of CPO for liposomes. Addition of Bcl-2 resulted in a substantial promotion of 670 nm fluorescence, and this was further enhanced by addition of 100 μM UDCA. These results indicate that Bcl-2 binds sufficiently close to liposomal sites of CPO so that FRET can take place, and that addition of UDCA results in yet closer contact between fluorogenic amino acids in the protein and liposome-bound CPO.
Hanada et al.31 reported homodimerization of Bcl-2, but Conus et al.24 were unable to confirm this observation and attributed the earlier observation to the use of a disrupted Bcl-2 structure. Xie et al.32 observed homodimers of a related protein, Bcl-xL, but this required the presence of 0.1% Tween 20. Using the gel exclusion chromatography system described by Conus et al., we found no evidence for the presence of Bcl-2 dimers under our experimental conditions, whether or not 100 μM UDCA was present. Moreover, we found no effects of UDCA on the fluorescence polarization of the flu-Bak peptide when bound to Bcl-2.
Steer's33 group recently reported that UDCA can prevent insertion of recombinant Bax into isolated rat liver mitochondria, thereby providing a mechanism for protection from apoptosis by UDCA. In studies to be reported elsewhere, we could not duplicate this effect with L1210 cells and recombinant Bax, unless the Bax was first ‘activated’,34 that is, by exposure to octyl glucoside. The ‘protective’ effect of UDCA on mitochondria must either be unique to a cell-free system, or a relatively minor effect, compared with the proapoptotic effects of UDCA in cell lines examined here and described in Kessel et al.18 The fluorescence polarization results are consistent with the hypothesis that UDCA initiates a conformational change that promotes binding of HA14-1 to Bcl-2. We propose that this change also leads to an increased sensitivity of Bcl-2 to photodamage by promoting proximity to membrane-bound CPO.
It is noteworthy that cell viability was not affected by 100 μM UDCA alone (Table 3), indicating that the postulated UDCA-induced change in Bcl-2 conformation has no long-term consequences unless a proapoptotic stimulus, for example, HA14-1 or Bcl-2 photodamage, is present. The proposed mechanism of action for UDCA predicts that this bile acid will promote PDT efficacy in vivo without adverse effects on the host. The results of a recent study involving a transplantable mouse tumor35 have confirmed this supposition.
Materials and Methods
Drugs and chemicals
The porphycene CPO6, 23 was obtained from Dr. Alex Cross, CytoPharm, San Francisco CA, USA. Both SnET236 and the water-soluble chlorin NPe637 were provided by Drs. Kevin Smith and Graça Vicente, Department of Chemistry, Louisiana State University in Baton Rouge. CPO and SnET2 were dissolved in DMSO to obtain 1 mM stock solutions. Stock solutions (200 mM) of UDCA and DCA (Sigma Chemical Co., St. Louis, MO, USA) were prepared in 200 mM NaOH.
Radioactive DCA and UDCA
The procedures outlined in Matern et al.38 and Tserng and Klein39 were followed, with minor modifications, to prepare UDCA and DCA labeled with 14C in the terminal COOH (Figure 1). UDCA or DCA (5 g) were converted to the corresponding formyl derivatives by treatment with 20 ml of 90% formic acid+0.5 ml of 70% HClO4. After heating to 55°C for 1.5 h, the solution was cooled to 40°C and acetic anhydride added dropwise, maintaining a temperature of 50–55°C, until a substantial evolution of CO2 was observed. The resulting mixture was allowed to cool and then poured into 200 ml of cold water. The formylated product was recovered by filtration, washed with water until the odor of acetic anhydride could no longer be detected, and dried over P2O5 in a vacuum desiccator.
The formylated bile acid (1 gm) was dissolved in 35 ml of benzene and dried azeotropically. Since Kochi40 reported that traces of water did not interfere with the subsequent lead tetraacetate oxidation, azeotropic distillation was limited to 30 min. The solution was then cooled to room temperature and 2.2 g of lead tetraacetate+200 mg of anhydrous LiCl added with constant stirring. The solution was immediately degassed with nitrogen (critical for a good yield), and heated under reflux for 6 h. During this time, additional 200 mg portions of LiCl were added at 1 h intervals. The solution was then stirred for 16 h at room temperature, filtered, and the filtrate was washed three times with 10 ml of 2% NaOH, four times with water and dried over anhydrous MgSO4. The product was evaporated to dryness in vacuum, taken up in 10 ml of absolute ethanol, evaporated under vacuum until crystals began to form, and then stored at −20° overnight. The yield of the chloro-diformylnorcholanes was 60–65%.
Conversion of the norcholanes to 14C-UDCA and -DCA was carried out by Moravec Biochemicals Inc., Brea, CA. USA. This involved heating a stirred suspension of 50 mg of the bile acid+1 mCi of Na14CN (500 mg) in 5 ml of dimethylformamide at 110°C for 5.5 h. After cooling to room temperature, 20 ml of water was added and the precipitated nitrile was removed by centrifugation. The product was dissolved in 5 ml of hot ethanol, and a solution of 630 mg of NaOH in 5 ml of water was added. The mixture was heated under reflux for 40 h, cooled and washed with ether. Residual ether was removed by brief heating, and the filtrate acidified with 1 M HCl to precipitate the 14C bile acid. After washing with water, the product was recrystallized from 2-propanol. Specific activities of 50–55 μCi/mmol were obtained. At each step in the reaction process, we confirmed that NMR and mass spectroscopy data (kindly performed by Professor M Graça Vicente, Department of Chemistry, Louisiana State University) were consistent with the expected products.
Cells and cell culture
Murine L1210 cells were maintained in culture using Fischer's growth medium+10% horse serum, 1 mM glutathione, 1 mM mercaptoethanol and gentamicin. Since Fischer's medium is no longer available, we supplemented α-MEM (GIBCO-BRL, Grand Island, NY, USA) to achieve the original Fischer's formulation. This involved addition of MgCl2 (45 mg/l), methionine (75 mg/l), phenylalanine (30 mg/l), valine (30 mg/l) and folic acid (9 mg/l).
Intracellular accumulation of bile acids
L1210 cells were incubated in growth medium containing 100 μM radioactive DCA or UDCA for 1–30 min at 37°C, and then washed with cold isotonic NaCl. Radioactivity was assessed by solubilizing cell pellets and determining radioactivity by scintillation counting. In some studies, nonradioactive DCA or UDCA was added 10 min before the radioactive substrates. In another series of studies, the cells were incubated with radioactive bile acids, and then washed in fresh medium for 10–30 min at 37°C to determine the stability of bile acid pools.
PDT protocols
Suspensions of L1210 cells (7 mg/ml wet weight=2 × 106 cells) were incubated in growth medium containing 2 μM CPO or SnET2 for 15 min at 37°C, or with 60 μM NPe6 for 4 h. The cells were subsequently washed and resuspended in fresh growth medium at room temperature. Irradiation was provided by a 600 W quartz-halogen source filtered with 10 cm of water and an 800 nm cutoff filter to remove IR. Bandwidth was further confined to 660±10 nm (SnET2, NPe6) or 610±10 nm (CPO) by narrow-band interference filters (Oriel, Stratford, CT, USA). The total light dose is specified for each experiment. Initial experiments indicated that a 90% loss of L1210 cell viability was produced by irradiation with 180 mJ/cm2 with CPO, 350 mJ/cm2 with NPe6 and 135 mJ/cm2 with SnET2. Different light doses were used in these experiments, as indicated above.
In some studies, cells were loaded with 100 μM UDCA and/or photosensitizer for 30 min prior to irradiation to assess the proapoptotic effects of the combination. To assess the reversibility of the UDCA effect, cells were incubated with the bile acid, then washed for 10 min at 37°C before loading with CPO and subsequent irradiation. Intracellular levels of UDCA were measured before and after washing over a range of drug levels (3–100 μM). Effects of washing on the subsequent ability of an LD50 PDT dose with CPO were also determined.
DEVDase assay
Cells were collected at varying times after PDT, washed and lysed in 200 μl of buffer containing 50 mM Tris pH 7.2, 0.03% Nonidet P-40 and 1 mM DTT. The lysate was briefly sonicated and the debris removed by centrifugation at 10 000 × g for 1 min. The supernatant fluid (100 μl) was mixed with 40 μM DEVD-R110, 10 mM HEPES pH 7.5, 50 mM NaCl and 2.5 mM DTT in a total volume of 200 μl. The rate of increase in fluorescence emission, resulting from the release of rhodamine-110 from the fluorogenic substrate,5, 6 was measured over 30 min at room temperature, using a fluorescence plate reader.
DEVDase activity is reported in terms of nmol product/min/mg protein. Control determinations were made on extracts of untreated cells. Each assay was performed with triplicate samples. The BioRad assay, using BSA as a standard, was used to estimate protein concentrations.
Western blots
L1210 cells were processed as described previously for Western blot analysis of Bax, Bcl-2, Bax or Bcl-xL levels.5 Equal protein amounts were loaded into each lane of the gels as determined by the BioRad procedure.
Viability studies
Cells were incubated for 60 min at 37°C in growth medium containing 100 μM DCA or UDCA. In attempts to reverse DCA toxicity, cells were incubated with 100 μM DCA+3–100 μM UDCA. The cells were then washed and used for a clonogenic growth assay.
Effects of UDCA on Bax association with mitochondria
L1210 cells were loaded with 2 μM CPO in the presence or absence of 100 μM UDCA for 30 min at 37°C. Cells were then washed, irradiated for varying lengths of time and then incubated at 37°C for an additional 30 min. A nitrogen cavitation technique41 was used to rupture the cell membrane, and mitochondria were collected at 1000 × g. Mitochondria were resuspended in lysis buffer and frozen. Proteins were separated on a 12% SDS-PAGE gel. After electrophoresis, the separated proteins were transferred to a PVDF membrane, blocked and incubated with a Bax antibody (PharMingen, product No. 554106). The membrane was subsequently incubated with an appropriate secondary antibody, and developed with an ECF Western blotting kit (Amersham Pharmacia) for analysis by fluorescence.
Fluorescence microscopy
Localization of SnET2, CPO and NPe6 in L1210 cells was determined after incubations with the drug levels described in the PDT protocol. A Nikon E600 series microscope was employed using 360–450 nm excitation, with fluorescence emission measured at 600–700 nm. For the detection of apoptotic nuclei, cells were loaded with drugs, washed and irradiated as described above, then diluted to a density of 4 × 105/ml and incubated for 60 min at 37°C. Thereafter, cells were collected and labeled with HO342 (2 μg/ml). A more detailed description is provided in Kessel et al.5 After 5 min at 37°C, the cells were collected by centrifugation and the percentage of apoptotic nuclei were determined by fluorescence microscopy (three fields of 100 cells each). HO342 fluorescence (420–450 nm) was detected upon excitation at 330–380 nm. A SenSys CCD camera (Photometrics) was used for image acquisition, and the resulting data were processed using MetaMorph software (Universal Imaging, Downingtown, PA, USA).
Fluorescence polarization
The binding affinity of organic compounds to Bcl-2 protein in vitro was determined by a competitive binding assay based on fluorescence polarization. The substrate was 5-carboxyfluorecein coupled to the N terminus of a peptide, GQVGRQLAIIGDDINR, derived from the BH3 domain of Bak (flu-Bak). This sequence has been shown to bind to the surface pocket of the Bcl-xL protein with high-affinity.19 The Bcl-2 used in studies reported in Wang et al.19 was a recombinant GST-fused soluble protein lacking the C-terminal membrane-traversing region (Bcl-2Δ21, Santa Cruz Biotechnology). We used a similar product provided by R&D Systems Inc, Minneapolis, MN, USA.
A mixture of 150 nM flu-Bak peptide (Peptidogenic Research, Livermore, CA, USA) and 280 nM Bcl-2 protein, containing HA14-1 and/or UDCA where specified, was incubated for 30 min at room temperature in 20 mM phosphate buffer pH 7.4 containing 50 mM NaCl and 1 mM EDTA. Binding of flu-Bak to Bcl-2 protein was measured with an LS-50 luminescence spectrometer equipped with polarizers, using a dual path length quartz cell (Perkin-Elmer). The fluorophore was excited with vertical polarized light at 480 nm (excitation slit width 15 nm), and the polarization value of the emitted light was observed through vertical and horizontal polarizers at 530 nm (emission slit width 15 nm). The binding affinity of HA14-1 for Bcl-2 was assessed by determining the ability of graded concentrations of the compound to inhibit flu-Bak binding to Bcl-2. Additional controls were carried out using fluorescein and flu-Bak in the presence versus absence of UDCA to assess the polarization of fluorescence in the absence of Bcl-2.
FRET analysis
These studies were carried out with full-length recombinant Bcl-2 generated by C&P Biotech Corp., Thornhill, Ontario, using cDNA provided by Dr. Stan Korsmeyer, Harvard Medical School. Dioleyl phosphatidylcholine liposomes were prepared as described in Schendel and Reed42, using a buffer composed of 125 mM sucrose+10 mM HEPES, pH 7.4. After evaporation of the solvent, the phospholipid residue was taken up in buffer, degassed with nitrogen and sonicated in a G112SP1 ultrasonic bath (Laboratory Supplies Inc., Hicksville, NY, USA) under nitrogen, until the solution was clear. An SLM 48000 instrument was used for acquisition of fluorescence spectra. FRET analysis was carried out by excitation at 280 nm, and the fluorescence emission spectra (625–675 nm) was recorded. Liposomes were incubated with 1 μM CPO for 10 min at room temperature at which time the fluorescence intensity of CPO emission (670 nm) had reached a steady state (excitation=400 nm). Additional studies were carried out using Bcl-2 (900 ng/ml) or CPO embedded in the liposomal preparation. In all such studies, the liposomal mixtures were held at room temperature for 10 min at which time a steady state was reached. Effects of Bcl-2 and/or UDCA on the fluorescence emission spectrum of CPO were determined using 400 nm excitation.
Effects of UDCA on Bcl-2 homodimerization
Bcl-2 dimerization was assessed by gel exclusion chromatography using a 0.8 × 25 cm2 Superose 12 column as described by Conus et al.24 The column was calibrated with a series of standards, which were detected by monitoring absorbance at 280 nm. Bcl-2 protein was detected using the NanoOrange reagent marketed by Molecular Probes, Eugene, OR, USA. Eluted fractions were mixed with NanoOrange and then heated to 90°C for 10 min, to denature proteins. Fluorescence was then read using a CCD camera and multichannel analyzer. Upon excitation at 470 nm, a fluorescence peak at ∼570 nm was recorded. To test the effect of UDCA on the elution profile, the column was equilibrated in a buffer containing 100 μM UDCA prior to the addition of a Bcl-2 preparation that also contained UDCA. The bile acid had no effect on the fluorescence emission intensity of NanoOrange.
Abbreviations
- CPO:
-
9-capronyloxy-tetrakis(methoxyethyl) porphycene
- DCA:
-
deoxycholic acid
- DEVD-R110:
-
asp–glu–val–asp–rhodamine 110 (fluorogenic caspase-3 substrate)
- ER:
-
endoplasmic reticulum
- flu-Bak:
-
5-carboxyfluorecein coupled to the N terminus of a peptide GQVGRQLAIIGDDINR derived from the BH3 domain of Bak
- HA14-1:
-
ethyl 2-amino-6-bromo-4-(1-cyano-2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate
- HO342:
-
Höchst dye HO33342
- mTHPC:
-
meta-(tetrahydroxyphenyl) chlorin
- NPe6:
-
N-aspartyl chlorin e6
- P :
-
fluorescence polarization value
- PDT:
-
photodynamic therapy
- SnET2:
-
tin etiopurpurin
- UDCA:
-
ursodeoxycholic acid.
References
Dougherty TJ, Gomer CJ, Henderson BW, Jori G, Kessel D, Korbelik M, Moan J and Peng Q (1998) Photodynamic therapy. J. Natl. Cancer Inst. 90: 889–905
Kim HR, Luo Y, Li G and Kessel D (1999) Enhanced apoptotic response to photodynamic therapy after Bcl-2 transfection. Cancer Res. 59: 3429–3432
Kessel D, Luo Y, Deng Y and Chang CK (1997) The role of subcellular localization in initiation of apoptosis by photodynamic therapy. Photochem. Photobiol. 65: 422–426
Xue LY, Chiu SM and Oleinick NL (2001) Photochemical destruction of the Bcl-2 oncoprotein during photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 20: 3420–3427
Kessel D, Castelli M and Reiners Jr JJ (2002) Apoptotic response to photodynamic therapy versus the Bcl-2 antagonist HA14-1. Photochem. Photobiol. 76: 314–319
Kessel D and Castelli M (2001) Evidence that Bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem. Photobiol. 74: 318–322
Usuda J, Chiu SM, Murphy ES, Lam M, Nieminen A and Oleinick NL (2003) Domain-dependent photodamage to Bcl-2: a membrane-anchorage region is needed to form the target of phthalocyanine photosensitization. J. Biol. Chem. 278: 2021–2029
Ljubuncie P, Fuhrman B, Oiknine J, Aviram M and Bomzon A (1996) Effect of deoxycholic acid and ursodeoxycholic acid on lipid peroxidation in cultured macrophages. Gut 39: 475–478
Mitsuyoshi H, Nakashima T, Sumida Y, Yoh T, Nakajima Y, Ishikawa H, Inaba K, Sakamoto Y, Okanoue T and Kashima K (1999) Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem. Biophys. Res. Commun. 263: 537–542
Nguyen TD, Oliva L, Villard PH, Puyoou F, Sauze C, Montet AM, Lacarelle B, Durand A and Montet JC (1999) CYP2E1 and CYP3A1/2 gene expression is not associated with the ursodeoxycholate effect on ethanol-induced lipoperoxidation. Life Sci. 65: 1103–1113
Rodrigues CM, Fan G, Wong PY, Kren BT and Steer CJ (1998) Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrane potential and reactive oxygen species production. Mol. Med. 4: 165–178
Rodrigues CM, Ma X, Linehan-Stieers C, Fan G, Kren BT and Steer CJ (1999) Ursodeoxycholic acid prevents cytochrome c release in apoptosis by inhibiting mitochondrial membrane depolarization and channel formation. Cell Death Differ. 6: 842–854
Rodrigues CM, Fan G, Ma X, Kren BT and Steer CJ (1998) A novel role for ursodeoxycholic acid in inhibiting apoptosis by modulating mitochondrial membrane perturbation. J. Clin. Invest. 101: 2790–2799
Rodrigues CM and Steer CJ (2001) The therapeutic effects of ursodeoxycholic acid as an anti-apoptotic agent. Expert Opin. Invest. Drugs 10: 1243–1253
Botla R, Spivey JR, Aguilar H, Bronk SF and Gores GJ (1995) Ursodeoxycholate (UDCA) inhibits the mitochondrial membrane permeability transition induced by glycochenodeoxycholate: a mechanism of UDCA cytoprotection. J. Pharmacol. Exp. Ther. 272: 930–938
Rolo AP, Palmeira CM and Wallace KB (2002) Interactions of combined bile acids on hepatocyte viability: cytoprotection or synergism. Toxicol. Lett. 126: 197–203
Schlottman K, Wachs FP, Krieg RC, Kullmann F, Scholmerich J and Rogler G (2000) Characterization of bile salt-induced apoptosis in colon cancer cell lines. Cancer Res. 60: 4270–4276
Kessel D, Caruso JA and Reiners Jr JJ (2000) Potentiation of photodynamic therapy by ursodeoxycholic acid. Cancer Res. 60: 6985–6988
Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES and Huang Z (2000) Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. USA 97: 7124–7129
Liu D and Huang Z (2001) Synthetic peptides and non-peptidic molecules as probes of structure and function of Bcl-2 family proteins and modulators of apoptosis. Apoptosis 6: 453–462
Wilton JC, Matthews GM, Burgoyne RD, Mills CO, Chipman JK and Coleman R (1994) Fluorescent choleretic and cholestatic bile salts take different paths across the hepatocyte:transcytosis of glycolithocholate leads to an extensive redistribution of annexin II. J. Cell Biol. 127: 401–410
Heyduk T (2002) Measuring protein conformational changes by FRET/LRET. Curr. Opin. Biotechnol. 3: 292–296
Toledano H, Edrei R and Kimel S (1998) Photodynamic damage by liposome-bound porphycenes: comparison between in vitro and in vivo models. J. Photochem. Photobiol. B 42: 20–27
Conus S, Kaufmann T, Fellay I, Otter I, Rosse T and Borner C (2000) Bcl-2 is a monomeric protein: prevention of homodimerization by structural constraints. EMBO J. 19: 1534–1544
Budihardjo I, Oliver H, Lutter M, Luo X and Wang X (1999) Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15: 269–290
Kessel D, Luo Y, Mathieu P and Reiners Jr JJ (2000) Determinants of the apoptotic response to lysosomal photodamage. Photochem. Photobiol. 71: 196–200
Reiners Jr JJ, Caruso JA, Mathieu P, Chelladurai B, Yin XM and Kessel D (2002) Release of cytochrome c and activation of pro-caspase-9 following lysosomal photodamage involves Bid cleavage. Cell Death Differ. 9: 934–944
Usuda J, Xue L-Y, Chio S-M, Azizuddin K, Morris RL, Mulvihill J and Oleinick NO (2003) From molecular PDT damage to cell and tumor responses: attempts at bridging the gap on the role of Bcl-2. Proc. SPIE 4952: 1–9
Xue L, Chiu S-M, Fiebig A, Andrews DW and Oleinick NO (2003) Photodamage to multiple Bcl-xL isoforms by photodynamic therapy with the phthalocyanine photosensitizer Pc 4. Oncogene 22: 9197–9204
Grebenova D, Kuzelova K, Smetana K, Pluskalova M, Cajthamlova H, Marinov I, Fuchs O, Soucek J, Jarolim P and Hrkal Z (2003) Mitochondrial and endoplasmic reticulum stress-induced apoptotic pathways are activated by 5-aminolevulinic acid-based photodynamic therapy in HL60 leukemia cells. J. Photochem. Photobiol. B 69: 71–85
Hanada M, Aime-Sempe C, Sato T and Reed JC (1995) Structure-function analysis of Bcl-2 protein. Identification of conserved domains important for homodimerization with Bcl-2 and heterodimerization with Bax. J. Biol. Chem. 270: 11962–11969
Xie Z, Schendel S, Matsuyama S and Reed JC (1998) Acidic pH promotes dimerization of Bcl-2 family proteins. Biochemistry 37: 6410–6418
Rodrigues CMP, Sola S, Sharpe JC, Moura JJG and Steer CJ (2003) Tauroursodeoxycholic acid prevents Bax-induced membrane perturbation and cytochrome c release in isolated mitochondria. Biochemistry 42: 3070–3080
Hsu YT and Youle RJ (1998) Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J. Biol. Chem. 273: 10777–10783
Garbo GM, Vicente MGH, Fingar V and Kessel D (2003) Effects of ursodeoxycholic acid on photodynamic therapy in a murine tumor model. Photochem. Photobiol. 78: 407–410
Morgan AR, Garbo GM, Keck RW and Selman SH (1988) New photosensitizers for photodynamic therapy: combined effect of metallopurpurin derivatives and light on transplantable bladder tumors. Cancer Res. 48: 194–198
Roberts WG, Shiau FY, Nelson JS, Smith KM and Berns MW (1988) In vitro characterization of monoaspartyl chlorin e6 and diaspartyl chlorin e6 for photodynamic therapy. J. Natl. Cancer Inst. 80: 330–336
Matern S, Marschall HU, Schill A, Schumacher B, Lehnert W, Sjovall J and Matern H (1991) Synthesis of 13C-labeled chenodeoxycholic, hyodeoxycholic, and ursodeoxycholic acids for the study of bile acid metabolism in liver disease. Clin. Chim. Acta 203: 77–89
Tserng KY and Klein PD (1977) An improved synthesis of 24-13C-labeled bile acids using formyl esters and a modified lead tetraacetate procedure. J. Lipid Res. 18: 400–403
Kochi J (1965) Formation of alkyl halides from acids by decarboxylation with lead (IV) acetate and halide salts. J. Org. Chem. 30: 3265–3271
Lee ST, Hoeflich KP, Wasfy GW, Woodgett JR, Leber B, Andrews DW, Hedley DW and Penn LZ (1999) Bcl-2 targeted to the endoplasmic reticulum can inhibit apoptosis induced by Myc but not etoposide in Rat-1 fibroblasts. Oncogene 18: 3520–3528
Schendel SL and Reed JC (2000) Measuring pore formation by Bcl-2 family proteins. Methods Enzymol. 322: 274–282
Acknowledgements
We thank Ann Marie Santiago and Brendan Leeson for excellent technical assistance. This study was supported by grants CA 92618, CA 23378 and ES009392 from the NIH and utilized the services of the Protein Interactions and Proteomics Facility Core, which is funded by P30 ES006639 from NIEHS.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Nunez
Rights and permissions
About this article
Cite this article
Castelli, M., Reiners, J. & Kessel, D. A mechanism for the proapoptotic activity of ursodeoxycholic acid: effects on Bcl-2 conformation. Cell Death Differ 11, 906–914 (2004). https://doi.org/10.1038/sj.cdd.4401433
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401433
Keywords
This article is cited by
-
CYD0281, a Bcl-2 BH4 domain antagonist, inhibits tumor angiogenesis and breast cancer tumor growth
BMC Cancer (2023)
-
Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function
Scientific Reports (2016)
-
Bcl2 inhibition of mitochondrial DNA repair
BMC Cancer (2015)
-
An Anionic Porphyrin Binds β-Lactoglobulin A at a Superficial Site Rich in Lysine Residues
The Protein Journal (2009)
-
Ruthenium red-mediated suppression of Bcl-2 loss and Ca2+ release initiated by photodamage to the endoplasmic reticulum: scavenging of reactive oxygen species
Cell Death & Differentiation (2005)
