
Abstract
NF-κB/Rel transcription factors have recently emerged as crucial regulators of cell survival. Activation of NF-κB antagonizes programmed cell death (PCD) induced by tumor necrosis factor-receptors (TNF-Rs) and several other triggers. This prosurvival activity of NF-κB participates in a wide range of biological processes, including immunity, lymphopoiesis and development. It is also crucial for pathogenesis of various cancers, chronic inflammation and certain hereditary disorders. This participation of NF-κB in survival signaling often involves an antagonism of PCD triggered by TNF-R-family receptors, and is mediated through a suppression of the formation of reactive oxygen species (ROS) and a control of sustained activation of the Jun-N-terminal kinase (JNK) cascade. Effectors of this antagonistic activity of NF-κB on this ROS/JNK pathway have been recently identified. Indeed, further delineating the mechanisms by which NF-κB promotes cell survival might hold the key to developing new highly effective therapies for treatment of widespread human diseases.
Similar content being viewed by others

Introduction
In the years immediately following its discovery in 1986,1 few could have predicted what a treasure trove for pharmacological therapies the NF-κB pathway would later turn out to be. NF-κB-targeting compounds have now become standard tools in immunosuppressive therapy and are part of the therapeutic regimen of various chronic inflammatory conditions and malignancies.2, 3, 4, 5 They are even being considered for use in metabolic and cardiovascular disorders such as type-II diabetes, atherosclerosis and myocardial infarction.6, 7, 8, 9 The widespread use of these agents is a reflection of the central role NF-κB plays in physiological processes such as immunity, inflammation, development and cell survival,3, 10, 11, 12, 13, 14 and – when deregulated – in human diseases.3, 4, 5, 6, 7, 8, 9, 15 From a therapeutic perspective, a major drawback of the involvement of NF-κB in such a wide range of biological activities is that its blockade, particularly over extended periods of time – as in the case of chronic illnesses – can cause serious adverse effects, among them immunosuppressive effects.2 Thus, an important therapeutic goal is to achieve selective inhibition of specific functions of NF-κB, such as prosurvival functions, rather than of NF-κB itself. Recent advances in understanding how NF-κB controls programmed cell death (PCD) now offer an opportunity for achieving this goal. Especially promising in this regard is the recent discovery that the NF-κB-mediated antagonism of PCD triggered by the so-called death receptors (DRs) involves suppression of the c-Jun-N-terminal (JNK) mitogen-activated protein kinase (MAPK) cascade and the activity of reactive oxygen species (ROS).16, 17 This suppression appears to be mediated by a specialized subset of NF-κB targets,17 thereby presenting the opportunity for selective inhibition of the prosurvival action of NF-κB, without compromising the capacity of NF-κB to serve in immunity, inflammation and tissue development. Further, because NF-κB, JNK and ROS activities are likely to be predominantly upregulated in diseased tissues, such as cancerous and inflamed tissues, therapeutic strategies aimed at targeting the antagonistic interplay between the NF-κB pathway and ROS and JNK signaling are likely to have inherent specificity for these tissues, compared to healthy ones.
The different aspects of the regulation and functions of NF-κB-family transcription factors and their role in the control of PCD are the subject of several other articles contained in this special issue of CDD and of excellent reviews published elsewhere.3, 10, 11, 12, 13, 14, 18, 19 Here, we focus on recent discoveries that have unveiled how NF-κB, stimulated by tumor necrosis factor (TNF)α, exerts a control on PCD by engaging in a negative regulation of the JNK pathway. We then go on to discuss the relevance of this negative regulation to health and disease.
The NF-κB Pathway
In vertebrates, NF-κB/Rel transcription factors stand out as master regulators of innate and adaptive immunity, inflammatory responses, organ development and cell survival.3, 10, 11, 12, 13, 14, 16, 17, 19 Similar functions have also been described in invertebrates.20 In mammals, the NF-κB family of polypeptides consists of NF-κB1 (p50/p105), NF-κB2 (p52/p100), RelA (p65), Rel (c-Rel) and RelB, and is characterized by the so-called Rel homology domain (RHD), which mediates DNA binding, dimerization and association with inhibitory proteins of the IκB group.11, 12, 14 Normally, NF-κB dimers are retained in the cytoplasm in an inactive state through binding to IκBs. A wide spectrum of stimuli are capable of triggering rapid activation of NF-κB from these cytoplasmic pools by inducing the sequential phosphorylation and proteolytic degradation of these inhibitors – actions usually dependent on the IκB kinase (IKK) complex (consisting of its three subunits, IKKα, IKKβ, and IKKγ/NEMO) and the ubiquitin/proteasome pathway, respectively.11, 12, 18 Upon being freed from the inhibitors, NF-κB dimers enter the nucleus where they activate expression of distinct sets of target genes encoding numerous cytokines, chemokines, adhesion molecules, growth factors, immune receptors and prosurvival proteins.3, 11, 12 Through the induction of these genes, NF-κB marshals and coordinates innate and adaptive immunity, inflammation, cell differentiation and cell survival.
Two main pathways for NF-κB activation have been described, both of which are capable of antagonizing PCD:11, 16 the canonical pathway, triggered in response to microbial products, stress and proinflammatory cytokines, depends on the activity of IKKβ and mainly results in the nuclear translocation of NF-κB1/RelA and NF-κB1/Rel dimers. The alternative pathway, activated by certain members of the TNF receptor (TNF-R) family such as lymphotoxin (LT)β and BAFF receptors, depends instead on IKKα and causes activation of NF-κB2/RelB complexes by inducing the proteolytic processing of the NF-κB2/p100 precursor.11, 12, 13 Under most circumstances, suppression of PCD is seemingly due to activation of the canonical pathway.3, 11, 12, 16
The prosurvival role of NF-κB in animal physiology
The prosurvival function of NF-κB has been implicated in a wide range of biological processes. In the B-cell lineage, NF-κB is constitutively activated by the triggering of TNF-R-family receptors, TACI, BCMA and BAFF-R,21 and this constitutive NF-κB activity is needed for differentiation and maintenance of mature IgMlow/IgDhigh B lymphocytes.11, 12, 13 NF-κB also plays an important protective role in the development of thymocytes,13, 22 and is required in the periphery for the productive responses of mature B and T lymphocytes to exposure to antigen and the costimulatory molecules CD40 ligand (CD40L) and B7-1, respectively.10, 11, 13, 19 Moreover, the canonical and alternative pathways of NF-κB activation antagonize PCD signaling triggered downstream of numerous other receptors found on the surface of cells of the immune system, including Toll-like receptors (TLRs) and other members of the TNF-R family such as DRs (see below).11, 12
The NF-κB-mediated control of PCD is also crucial to the physiology of organs outside the immune system.3, 16 The first direct evidence that NF-κB inhibited PCD was provided by the observation that knockout ablation of RelA in mice causes embryonic lethality due to massive apoptosis in the liver.23 Similar phenotypes were later described in IKKβ−/− and IKKγ−/− animals.3, 24, 25 Notably, both liver damage and embryonic lethality in mutant mice were found to be reversed by compound mutation of TNF-R1.26, 27 The hepatoprotective activity of NF-κB was recently confirmed in adult animals with the demonstration that this activity is essential for antagonism of TNF-R-mediated damage also in these animals (further discussed below).16, 28, 29 Another paradigmatic example of a protective function of NF-κB in the context of a biological response to stimulation of a TNF-R-family member is provided by RANK, a receptor required for survival and maturation of osteoclasts – the cells that resorpt bone.30, 31, 32 The protective action of NF-κB has been implicated in several other processes, including epidermal homeostasis, hair follicle development, and development and function of the central nervous system.3, 33, 34
NF-κB-Mediated Inhibition of PCD in Human Disease
Roles in tumorigenesis and cancer cell survival
In addition to playing these important physiological roles, when deregulated, the ability of NF-κB to control PCD can contribute to human disease. This pathogenetic role of NF-κB has arguably been best characterized in cancer. Genes encoding NF-κB/IκB-family members are frequently amplified, rearranged or mutated in human cancers, and most viral and cellular oncogene products, including HTLV-I's Tax, EBV's EBNA2 and LMP-1, Bcr-Abl, Her-2/Neu and oncogenic H- and K-ras, are capable of eliciting NF-κB activation.3, 5, 35, 36, 37, 38 Direct evidence from various in vivo and in vitro models now indicates that the NF-κB-mediated suppression of PCD is crucially involved in various aspects of cancer biology, including malignant transformation, tumor progression and resistance to anticancer therapy (reviewed elsewhere in this issue of CDD).3, 4, 35, 37 Indeed, a constitutive NF-κB activity is required for survival of several cancerous cells, such as those in Hodgkin's lymphoma (HL), diffuse large B-cell lymphoma (DLBCL), chronic myelogenous leukemia (CML), acute lymphoblastic leukemia (ALL), multiple myeloma (MM), breast cancer and other solid tumors.3, 5, 36, 37
Roles in tumor progression: a link between cancer and inflammation
A novel role for NF-κB in cancer viability has recently been unveiled with the discovery that an NF-κB-driven inflammatory reaction is a key requirement for tumor progression and metastatic growth (reviewed elsewhere in this issue of CDD).4, 16, 38, 39 In the link between inflammation and cancer, NF-κB seemingly serves two distinct roles: one in the cancerous cell and one in proinflammatory cell. Using a mouse model of inflammation-induced colon carcinogenesis, it was shown that conditional deletion of IKKβ in enterocytes hinders tumor initiation, in part, by enhancing apoptosis in these cells.39 Conversely, an inactivation of IKKβ in myeloid cells impaired secretion of proinflammatory cytokines, such as IL-6 and TNFα, by these cells, thereby impeding their ability to stimulate tumor growth.38, 39 Similar observations were made using a mouse model of hepatocarcinogenesis triggered by the targeted deletion of Mdr2, an event causing a chronic cholestatic hepatitis.38, 40 Another compelling study demonstrating this key role of NF-κB in linking cancer to inflammation has recently been provided by analyses of mice with colon carcinoma cell metastases in the lung.41 Remarkably, inactivation of NF-κB in metastatic cells had no effect on the ability of these cells to colonize and grow in the lungs, but completely abolished their ability to proliferate in response to systemic administration of lipopolysaccharide (LPS) – a process dependent upon TNFα synthesis by the host. LPS caused rapid tumor regression instead by inducing death of NF-κB-deficient cancer cells.38, 41 This association between inflammation and cancer is well established also in humans, where a paradigmatic example is offered by MALT lymphoma – a B-cell malignancy often caused by chronic gastritis resulting from persistent Helicobacter pilori infection.4, 38, 42 Chronic inflammation has also been implicated in pathogenesis of a wide range of carcinomas – the most frequent type of cancers – arising from various tissues.
Roles in cancer chemoresistance
In addition to these roles in malignant transformation and tumor progression, the NF-κB protective activity can hamper tumor cell killing inflicted by radiation or chemotherapeutic drugs such as topisomerase inhibitors, and in so doing, promote resistance to anticancer treatments.2, 3, 5, 35, 37By blocking TNF-R-, TRAIL-R- and Fas-triggered death, NF-κB also helps malignant cells evade immune surveillance.4, 38, 41 Consistent with the relevance of NF-κB in cancer, pharmacological agents targeting NF-κB such as proteasome inhibitors (e.g. PS-341), glucocorticoids and nonsteroidal anti-inflammatory drug (NSAIDs) such as aspirins, are currently being used or are being tested with promising results in clinical trials for the treatment of various malignancies, including HL, non-HL, MM and solid cancers such as prostate and lung cancers.2, 3, 43, 44
Roles in other diseases
Accumulating evidence indicates that, in addition to cancer, the NF-κB protective activity is critically involved in the pathogenesis of chronic inflammatory diseases such as rheumatoid arthritis (RA) and inflammatory bowel disease (IBD)3, 6 – sustained by a positive feedback regulation between TNFα and NF-κB.3, 6, 45 An aberrant NF-κB-mediated inhibition of PCD might also contribute to the onset of widespread metabolic disorders such as type-II diabetes and atherosclerosis.6, 7, 8, 9 In addition, this aberrant activity of NF-κB may participate in viral pathogenesis. Indeed, many viruses have evolved to adapt to the host defense mechanisms by developing their own protective strategies, often utilizing genes encoding factors that either induce or mimic NF-κB signaling, such as Tax of HTLV-1, LMP and EBNA2 of HBV and v-Rel of the avian retrovirus, REV-T.3, 36 Finally, an inappropriate resistance to cell death is a contributing cause in the formation of tumors in cylindromatosis patients, who carry hereditary defects of the CYLD gene – encoding a de-ubiquitinase that downregulates NF-κB activation downstream of TNF-R1.15, 46 Other inherited conditions characterized by constitutive activation of NF-κB include familial expansile osteolysis (FEO) and familial Paget disease of bone (PDB), both caused by hypermorphic mutations of RANK and exaggerated survival and function of osteoclasts.15
Conversely, an impairment in cell survival is a key pathogenetic element in incontinentia pigmenti (IP) – a rare X-linked disorder usually caused by rearrangements of the IKKγ/NEMO gene15, 46 – and other genetic illnesses characterized by various defects in the NF-κB activation pathway, including primary immunodeficiencies (ID) and anhidrotic ectodermal dysplasia (AED) syndromes.15, 46, 47
The NF-κB-Mediated Control of PCD Induced by TNF-Rs
The duality of TNF-R1
The protective action of NF-κB was first discovered and is best understood in the context of the response to TNFα,3, 16 a pleiotropic cytokine that plays a key role in inflammation, immunity, cell growth, differentiation and induction of PCD.30 NF-κB was also later found to promote survival in response to the triggering of members of the TNF-R group other than TNF-R1 (the primary receptor of TNFα), including CD40, EDAR, BAFF-R and RANK, as well as of DRs in this group, such as Fas (CD95), DR4 (TRAIL-R1) and DR5 (TRAIL-R2).3, 13, 16, 30 Notably, the NF-κB-mediated control of cell killing induced by these receptors appears to be a central element in a vast array of both physiological and pathological processes (discussed in previous sections).16, 30, 48 Signaling through TNF-R1 has the potential for triggering PCD (Figure 1) – an activity that is evident, for instance, during activation-induced cell death (AICD) in T lymphocytes30, 48 and ischemic death in neurons.30, 48, 49 Normally, however, despite this well-documented ability to kill cells, stimulation of this receptor by TNFα does not result in PCD, unless NF-κB activation or new RNA/protein synthesis is blocked (Figure 1).3, 48

Death and survival pathways activated downstream of TNF-R1. Normally, engagement of TNF-R1 leads to rapid activation of NF-κB, which promotes cell survival, at least in part, by interfering with induction of ROS and JNK signaling (right). Blockade of NF-κB, however, results in an accumulation of ROS and sustained activation of the JNK pathway leading to cell death via either a necrotic or an apoptotic pathway (left). In most models, ROS lie upstream of JNK in the death pathways induced by TNF-R1, due to their ability to activate MAP3Ks (such as ASK1) and to inactivate MKP phosphatases
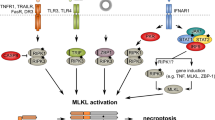
Recent studies offer important insights into the bases for these seemingly opposing abilities of TNF-R1 for inducing PCD, while concurrently blocking it. Stimulation of this and other DRs, such as DR3, DR6 and ectodysplasin A1 receptor (EDAR), promotes formation of complex I, consisting of the adaptor molecule TNFR1-associated death-domain protein (TRADD) and the signaling proteins TNF-R-associated factor 2 (TRAF2), TRAF5 and receptor-interacting protein 1 (RIP1). This event then leads to activation of NF-κB and, ultimately, cell survival (Figure 1).16, 50 Upon dissociating from the plasma membrane, TRADD and RIP1 associate with Fas-associated death domain (FADD), which in turn binds to and activates procaspase-8 and procaspase-10, thereby forming complex II, which ultimately triggers cell death50 (reviewed elsewhere in this issue of CDD; see also Wajant et al.48). Another recent study suggests that complex II might assemble on endocytic vesicles (termed ‘receptosomes’), rather than in the cytosol.51 Finally, it is worth noting that a distinct class of DRs, including Fas, DR4 and DR5, has the ability to interact with FADD directly, forming the so-called death-inducing signaling complex (DISC)16, 48 – the functional equivalent of complex II.
TNF-R1-induced pathways of PCD: necrosis versus apoptosis
TNFα is capable of eliciting PCD through both the apoptotic and the caspase-independent, necrotic pathway (Figure 2).16, 52 Owing to a limited understanding of the biochemical events involved in necrosis, these pathways are usually distinguished based upon morphological criteria:52, 53, 54 Apoptotic cells are typically characterized by chromatin condensation, picnosis, cytoplasmic shrinkage and plasma membrane blebbing; whereas necrotic cells usually exhibit nuclear degradation, organelle swelling and disintegration and loss of plasma membrane integrity. Indeed, these latter cells are virtually indistinguishable from those dying of accidental necrosis. Another form of PCD described more recently is termed ‘autophagy’ and is characterized by a slow ‘metabolic’ death and the presence of large, lysosome-derived vacuoles.53

Possible alternate positioning of ROS and JNK signaling in the apoptotic and necrotic pathways of PCD induced by TNF-R1. These two pathways of PCD seem to diverge immediately downstream of TRADD, with FADD signaling to apoptosis and RIP1 signaling to necrosis. Most available evidence is consistent with a model whereby ROS mediate sustained activation of the JNK pathway by TNF-R1, and this sustained activation of JNK then leads to induction of apoptosis (a); albeit, it can also participate in induction of necrosis in some systems. Some recent evidence, however, suggests that JNK activation may also lie upstream of the TNF-R1-induced formation of ROS, and that this JNK-dependent induction of ROS is critical for the triggering of necrosis (b). Regardless of the positioning of ROS relative to JNK, however, most studies seem to agree that NF-κB can effectively block both necrosis and apoptosis signaling
The pathways of apoptosis and necrosis induced by TNF-R1 appear to diverge immediately downstream of TRADD, with FADD/caspase-8 promoting apoptosis and RIP1 promoting necrosis.52, 54 The key event in apoptosis signaling is the activation of caspase proteases.48, 55 Upon recruitment to complex II, caspase-8 is activated via autocatalytic cleavage and begins processing Bid (a ‘BH3-only’ member of the Bcl-2 family) into tBid, which then targets mitochondria to engage the Bcl-2-like factors, Bax and Bak.48, 55 This event induces the release of mitochondrial proteins such as cytochrome c and Smac/Diablo into the cytosol, causing activation of caspase-9 and downstream effector caspases and, ultimately, apoptosis.48, 55 Notably, Bax and/or Bak can be also engaged by the other major pathway for caspase activation (distinct from the aforementioned DR-induced, ‘extrinsic’ pathway), that is the ‘intrinsic’ pathway – activated by stress and developmental cues.55 Finally, it is worth noting that caspase proteases are not solely involved in death signaling;56 in fact, in some cases (caspase-8, e.g.), they can promote survival and inflammation, actions mediated in part by an induction of NF-κB.56, 57
Although its molecular basis is poorly understood, necrosis signaling is believed not to involve caspase function, and in fact, necrosis can even be potentiated by an inhibition of caspases (Figure 2).52, 53, 54, 58 Moreover, ROS and certain mitochondrial factors, such as apoptosis-inducing factor (AIF), endonuclease G (EndoG) and the serine protease Omi, have been implicated as critical effectors of necrosis downstream of RIP1 and/or TNF-R1.52, 54, 58 Another important element participating in both the necrotic and apoptotic pathways triggered by TNFα is the lysosome.52, 58 Upon formation of ceramide lipids by acidic sphingomyelinase (A-SMase), this organelle leaks into the cytosol cathepsin-group proteases, which can then promote PCD.51, 52, 58 The elements controlling the choice between induction of necrosis or apoptosis are presently unclear, but recent studies suggest that the metabolic status of the cell is a key element in dictating this choice (discussed below).53 A physiologically relevant consequence of making this choice, however, is that whereas necrotic cells release factors such as chromatin-associated HMGB1 that serve as potent stimuli for inflammation, apoptotic cells are rapidly engulfed by professional phagocytes and release signals that instead suppress inflammation, in vivo.52, 53, 58
Basis for the cytoprotective activity of NF-κB
It is now clear that both necrosis and apoptosis can be effectively blocked by NF-κB (Figure 1). By and large, NF-κB exerts its protective effects by upregulating expression of target genes.3 Among these are Bcl-2-family members, A1/Bfl-1 and Bcl-XL, the caspase-8 regulator FLIPL, the caspase inactivators, cellular inhibitor of apoptosis proteins (c-IAP)1 and c-IAP2 and X chromosome-linked IAP (XIAP) and TRAF1 and TRAF2 – which have been predominantly implicated in inhibition of apoptosis signaling.3 The NF-κB-inducible serpin SPI2a, however, was recently shown to also block programmed necrosis.59 These proteins were shown to play important roles in mediating the NF-κB prosurvival function in certain contexts.3 For instance, Bcl-xL and Bfl-1/A1, and possibly Bcl-2 itself, appear to mediate protection afforded by the triggering of CD40 and CD28 receptors in B and T lymphocytes, respectively.3, 49 However, as discussed previously, these factors appear insufficient alone to account for the complete NF-κB-mediated blockade of PCD.3, 49, 60
The NF-κB-Mediated Suppression of the JNK Cascade in Prosurvival Signaling Downstream of TNF-Rs
The JNK MAPK cascade
The protective activity of NF-κB against TNF-R-inflicted killing has been recently associated with suppression of the JNK cascade (Figure 1).60, 61, 62 This is conserved throughout eucaryotes and is one of the major MAPK pathways found in mammalian cells.63, 64, 65 Like other MAPK pathways, such as the p38 and the extracellular-regulated kinases (ERK), it transduces signals triggered by diverse stimuli through the sequential phosphorylation of hierarchically arranged, kinase modules, in order to elicit an appropriate organismal response.63, 64, 65 The most distal of these modules is encoded in mammals by three separate genes: ubiquitously expressed JNK1 and JNK2, and neuronal-specific JNK3.63, 64 In these organisms, JNK kinases are primarily activated upon exposure to proinflammatory cytokines such as TNFα and IL-1β or stress stimuli such as UV radiation and genotoxic, osmotic, oxidative and hypoxic stress.63, 64 Induction of JNK by these stimuli has been linked to different biological responses, including growth and differentiation.63, 64, 65 By and large, however, activation of JNK appears to be a trigger for cell death.63, 64, 65
The involvement of JNK in PCD signaling
This involvement of JNK in PCD signaling is most clearly demonstrated by the analyses of JNK null mutations in mice. Mouse embryonic fibroblasts (MEFs) lacking both JNK1 and JNK2 are refractory to cytochrome c release and apoptosis elicited in response to various stress stimuli, including UV radiation;66 and thymocytes and peripheral T cells from either JNK1−/− or JNK2−/− animals are seemingly protected against anti-CD3-induced death and AICD, respectively.63, 67 Moreover, JNK3−/− neurons display a severe defect in their apoptotic response to excitotoxins,64, 68 and T cells deprived of either JNK1 or the JNK kinase, MKK7/JNKK2, mount an exaggerated proliferative reaction to stimulation with antigen.63, 67, 69, 70
The suppression of JNK as a critical protective mechanism mediated by NF-κB
Despite these findings, however, the role of JNK in TNF-R-induced PCD has been, for years, a matter of intense debate. Some initial studies had suggested that JNK activation by TNFα either enhanced or did not affect cell survival.71, 72 Others had instead found a death-promoting effect of this activation by TNFα.73 This issue has now been clarified by the analyses of JNK- and NF-κB-deficient models. The use of these models has now shown that JNK signaling plays an obligatory role in TNF-R-inflicted killing, and that the targeting of this signaling is a key means by which NF-κB counters PCD.16, 49 Normally, TNFα triggers potent, but only transient activation of the JNK cascade, with basal levels being re-established rapidly, within 30–60 min.60, 61, 62 Blocking NF-κB activity, however, by either knockout deletion of RelA or IKKβ, or ectopic expression of IκBαM, a degradation-resistant variant of IκBα, markedly impairs the normal shutdown of TNFα-induced JNK signaling (see Figure 3), thereby unveiling an additional, prolonged phase of this signaling.60, 61, 62 It is in fact this prolonged phase of the JNK induction that has been implicated in activation of PCD.16, 49, 60, 61, 62 In support of this view, constitutive JNK activity caused by ectopic expression of chimeric MKK7-JNK1 proteins appears to be sufficient alone to trigger cell death.74 In the presence of NF-κB, JNK activity is elevated by TNFα only transiently,60, 61, 62 which explains why this transient elevation occurs without significant cytotoxicity.49, 64 Caspases have the ability to activate MAPK kinase kinases (MAP3Ks).16, 64 Yet, the effects of NF-κB on the JNK pathway do not appear to be a secondary consequence of an inhibition of caspases, because the activity of this MAPK pathway in NF-κB-deficient cells is unaffected by treatment with caspase blockers such as z-VADfmk.49, 62 Hence, NF-κB factors behave as genuine regulators of the JNK cascade, and this ability of NF-κB to control sustained activation of JNK signaling appears to be a pivotal protective mechanism against TNF-R-inflicted cytotoxicity.

The effectors of the NF-κB-mediated suppression of ROS and JNK activities and their proposed modes of action. For most part, the protective activity of NF-κB against TNF-R1-induced killing is mediated through upregulation of target genes. One subset of NF-κB-inducible targets, including A20, XIAP and Gadd45β, appears to act directly on the JNK cascade; another subset of targets, including FHC, Mn-SOD and possibly other genes, appears to block JNK signaling indirectly, by suppressing accumulation of ROS
Consistent with this notion, suppression of JNK signaling by either pharmacological means or expression of dominant negative kinase mutants effectively rescues NF-κB null cells from TNFα-induced killing.60, 61, 62 Likewise, either compound deletion of JNK1 and JNK2 or silencing MKK7 expression virtually abrogates this killing in RelA−/−-, IKKβ−/−- or IκBαM-expressing cells.75, 76, 77 Further evidence of a critical role of JNK in promoting TNFα-induced PCD comes from in vivo studies demonstrating that both JNK1−/− and JNK2−/− knockout mice are markedly protected against TNF-R-mediated liver damage caused by systemic administration of concavalin A (Con A) (see below).78 Hence, available evidence now indicates that an abrogation of sustained activation of the JNK cascade is crucial for control of TNFα-induced PCD, and furthermore, that this abrogation is critically dependent upon NF-κB.16, 49 Despite this indisputable role of JNK in TNFα-inflicted killing, however, there is still some remaining debate as to whether the NF-κB-mediated targeting of this MAPK is more relevant to the control of necrosis or that of apoptosis75 (discussed below; see also Figure 2).
Evolutional Conservation of the NF-κB-Mediated Control of the JNK Pathway
The biological relevance of the NF-κB-mediated restraint of JNK signaling is underscored by the evolutionary conservation of this restraint. In Drosophila, the duration of JNK activation by lipopolysaccharide (LPS) is directly controlled by the NF-κB protein, Relish.79 In this organism, LPS signals through the so-called Imd pathway, named after the insect ortholog of RIP, to coordinate immunity to Gram-negative bacteria via its two main branches: the JNK and IKK/Relish branches.20, 80 These diverge immediately downstream of the MAP3K, TAK1, to direct transcription of distinct subsets of target genes in a temporally coordinated manner.80, 81 Whereas Relish targets display a pattern of prolonged expression, JNK-responsive genes are characterized by transient activation, owed to a Relish-imposed shutdown of JNK signaling. The process appears to involve proteosomal degradation of TAK1, promoted by upregulation of Relish target genes.79
Notably, in addition to immune responses, the Relish-mediated attenuation of JNK signaling might control PCD. The Drosophila homolog of TNFα, Eiger, depends on JNK – rather than on the caspase-8 homolog, Dredd – to trigger cell death.80, 82, 83 Furthermore, JNK-induced apoptosis in flies is mediated by Reaper, Hid and Grim, the functional counterparts of mammalian Smac/Diablo (see below).80, 83 Thus, the participation of JNK in apoptosis signaling appears to be a remnant of an ancestral death-inflicting mechanism engaged by TNF-Rs. Indeed, only later in evolution did TNF-Rs begin to exploit the FADD/caspase-dependent pathway for triggering PCD.48
The NF-κB-Mediated Suppression of ROS as a Means for Controlling the JNK Cascade
The NF-κB-mediated survival also involves a suppression of ROS
Recent studies from several groups, including our own, now indicate that NF-κB also protects cells against TNF-R-induced cytotoxicity by controlling the accumulation of ROS induced downstream of these receptors (Figures 1 and 2).76, 84, 85 ROS are emerging as key mediators of death signaling initiated by the triggering of TNF-R1 (Figures 1 and 2).16, 48, 52, 54, 75, 76, 84, 85, 86 They have been known to play critical roles as second messengers in the PCD pathways initiated by other cytotoxic stimuli such as ceramide, ionizing radiation and chemotherapeutic drugs, and indeed, oxidative stress is sufficient alone to induce cell death.86, 87 Unlike what is seen in normal cells, treatment with TNFα leads to aberrant accumulation of ROS in NF-κB-deficient cells, indicating that these cells are defective in their ability to maintain redox homeostasis.76, 84, 85 Hence, the restraint of ROS accumulation induced downstream of TNF-Rs is a new physiological function ascribed to NF-κB (Figures 1 and 2).16, 76, 84, 85 Remarkably, exposure to antioxidant agents such as N-acetyl-cysteine (NAC), butylated hydroxyanisole (BHA) or pyrrolidine dithiocarbamate (PDTC) virtually abrogates TNFα-inflicted killing in NF-κB-deficient cells.76, 84, 85 Hence, suppressing ROS formation represents a key protective mechanism mediated by NF-κB (Figures 1 and 2).16, 75, 76, 84, 85
Mechanisms of ROS-induced PCD: the role of JNK
The precise mechanism(s) by which ROS trigger PCD is unclear. Accumulating evidence, however, suggests that ROS-inflicted cytotoxicity is mediated in part by prolonged activation of the JNK cascade (Figure 1).16, 76, 85, 86 Indeed, the activities of ROS and JNK induced by TNF-Rs appear to be linked, both being obligatory participants in the same death-inducing pathway triggered by these receptors (Figure 1).16, 76, 85, 86 In RelA−/− fibroblasts and other NF-κB-deficient cells, sustained induction of JNK signaling downstream of TNF-Rs is virtually abolished by treatment with ROS-neutralizing agents such as NAC and BHA.76, 84, 85 Interestingly, this treatment has only modest effects on the transient phase of this induction.76, 84, 85 ROS may enhance PCD also via other mechanisms, for instance, via inhibition of NF-κB.16 Indeed, whereas ROS have been found to trigger NF-κB activation in certain systems,88, 89 in others, they have been shown to interfere with RelA binding to DNA and the activity of IKKβ by oxidizing critical cysteine residues in the RHD and activation loop, respectively, of these proteins.16, 88
Mechanisms for the ROS-mediated activation of JNK
Exactly how ROS promote sustained induction of the JNK pathway by TNFα is not known. Recent reports, however, have provided important insights into the mechanisms of this induction. One study by M Karin and co-workers shows evidence that phosphatases of the MAP kinase phosphatase (MKP) family, such as MKP-1, MKP-3, MKP-5 and MKP-7 – known to be involved in downmodulation of MAPK activities63– are crucial molecular targets of ROS in the TNFα-induced pathway for JNK activation and PCD (Figure 1).76 The authors propose that ROS induce oxidation of a highly reactive, cysteine residue in the catalytic domain of MKPs, thereby causing their inactivation.76 This ROS-mediated inactivation of MKPs seemingly impairs shutdown of JNK signaling following exposure to TNFα, prompting persistent activation of this signaling and, ultimately, cell death (see Figure 1).76 According to this view, by preventing oxidation of MKPs, antioxidants ensure transient activation of JNK, and in so doing, impede induction of PCD by TNF-Rs.76 This provocative model, however, is mainly based on evidence obtained with pharmacological and dominant-negative MKP inhibitors, and so, awaits validation through studies with knockout and/or knockdown systems.
Further, inactivation of MKPs does not appear to be the sole mechanism for ROS-mediated induction of JNK signaling. Earlier studies by Matsuzawa and Ichijo86 have shown that ROS also triggers activation of ASK1/MEKK5, a TRAF2-associating MAP3K that is essential for prolonged activation of JNK (and p38) downstream of TNF-R1.64, 86 This model is supported by genetic evidence, because ASK1−/− fibroblasts exhibit a severe (albeit incomplete) defect in the activation of JNK signaling and PCD in response to TNFα.90 Interestingly, as with MKPs,76 ROS-mediated induction of ASK1 activity depends on inhibition of the redox-regulated factor, thioredoxin.86 Hence, ROS can influence JNK signaling both via activation of upstream MAP3Ks and via inhibition of regulatory MKPs (Figure 1). The relative importance of each mechanism for control of TNFα-induced JNK activity is likely to depend upon the biological context. Despite the fact that several MAP3Ks such as MEKK1, GCK, GCKR, MLK3 and ASK1 have been implicated in activation of JNK by TNF-Rs,63, 64, 86 the precise mechanism underlying this activation is not known. Thus, delineating this mechanism will further help clarify the basis for the ROS-mediated regulation of the JNK cascade. Regardless of this basis, however, several studies now seem to concur that ROS lie upstream of JNK in the TNF-R-induced pathways of PCD (Figure 1),76, 84, 85, 86 and therefore that attenuation of ROS represents another, indirect mechanism through which NF-κB exerts a restraint on sustained activation of the JNK cascade and subsequent induction of PCD (Figure 1).16, 84, 85 Nevertheless, there is evidence suggesting that the crosstalk between JNK and ROS might be more complex75 (see below; see also Figure 2).
The origin of TNFα-induced ROS
The origin of the ROS produced downstream of TNF-Rs is unknown. Nevertheless, it is normally assumed that this production occurs primarily in mitochondria,16, 52, 76 the main source of ROS in eucaryotes.87 Here, electron leakage from the electron transport chain causes formation of superoxide radical (•O2−), a moderately reactive species that can generate hydrogen peroxide (H2O2), which in turn can produce highly reactive hydoxyl radicals (•OH), especially in the presence of transition metals such as iron.87 Previous studies, however, have failed to differentiate between the ROS acting as second messengers in TNF-R-induced signaling and those produced as a consequence of the oxidative burst that typically follows mitochondrial outer membrane permeabilization (MOMP),48, 52, 75, 76, 84, 85 a sign that the cell might have already committed to die.48, 58 Thus, whether the mitochondrial ROS detected in previous studies were a cause or a secondary effect of cell death remains unclear. Indeed, the observations that ROS are not elevated following exposure to IL-1β76, 84 and that their induction by TNFα is blocked by compound mutation of JNK 1 and JNK275 could simply reflect an absence of cell death in these systems. In further agreement with a primary extra-mitochondrial origin of the signal-transducing ROS induced by TNFα, ectopic expression of the mitochondrial antioxidant enzyme, Mn2+ superoxide dismutase (Mn-SOD), was shown to afford only limited protection against TNF-R-mediated cytotoxicity in NF-κB null cells.84, 85 Indeed, extra-mitochondrial sources of ROS have been identified and previously implicated in cytotoxic signaling induced by TNF-Rs.16, 52 These include cytosolic phospholipase A2 (cPLA2), plasma membrane NADPH oxidase and ER-associated, NADPH cytochrome P450 reductase.16, 52, 91 Ultimately, the characterization of the precise mechanisms through which ROS are formed during stimulation with TNFα will require the application of genetic tools and the development of more sophisticated methods of detection.
The TNF-R-induced signaling pathways leading to ROS formation
The TNFα-induced pathways leading to ROS formation are also unknown. It appears, however, that both RIP1 and FADD are required for promoting this formation downstream of TNF-Rs (Figure 2).16, 52, 92 Interestingly, as with programmed necrosis, the ability of RIP1 to participate in the induction of ROS depends on its kinase activity52, 93 – whereas this activity is not required for its ability to induce NF-κB.52, 93 In certain systems, neither the RIP1- nor the TNF-R-triggered accumulation of ROS seems to involve a participation of caspases (Figure 2). Paradoxically, both this accumulation and necrosis signaling are even potentiated by either mutation of caspase-8 or treatment with the pan-caspase inhibitor, zVAD-fmk16, 52, 93, 94, 95 (see Figure 2). Yet, in other systems, caspases appear to be themselves potent effectors in the DR-induced pathways leading to ROS elevation (Figure 2).16, 52, 54 Thus, ROS accumulation induced downstream of DRs appears to depend on both caspase-dependent and caspase-independent mechanisms.
Possible Distinct Relationship Between ROS and JNK in Necrosis and Apoptosis Signaling Downstream of TNF-R1
ROS-mediated activation of JNK
Most studies indicate that ROS reside upstream of JNK in the TNF-R-induced pathways of PCD (Figure 2).16, 76, 84, 85, 86 Yet, some new data suggest that ROS induction in these pathways can also occurs downstream of JNK activation (Figure 2b),75 indicating that there might be a reciprocal interplay between ROS and JNK signaling induced by the triggering of TNF-Rs (Figure 2). Compound mutation of JNK1 and JNK2 in NF-κB-deficient MEFs was found to prevent accumulation of ROS elicited upon exposure to TNFα.75 This accumulation was detected instead in JNK1+/+ JNK2+/+, control cultures, also lacking NF-κB.75 This led to the conclusion that ROS and JNK activities participate in a positive feedback mechanism that serves to amplify death signaling downstream of TNF-Rs (Figure 2). Yet, while a dynamic relationship between these activities is likely to exist,16, 52, 54, 86, 89 it should be cautioned that, as discussed earlier, the antioxidant effects of JNK inactivation in this system might be owed in part to a suppression of PCD. The mechanism(s) by which TNFα-induced JNK activity promotes ROS formation is also uncertain. However, both extra-mitochondrial and mitochondrial mechanisms, such as an action on cPLA2 and mitochondrial complex III, respectively, have been proposed to explain the JNK-mediated enhancement of ROS formation.16, 52, 54, 75, 89
Possible distinct ordering of ROS and JNK signaling in TNF-R-induced necrosis versus apoptosis
While increasing apoptosis, ablation of JNK in the aforementioned study was found to markedly protect NF-κB-deficient fibroblasts against TNFα-induced necrosis,75 suggesting that in the system used, JNK and ROS activities are predominantly involved in transducing necrosis signaling (Figure 2b).75 Additional studies have confirmed this role of JNK and ROS in TNFα-induced necrosis.52, 54, 76, 84 Yet, in other NF-κB null systems, the JNK and ROS activities elicited by TNF-Rs have been primarily implicated in induction of apoptosis (Figure 2a).49, 60, 61, 62, 85 In both RelA−/− MEFs and IκBαM-expressing cells, treatment with TNFα caused caspase activation and nucleosomal fragmentation,60, 62, 76, 85 two hallmarks of apoptosis,48, 55 and exposure to z-VADfmk virtually abrogated JNK/ROS-mediated killing induced by TNFα in these cells.85 Thus, experimental conditions and cell system seem to determine which pathway of cell death is ultimately activated during stimulation of TNF-Rs (Figure 2). An intriguing possibility presented by previous studies is that the cellular decision between necrosis and apoptosis is carried out in part through a control of the relative ordering of ROS and JNK signaling (Figure 2). Indeed, a positioning of JNK activation upstream of ROS has been associated with TNFα-induced necrosis,75 whereas a reciprocal positioning of JNK downstream of ROS has been linked to the induction of apoptosis (Figure 2).85, 86 Regardless of the nature of the death mechanism that is triggered by TNFα, however, in each of these studies, the net effect of inhibiting JNK or ROS activities was an enhancement of cell survival,60, 61, 62, 75, 76, 84, 85 indicating that these activities are essential for induction of cytotoxicity by TNF-Rs and that they are capable of eliciting this cytotoxicity via both a necrotic and an apoptotic mechanism (Figure 2). Moreover, most studies agree that both types of TNFα-induced PCD responses are effectively halted by activation of NF-κB, regardless of the relative positioning of ROS with respect to JNK (Figure 2) (see below).16, 59, 60, 61, 62, 75, 76, 84, 85
Effects of the metabolic state in dictating which pathway of PCD is activated in the cell
The mechanisms controlling which form of PCD is triggered by TNF-Rs are presently poorly understood. One emerging view, however, is that a critical element influencing this choice is the cell metabolic state.53, 96, 97 Cells depending on glycolysis, such as tumor and rapidly growing cells, appear to be more prone to necrosis. In contrast, cells depending on oxidative phosphorylation (OP), such as resting cells, have the ability to quickly replenish their cytosolic pools of NAD and ATP pools, upon depletion by cytotoxic stimulation, and because of this property, appear to die predominantly via apoptosis53, 96, 97 – a dominant and energy-consuming process.55, 97 Thus, it is possible that the metabolic status of the cell may influence qualitative and/or quantitative aspects of the ROS activity triggered by TNF-Rs, and that this activity may then contribute to dictate the type of death response that is induced downstream of these receptors.87
Mechanisms for TNF-R-Induced, Proapoptotic JNK Signaling
The mechanism(s) through which activation of JNK by ROS promotes apoptosis signaling is not known. In certain system, JNK-mediated induction of PCD involves modulation of gene expression.63, 64 In neurons, for instance, excitotoxin-inflicted apoptosis was shown to involve phosphorylation of the c-Jun transcription factor by JNK3 and regulation of c-Jun target genes.63, 64, 68 For promotion of apoptosis triggered by stress stimuli or TNFα, however, the effectors of cytotoxic JNK signaling appear to be already present in the cell.49, 64, 66 It was recently suggested that activation of JNK by TNF-Rs causes cell death by inducing caspase-8-independent, proteolytic processing of Bid into jBid (a product distinct from tBid), which then translocates to mitochondria to promote the selective release of Smac/Diablo (but not cytochrome c) into the cytosol, resulting in activation of caspase-8 and, ultimately, apoptosis.48, 55, 77 Interestingly, as mentioned earlier, Eiger-induced JNK activation in flies also triggers PCD through Hid, Reaper and Grim, the functional equivalents of mammalian Smac/Diablo.82, 83 This JNK-jBid-Smac/Diablo cascade appears therefore to be an additional link between the intrinsic and extrinsic pathways of PCD, albeit – in contrast with what is seen with the classical caspase-8-tBid-cytochrome c cascade48 – in this other link, caspase-8 lies downstream (rather than upstream) of mitochondria.77
Several important questions, however, remain unanswered. For instance, how JNK promotes formation of jBid is unknown. Further, it is likely that there are also other means by which JNK mediates apoptosis signaling downstream of TNF-Rs. In support of this notion, cytotocity induced by ectopic MKK7-JNK1 proteins, causing constitutive JNK activity, requires Bax-like factors, but interestingly, does not require Bid.74 Additionally, following exposure to UV light, JNK was shown to promote caspase activation and PCD by enhancing the mitochondrial release of cytochrome c, rather than that of Smac/Diablo alone.64, 66 Finally, although JNK (and ROS) are known participants in TNFα-induced programmed necrosis, the basis for this participation is unknown.
The NF-κB-Mediated Suppression of JNK Signaling in Enhancement of PCD
NF-κB as an inducer of PCD
Although activation of NF-κB is usually a potent signal for survival (Figure 1),3, 16, 17 in specific circumstances, this activation can promote PCD.3, 16 NF-κB was shown to enhance cytotoxicity induced by the triggering of CD3 in double-positive thymocytes,98 and by treatment with UV radiation or certain chemotherapeutic agents such as doxorubicin, etoposide and 1-β-D-arabinofuranosylcytosine in certain cancer cells.3, 16, 99, 100 In some circumstances, this cytotoxic activity of NF-κB has been validated in in vivo models, as with models of thymocyte development and negative selection,13, 98 and so, it appears to be biologically significant, at least in these circumstances. The basis for the participation of NF-κB in PCD signaling is unclear. It was shown, however, that certain cytotoxic stimuli, including UV light and anticancer drugs, induce a form of NF-κB activity that is qualitatively different from that elicited by TNFα, because it appears that this activity causes repression, rather than activation of protective genes.101 In other systems, NF-κB was shown to enhance p53 activity and expression of p53 target genes,3, 16, 102 or to promote direct transcriptional upregulation of proapoptotic genes, such as those encoding Fas/CD95, FasL, DR4 and DR5.3, 16
The NF-κB-mediated targeting of JNK as a means for inducing PCD
It is plausible that, in certain contexts, this action of NF-κB in promoting PCD can also be due to an interference with the JNK pathway. The notion that this pathway is involved in the induction of cell death is not without exceptions.63, 64 JNK has, in fact, also been implicated in cell differentiation, cell growth and even cell survival in certain systems.63, 65 In one study, MEFs from JNK1−/− JNK2−/− mice were found to be abnormally sensitive to TNFα apoptosis.103 It was therefore proposed that, in the presence of functional NF-κB dimers, transient activation of JNK by TNFα is a signal for cell survival.103 The prosurvival action of JNK in this system was associated with induction of the c-Jun-like factor, JunD, which seemingly cooperates with NF-κB to upregulate transcription of c-IAP2.103 In a later study, however, JNK null MEFs were found to be abnormally susceptible to apoptosis even in the absence of NF-κB. Of note, these cells were found to be concomitantly resistant to TNFα-inflicted necrosis, with the overall result of JNK activation being a net increase in cell death (discussed earlier).75 Finally, while confirming the existence of an antagonistic activity of NF-κB on JNK signaling, another report has indicated a role for this activity of NF-κB in promotion of TNFα-induced cell death.104 Hence, a suppression of PCD might not be the sole possible outcome of the negative regulation that NF-κB imposes on the JNK pathway.
Other determinants of the biological outcome of JNK activation and the interference by NF-κB
Most likely, the biological effects of JNK activation also depend on stimulus- and tissue-specific elements, including an interplay with other signaling pathways such as the Akt/PKB, ERK and NF-κB pathways that might be concomitantly active in the cells.63, 64, 65, 103 Notably, the biological activity of JNK signaling appears to depend in fact on the duration of this signaling, and so context-specific factors such as NF-κB, MKPs and ROS can modulate the functional outcome of the activation of JNK by controlling the duration of this activation.16, 49, 64 The reason why only prolonged JNK induction is a potent signal for cell death is not known. It is plausible, however, that while transient activation of JNK can promote survival via a modulation of gene expression, upon prolonged activation, JNK may acquire the ability to engage a transcription-independent, cytotoxic mechanism.16, 49, 64, 77, 103 This temporal requirement for JNK-mediated cytotoxicity might depend for instance on substrates that become available only with time after stimulation, on death-signaling effectors (e.g. jBid or Smac/Diablo) that accumulate only slowly prior to reaching the threshold needed for triggering PCD or on amplification of the JNK-mediated death signal through feed-forward loops (e.g. with caspases).16, 64 It is also worth noting that because of the complex biological actions of the JNK cascade, a restraint of this cascade might enable NF-κB to also influence biological processes other than cell survival, including cell growth and differentiation, and gene expression. In the skin, for instance, the NF-κB-mediated control of JNK activation appears to block JNK-driven proliferation and neoplastic outgrowth of the epithelium.105 Further, interactions between the NF-κB and JNK pathways are also likely to occur at a downstream level – that is, at promoters of genes that are jointly controlled by NF-κB and JNK-regulated transcription factors, such as c-Jun and JunD63, 64 – in order to coordinate gene expression.
Direct Means for the NF-κB-Mediated Control of JNK Signaling
Gadd45β
The NF-κB-mediated suppression of the JNK cascade appears to involve induction of target genes3, 16, 17 (Figure 3). Thus far, few such targets have been characterized. Screens for cDNAs capable of blocking TNFα-induced PCD in RelA−/− fibroblasts identified Gadd45β/Myd118 – a member of a family of structurally related factors also comprising Gadd45α and Gadd45γ49, 106 – as an NF-κB-inducible, bona fide inhibitor of the JNK cascade (Figure 3).60 Gadd45 proteins have been implicated in various biological activities, including DNA repair, cell cycle control and regulation of several MAPK cascade kinases.49, 106 Upregulation of Gadd45β by TNFα requires NF-κB, and ectopic expression of this factor in NF-κB-deficient cells inhibits JNK activation and apoptosis downstream of TNF-Rs.60 The Gadd45β-mediated inhibition of this JNK activation appears to involve a blockade of MKK7/JNKK2 (Figure 3),107 an essential and specific inducer of JNK during stimulation of TNF-Rs.64, 108 Gadd45β directly associates with this MAP2K, blocking its catalytic activity, most likely, by precluding access to ATP.107 Although Gadd45β also has the ability to interact with other MAPK-pathway kinases, such as the MAP3Ks, MEKK4/MTK1107, 109 and ASK1/MEKK5,107 it appears that these other interactions of Gadd45β are not involved in control of JNK activation downstream of TNF-Rs.49, 107
Of interest, inactivation of endogenous Gadd45β by either expression of antisense mRNAs or targeted deletion of the Gadd45β gene hampers cell survival and downmodulation of JNK signaling following exposure to TNFα,60, 107 suggesting that in some tissues Gadd45β upregulation is required for the NF-κB-mediated antagonism of JNK activation and PCD. The Gadd45β–MKK7 interaction appears in fact to be a critical link between the NF-κB and JNK pathways (Figure 3) – confirmed by the use of cell-permeable peptides that impede Gadd45β binding to MKK7.107 Another study using Gadd45β-deficient fibroblasts, however, has recently suggested that Gadd45β is not involved in the NF-κB-mediated control of TNFα-induced PCD in MEFs, as these cells were found to have no defect in the suppression of JNK induction or PCD downstream of TNF-Rs.110 The apparent discrepancies between published reports are likely owed, at least in part, to the different sensitivities and experimental conditions of the assays used.49, 111 Nevertheless, from both studies, it seems clear that the effects of knocking out Gadd45β in fibroblasts are not complete (i.e. not as profound as those of blocking NF-κB).16, 49, 60, 107, 110 There is in fact evidence that the function of Gadd45β in these cells is redundant, as factors other than Gadd45β were also found to contribute to suppression of the TNFα-mediated induction of MKK7.49, 107 Yet, despite this functional redundancy, data with Gadd45β-inactivating peptides revealed that, even in this system, Gadd45β plays an obligatory role in complete inhibition of TNF-R-induced killing.107 Additionally, the mechanisms of MKK7 inactivation downstream of TNF-Rs appear to be tissue specific,49, 107 and so, it is possible that Gadd45β has a more prominent role in this inactivation in other tissues.
This view is supported by recent studies with Gadd45β−/− knockout mice that have shown that Gadd45β is an essential participant in skeleton morphogenesis,112 the resistance of hematopoietic cells to stress- and anticancer drugs-induced killing,113 differentiation and function of T-helper 1 (TH1) T cells and immunity against infection with the intracellular bacterium, Listeria monocytogenes.114 In some studies, these functions of Gadd45β have been linked to a regulation of MAPK signaling. For instance, Gadd45β−/− TH1 T cells were found to exhibit a severe defect in the control of JNK and p38 activation downstream of both antigen and cytokine receptors.114 Whether this defect is primarily mediated through a deregulation of MKK7 activity, however, is presently unknown. Indeed, whereas a deregulation of MEKK4, an MAP3K implicated in p38 and JNK induction in response to stress,64, 109 has been implicated in some of the signaling abnormalities observed in Gadd45β null cells,115 it is evident that these abnormalities also depend upon MEKK4-independent mechanisms.49
XIAP
The inhibitory effects of NF-κB on the JNK cascade have also been associated with upregulation of the caspase inhibitor XIAP61 (Figure 3). This is a downstream target of NF-κB3, 49 and a well-characterized inactivator of caspase-3 and -7.116 Upon overexpression, XIAP attenuates TNFα-induced PCD in NF-κB-deficient cells,117 and moreover, thymocytes from XIAP transgenic animals are protected against killing induced by various triggers.118 Ectopic expression of XIAP was also shown to reduce TNFα-induced JNK signaling in RelA−/− cells, but to have no effect on p38 and ERK activities.61 Despite its ability to block caspases,116 XIAP seems to be an authentic inhibitor of the JNK cascade, because when overexpressed in RelA null cells, it is capable of interfering with both the caspase-dependent and the caspase-independent phases of the activation of this cascade by TNFα (Figure 3).16, 49, 61 Notably, however, XIAP null mice exhibit no obvious apoptosis-related phenotype, and MEFs from these mice appear to be unaffected in their ability to control JNK activation and PCD induced by the triggering of TNF-Rs.3, 16, 49 The mechanism by which XIAP mediates a restraint on the JNK cascade also remains unclear. Indeed, whereas previous studies have shown that XIAP is capable of interacting with kinases in the JNK pathway,49, 119 these studies have found that the interactions of XIAP with these kinases lead to an enhancement, rather than an inhibition of JNK signaling.49, 119 Thus, further studies are needed to determine the physiological relevance of XIAP to the NF-κB-mediated control of JNK signaling and PCD.
A20
Another target of NF-κB that is seemingly involved in mediating the inhibitory activity of NF-κB on the JNK pathway is the zinc-finger protein, A20 (Figure 3).120, 121This is rapidly induced by TNFα through a mechanism that depends on NF-κB,3, 121 and this induction by NF-κB plays an essential role in downmodulation of JNK signaling activated downstream of TNF-Rs (Figure 3).120, 121 Accordingly, A20−/− MEFs exhibit persistent activation of JNK and exaggerated PCD following exposure to TNFα.120, 121 Notably, however, overexpressed A20 is incapable of protecting NF-κB-deficient cells against TNFα-induced killing.3, 121 Hence, while being required, upregulation of A20 alone appears to be insufficient to account for the NF-κB-mediated suppression of TNFα-induced JNK signaling and PCD. The precise mechanism(s) by which A20 blunts activation of the JNK cascade are not known. Interestingly, A20 is also necessary for downregulation of NF-κB activation by TNFα,120, 121 a function that seemingly involves an enhancement of the ubiquitin-mediated inactivation of RIP1,122 an essential inducer of NF-κB. RIP1, however, does not participate in the induction of JNK by TNF-Rs,48 and so, the A20-mediated suppression of the JNK pathway is likely to be mediated through a different mechanism. It is also worth noting that A20 is capable of interacting with TRAF2,48, 121 a molecule required for activation of JNK signaling by TNFα.48, 64 In fact, some biochemical evidence suggests that the inhibitory effects of A20 on this signaling might involve an action at the level of either TRAF2 or of another molecule positioned immediately downstream of TNF-R1 (Figure 3).48, 120, 121, 123 Accordingly, A20 appears to selectively hinder NF-κB and JNK activations induced by TNF-Rs, and to have no effect on these activations downstream of IL-1β receptor – shown by analyses of A20−/− MEFs.120
The NF-κB-Mediated Targeting of ROS: an Indirect Means for Control of the JNK Pathway
FHC
Interestingly, a subset of NF-κB target genes is involved in the control of ROS (Figure 3).16, 17 Using a gene array-based screen, we have recently identified Ferritin heavy chain (FHC) as a critical mediator of the antioxidant and antiapoptotic activities of NF-κB downstream of TNF-Rs.85 Together with light chains (FLC), FHC constitutes Ferritin – a large heteropolymer that represents the major iron storage mechanism within cells.124, 125 In eucaryotes, iron is required both for production of mitochondrial •O2− and for Fenton and Haber-Weiss reactions, generating highly toxic •OH radicals,87, 124, 125 and therefore limiting availability of this metal, through an upregulation of Ferritin, which is a critical mechanism by which cellular ROS can be controlled (Figure 3). FHC is induced by TNFα through a mechanism dependent upon NF-κB, is required for antagonism of TNFα-induced killing, and upon overexpression, counters apoptosis in NF-κB-deficient cells (Figure 3).85 The protective action of FHC against TNFα-inflicted PCD is mediated through iron sequestration, which prevents ROS accumulation and subsequent sustained JNK activation downstream of TNF-Rs (Figure 3).85 The relevance of FHC to the NF-κB-activated mechanism for restraint of TNF-R-induced JNK signaling and PCD has been validated in FHC-deficient cells and in vivo systems.85, 126 Indeed, knockdown of FHC in fibroblasts results in persistent activation of JNK by TNFα and hypersensitivity to TNF-R-induced cytotoxicity.85 Induction of FHC appears to be a critical element for control of the TNFα-induced fluctuations of the intracellular labile iron pool (LIP).85, 124, 127 Remarkably, systemic administration of the iron chelator, deferoxamine (DFO), protects mice against TNF-R-mediated lethality and tissue damage.126
A new study indicates that, in addition to blocking caspase activation and apoptosis,85 FHC halts TNFα-induced programmed necrosis.127 Thus, unlike other NF-κB-inducible genes, FHC seems capable of suppressing both pathways of PCD activated downstream of TNF-Rs, and thereby, recapitulates the effects of NF-κB on PCD (see Figure 1). It is possible that the iron-mediated control of ROS formation might even contribute to determining which pathway of PCD is ultimately triggered by TNFα.
Notably, FHC is one of several proteins induced in the liver during a systemic acute-phase response to injury, stress and microbial infection.124 This induction of FHC in the liver is triggered by proinflammatory cytokines such as TNFα and contributes to hypoferremia – a common condition in patients with chronic inflammatory illnesses.124, 128 Interestingly, this generalized restriction of iron availability might enable NF-κB to contain ROS-inflicted injury at distant sites, such as sites of inflammation, where there are elevated, potentially harmful concentrations of both ROS and TNFα.4, 6, 16, 17, 45 Accumulating evidence suggest that FHC also plays an important role in cancer.124, 129 An elevation of ROS is often required for oncogene-driven transformation;124, 129 on the other hand, this elevation appears to also enhance the propensity of certain malignant cells to die.17, 87, 124, 129 With tumor progression, however, growth becomes independent of ROS,129 which enables certain cancers to upregulate FHC.124 Notably, the protective action of FHC extends to oxidative and genotoxic stress,124, 125, 130 and in certain human tumors, high expression of FHC has been linked to anaplasia, metastatic spread and poor prognosis.130, 131 Thus, upregulation of FHC may represent a means by which NF-κB promotes oncogenesis, cancer progression and resistance to anticancer treatment.3, 16, 44, 85
Mn-SOD
The antioxidant activity of NF-κB has also been associated with upregulation of the ROS scavenger, Mn-SOD (Figure 3),3, 16, 17, 132, 133 which catalyzes dismutation of •O2− into H2O2.87 Mn-SOD is a TNFα-inducible target of NF-κB, and upon overexpression, attenuates TNFα-mediated cytotoxicity in certain systems (see Figure 3).3, 16, 48, 85, 132, 133 Its significance to the protective function of NF-κB, however, remains uncertain. This is because in various NF-κB-deficient systems, ectopic expression of Mn-SOD affords little or no protection against PCD caused by exposure to TNFα.32, 84, 85 This enzyme also scored poorly in a screen for factors capable of rescuing RelA−/− cells from TNFα-induced killing.85 Moreover, in various cell –types, its levels are seemingly independent of either TNFα or NF-κB,17, 85 suggesting that in these cell types, Mn-SOD does not contribute to the prosurvival action of NF-κB.
Despite the fact that it might be insufficient for control of TNF-R-triggered redox changes and PCD, however, Mn-SOD might still represent a critical effector of the antioxidant and protective activities of NF-κB, in certain contexts. Indeed, there is suggestive evidence that it might also contribute to the tumorigenic activity of NF-κB, for instance in some leukemias.16, 134 For an effective control of ROS levels, synergism between FHC and Mn-SOD might in fact be crucial. While induction of Mn-SOD promotes dismutation of •O2− into H2O2, FHC-mediated iron sequestration may facilitate disposal of H2O2 by peroxidases and catalases.87, 124 In NF-κB-deficient cells, FHC levels are usually low,85 and as a consequence, free iron remains available to catalyze the Fenton reaction reducing H2O2 into highly reactive •OH radicals. This could explain the inability of ectopic Mn-SOD to inhibit, alone, TNFα-induced cytotoxicity in NF-κB null cells.84, 85, 132
Complexity of the NF-κB-activated program for JNK inhibition
Combined upregulation of FHC and Mn-SOD not only provides a basis for the antioxidant action of NF-κB but also provides an additional, indirect link between the NF-κB and JNK pathways (Figure 3). Hence, NF-κB appears to halt the TNFα-mediated induction of the JNK cascade via at least two separate mechanisms: directly, through upregulation of Gadd45β, A20 and XIAP (Figure 3); and indirectly, through upregulation of FHC, Mn-SOD and possibly other factors – which suppress the elevation of ROS (Figure 3). Most likely, to ensure effective inhibition of JNK signaling, these factors must act cooperatively. Indeed, a recent study suggests that the NF-κB-mediated control of redox homeostasis might involve upregulation also of other antioxidant factors.16, 135 In this regard, it is also noteworthy that the specific JNK-inhibiting program activated by NF-κB depends upon tissue- and stimulus-specific elements,3, 17 and therefore the contribution of each target gene might be essential only within specific biological contexts. Interestingly, this adaptable network of genes that is induced by NF-κB might enable an organism to orchestrate a suitable biological response to each type of apoptotic stimulus and challenge (see below).
In vivo Relevance of the NF-κB-Mediated Control of JNK Signaling
The response of the liver to stress and injury
The biological relevance of the NF-κB-mediated restraint of JNK signaling has been amply documented in animal models, most notably in the liver.16, 28, 76, 136 Recent studies have shown that NF-κB is required to counter hepatotoxicity caused by systemic administration of ConA, an agent provoking massive liver damage through a mechanism that depends on TNF-Rs.28 Conditional deletion of IKKβ in hepatocytes resulted in prolonged activation of JNK signaling following treatment with ConA, and suppression of this signaling by ablation of either JNK1 or JNK2 markedly attenuated ConA-induced injury in the NF-κB-deficient liver.28 Thus, the NF-κB-mediated restraint of JNK activation is required to prevent hepatic damage, in vivo. Indeed, a similar mechanism might account for the NF-κB protective action in the fetal liver.3, 23, 49
TNF-R1, JNK and NF-κB have been shown to play important roles also during liver regeneration post-partial hepatectomy,16, 76, 137, 138 and so, NF-κB may exert its critical influence on this process, at least in part, through a control of the duration of JNK activation.16, 76 Interestingly, it was reported that, whereas in the ConA response model, both sustained JNK activation and hepatic injury are critically dependent on ROS formation, in the partial hepatectomy model, neither the induction of JNK nor the liver compensatory reconstitution appears to be reliant on this ROS formation76 – despite that both processes are controlled by an integration of TNF-Rs, JNK and NF-κB activities.16, 49, 76, 137, 138 Hence, it appears that during different liver responses, both the TNF-R-elicited pathway leading to JNK activation and the NF-κB-induced program that controls this activation of JNK are mediated through distinct mechanisms (also discussed below).
Chronic inflammatory and hereditary disorders
Disturbances of the NF-κB-imposed blockade of TNF-R-induced PCD play a central role in pathogenesis of a wide spectrum of human diseases.3, 4, 5, 6, 7, 8, 45 Notably, this detrimental action of NF-κB appears to depend, at least in part, on a deregulation of ROS and JNK activities. This is evident, for instance, in diseases such as IBD and RA where chronic inflammation is perpetuated by a positive feedback regulation between TNFα and NF-κB (Figure 4).3, 6, 30, 45, 48 Other examples of a pathogenetic role of an NF-κB-dependent deregulation of JNK signaling could be provided by certain hereditary disorders. Indeed, the impaired survival of lymphocytes and keratinocytes from patients with primary deficiencies in the NF-κB pathway such as IP, AED and select ID syndromes might be due in part to an exaggerated activity of the JNK pathway.15, 46, 47 Likewise, an NF-κB-mediated deregulation of JNK activity, either by defect or excess, is likely to be an important contributing factor in the abnormalities of cell survival observed in patients with disorders caused by mutations or rearrangements in genes encoding either TNF-R-family receptors (such as RANK) or signaling molecules in the pathways activated by the these receptors.15, 46, 47 Indeed, not only are these receptors key regulators of cell survival – a function invariably dependent on NF-κB – but they also are potent inducers of both NF-κB and JNK signaling (discussed earlier).3, 15, 30, 48

Potential therapeutic implications of the NF-κB-mediated blockade of TNFα-induced ROS and JNK signaling. A positive feedback loop between TNFα and NF-κB often drives tumor progression and chronic inflammation. Several pharmacological agents could be developed to treat these conditions, without causing a global inhibition of NF-κB. Such agents could in fact induce death of diseased cells by blocking the activity of either MKP phosphatases or downstream effectors of the NF-κB-mediated suppression of ROS and JNK signaling. This therapeutic approach would not directly impact NF-κB functions in immunity and development
Cancer
Finally, the relevance of the NF-κB-mediated control of JNK and ROS activities might apply to cancer (Figure 4).3, 16, 49, 139 JNK and NF-κB appear to have opposing actions in various tumors. Whereas inducers of the JNK pathway (e.g. BRCA1, MKK4 and JNK3) and JNK-activated transcription factors (e.g. JunB and JunD) often behave as tumor suppressors,49, 64, 139, 140 activation of NF-κB seems to be involved in antagonism of transformation-associated apoptosis and enhancement of tumor cell survival in various late-stage cancers.3, 35, 36, 37 JNK and ROS also mediate cancer cell killing induced by radiation and certain anticancer drugs,16, 49, 64, 87, 129 whereas NF-κB often contributes to blunt this killing.3, 5, 6, 16, 35, 44 Hence, because many oncogene products, such as Her-2/Neu and oncogenic ras, as well as chemotherapeutic drugs such as topoisomerase inhibitors, trivalent arsenicals and proteasome inhibitors, are potent elicitors of JNK,16, 49, 64, 139 the dependence of certain cancerous cells on a constitutive NF-κB activity for their survival might lie in their need to suppress JNK-mediated PCD promoted by these oncogene products or anticancer agents.
Another link between cancer and the inhibitory activity of NF-κB on the JNK pathway has been unveiled with the recent discovery that progression and growth of both primary and metastatic tumors are often critically dependent upon the inflammatory reaction that pervades the cancerous tissue.4, 16, 38, 39, 40, 136 In various models, this reaction is driven by NF-κB and proinflammatory cytokines such as TNFα (Figure 4).38, 39, 40, 41 Thus, it is plausible that the mandatory role of NF-κB in progression, growth and survival of these inflammation-driven tumors is also owed, at least in part, to a need to restrain the cytotoxic activities of ROS and JNK – invariably elevated at sites of inflammation.16, 30
New Therapeutic Challenges and Opportunities
Utility and limitations of global blockers of NF-κB in treatment of human diseases
The pivotal importance of the NF-κB pathway in human diseases, including chronic inflammatory disorders, is highlighted by the fact that blockers of this pathway, such as aspirin and glucocorticoids, or of its inducer, TNFα, are presently standard therapy for the treatment of these diseases (Figure 4).2, 3, 5, 6, 16, 43, 44 NF-κB-targeting agents, including glucocorticoids and proteasome inhibitors, are also being used to treat human malignancies, such as MM, HL and certain solid tumors (Figure 4).3, 5, 16, 43, 44
Unfortunately, the clinical utility of these drugs is undermined by their severe adverse effects, including their immunosuppressive effects (see Figure 4). Moreover, available NF-κB blockers, such as corticosteroids, NSAIDs, cyclopentenone prostaglandins, trivalent arsenicals, proteasome inhibitors and sulfasalazine, are not specific for the NF-κB pathway,2, 5, 16, 43, 44 and this causes a set of additional adverse effects. This issue will likely be addressed in part by the introduction of more selective blockers of NF-κB activation, such as IKKβ inhibitors – which are currently being tested in vitro and in preclinical trials.16, 141, 142 These agents will also be likely to exert their pharmacological effects by inhibiting infiltrating inflammatory cells, an important source of tumor growth factors,4, 16, 38 and be effective especially when used in combination with radiation and/or standard chemotherapeutic drugs.3, 16, 43, 44
Yet, it should be cautioned that, even with drugs that are highly specific for the NF-κB pathway, serious side effects beyond those caused by immunosuppression are likely to arise, particularly upon chronic use. This is because the biological activity of NF-κB is subject to tissue-specific elements.3, 12, 16 For instance, despite its well-established role in oncogenesis and tumor progression in various tissues, NF-κB promotes growth arrest, differentiation and tumor suppression in epidermal keratinocytes.16, 105, 143, 144 Indeed, a prolonged inhibition of NF-κB in these cells results in formation of squamous cell carcinoma and can act synergistically with oncogenic H-ras to induce malignant transformation.16, 105, 143, 144 NF-κB might exert a similar tumor-suppressor function also in the liver, because (while impeding tumor progression) conditional deletion of IKKβ in hepatocytes appears to enhance tumor initiation in a model of chemical carcinogenesis.16, 136 Thus, another major problem associated with long-term treatment with NF-κB-targeting agents is the potential of this treatment for promoting oncogenesis in certain tissues. The immunosuppressive effects of these agents could in fact further exacerbate these cancer-inducing effects through a repression of immunosurveillance mechanisms.
Therapeutic agents that selectively interfere with the NF-κB-mediated suppression of JNK signaling
Thus, a preferable therapeutic approach is to develop drugs that enable targeting of the downstream prosurvival effectors of NF-κB, rather than of NF-κB itself (Figure 4). The discovery that the suppression of ROS and JNK signaling is a key protective mechanism mediated by NF-κB now offers an opportunity for developing such drugs (Figure 4). Indeed, selective blockade of the ability of NF-κB to shutdown ROS and JNK activation is likely to enhance PCD preferentially in self-reactive and proinflammatory cells at sites of inflammation (see Figure 4) – where there are elevated levels of TNFα and JNK and ROS activities.6, 16, 30, 45 Drugs targeting the inhibitory action of NF-κB on the JNK cascade might in fact also cause augmentation of JNK and ROS cytotoxic signaling in cancerous cells, particularly in those engulfed within an inflammatory reaction, and therefore provide a powerful adjuvant for anticancer therapy (see Figure 4).4, 16, 38 Agents that disrupt Gadd45β binding to MKK7 or that hinder iron sequestration by FHC or the activity of MKPs might indeed succeed in accomplishing these tasks (Figure 4).
Notably, compounds that selectively interfere with the negative regulation of ROS/JNK activities by NF-κB might also enable dissociation of the prosurvival and proinflammatory actions of NF-κB (Figure 4), and so, minimize the deleterious side effects of global NF-κB inhibitors, for instance their immunosuppressive effects (Figure 4).2, 3, 43 Thus, a capital future challenge is determining the precise mechanisms by which NF-κB counters ROS accumulation and JNK signaling in specific pathophysiological contexts. Indeed, because the NF-κB-activated program for JNK and ROS inhibition manifests a degree of cell-type specificity, it might be possible to achieve selective targeting of this program in diseased tissues. Another important challenge in anticancer therapy will be to determine which targets of NF-κB are most critical for suppression of programmed necrosis, because it is becoming increasingly clear that, while often possessing mechanisms that suppress apoptosis, these cells are usually highly susceptible to undergo necrosis, due to their energy dependence on glycolysis.53, 96, 97 Given the roles of JNK and ROS activities in necrosis signaling (Figure 2), it is plausible that the targeting of these activities also represents a means for the NF-κB-mediated suppression of programmed necrosis.
Concluding Remarks and Future Perspectives
The NF-κB-imposed restraint of ROS and JNK activities plays key roles in both health and disease. In recent years, much progress has been made towards gaining an understanding of the mechanisms of this restraint. The identification of NF-κB-inducible blockers of ROS and JNK signaling has been an important step in this direction. Multiple such blockers have been characterized thus far, and this redundancy may serve to ensure an effective shutdown of ROS and JNK signaling. It might also enable an organism to finely tune its prosurvival response according to specific contexts and needs. Undoubtedly, major future challenges include delineating the precise mechanisms responsible for the mutual positive interplay between ROS and JNK activities in the induction of PCD, and determining which NF-κB-inducible genes are most critical for control of these activities in specific pathophysiological contexts. Establishment of conditional knockout models will be key for addressing these issues. As an inappropriate blockade of PCD by NF-κB appears to underlie various human diseases, these efforts will likely enable development of new therapies that are both more effective and more selective for treatment of these diseases. Since functions of NF-κB in immunity, development and cell survival are seemingly executed through distinct subsets of target genes, the possibility now exists for developing agents that enable selective blockade of the prosurvival action of NF-κB, without significantly compromising its capacity to serve in immunity and/or development. Such selective blockade could help to limit the side effects inherent in global NF-κB blockers. Additionally, because NF-κB, JNK and ROS activities are likely to be predominantly upregulated in inflamed and cancerous tissues, therapeutic strategies aimed at interfering with the interplay between these activities are likely to have inherent specificity for diseased tissues. Indeed, achieving this specificity represents a major therapeutic goal.
Abbreviations
- PCD:
-
programmed cell death
- NF- κB:
-
nuclear factor-κB
- TNF-α:
-
tumor necrosis factor-α
- TNF-Rs:
-
tumor necrosis factor-receptors
- ROS:
-
reactive oxygen species
- JNK:
-
Jun-N-terminal kinase
- DRs:
-
death receptors
- MAPK:
-
mitogen-activated protein kinase
- IKK:
-
I κ B kinase
- LPS:
-
lipopolysaccharide
- TRADD:
-
TNFR1-associated death-domain protein
- TRAF:
-
TNF-R-associated factor
- RIP:
-
receptor-interacting protein
- FADD:
-
Fas-associated death domain
- MAPK:
-
mitogen-activated protein kinase
- MAP2K:
-
MAPK kinase
- MAP3K:
-
MAPK kinase kinase
- MKP:
-
MAP kinase phosphatase
- Gadd45:
-
growth arrest and DNA damage-inducing 45 protein
- Mn-SOD:
-
manganese-dependent superoxide dismutase
- FHC:
-
ferritin heavy chain
- XIAP:
-
X chromosome-linked inhibitor of apoptosis
- MEFs:
-
murine embryonic fibroblasts
- Con A:
-
concavalin A
- BHA:
-
butylated hydroxylanisole
- NAC:
-
N-acetyl cystein
- PDTC:
-
pyrrolidine dithiocarbamate
- MM:
-
multiple myeloma
- HL:
-
Hodgkin's lymphoma
- DLBCL:
-
diffuse large B-cell lymphoma
- CML:
-
chronic myelogenous leukemia
- ALL:
-
acute lymphoblastic leukemia
- IBD:
-
inflammatory bowel disease
- RA:
-
rheumatoid arthritis
References
Sen R and Baltimore D (1986) Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a post-translational mechanism. Cell 47: 921–928
Karin M, Yamamoto Y and Wang QM (2004) The IKK NF-κB system: a treasure trove for drug development. Nat. Rev. Drug Discov. 1: 17–26
Kucharczak J, Simmons MJ, Fan Y and Gelinas C (2003) To be, or not to be: NF-κB is the answer – role of Rel/NF-κB in the regulation of apoptosis. Oncogene 56: 8961–8982
Li Q, Withoff S and Verma IM (2005) Inflammation-associated cancer: NF-κB is the lynchpin. Trends Immunol. 26: 318–325
Greten FR and Karin M (2004) The IKK/NF-κB activation pathway – a target for prevention and treatment of cancer. Cancer Lett. 206: 193–199
Kumar A, Takada Y, Boriek AM and Aggarwal BB (2004) Nuclear factor-κB: its role in health and disease. J. Mol. Med. 82: 434–448
Shoelson SE, Lee J and Yuan M (2003) Inflammation and the IKKβ/IκB/NF-κB axis in obesity- and diet-induced insulin resistance. Int. J. Obes. Relat. Metab. Disord. 3: S49–S52
Dandona P, Aljada A, Chaudhuri A, Mohanty P and Garg R (2005) Metabolic syndrome: a comprehensive perspective based on interactions between obesity, diabetes, and inflammation. Circulation 111: 1448–1454
Nichols TC (2004) NF-κB and reperfusion injury. Drug News Perspect. 17: 99–104
Weil R and Israel A (2004) T-cell-receptor- and B-cell-receptor-mediated activation of NF-κB in lymphocytes. Curr. Opin. Immunol. 16: 374–381
Bonizzi G and Karin M (2004) The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 25: 280–288
Hayden MS and Ghosh S (2004) Signaling to NF-κB. Genes Dev. 18: 2195–2224
Siebenlist U, Brown K and Claudio E (2005) Control of lymphocyte development by NF-κB. Nat. Rev. Immunol. 5: 435–445
Chen LF and Greene WC (2004) Shaping the nuclear action of NF-κB. Nat Rev. Mol. Cell Biol. 5: 392–401
Orange JS, Levy O and Geha RS (2005) Human disease resulting from gene mutations that interfere with appropriate NF-κB activation. Immunol. Rev. 203: 21–37
Luo JL, Kamata H and Karin M (2005) IKK/NF-κB signaling: balancing life and death – a new approach to cancer therapy. J. Clin. Invest. 115: 2625–2632
Bubici C, Papa S, Pham CG, Zazzeroni F and Franzoso G (2004) NF-κB and JNK: an intricate affair. Cell Cycle 3: 1524–1529
Chen ZJ (2005) Ubiquitin signalling in the NF-κB pathway. Nat. Cell Biol. 7: 758–765
Li Q and Verma IM (2002) NF-κB regulation in the immune system. Nat. Rev. Immunol. 2: 725–734
Silverman N and Maniatis T (2001) NF-κB signaling pathways in mammalian and insect innate immunity. Genes Dev. 18: 2321–2342
Mackay F and Browning JL (2002) BAFF: a fundamental survival factor for B cells. Nat. Rev. Immunol. 2: 465–475
Voll RE, Jimi E, Phillips RJ, Barber DF, Rincon M, Hayday AC, Flavell RA and Ghosh S (2000) NF-κB activation by the pre-T cell receptor serves as a selective survival signal in T lymphocyte development. Immunity 13: 677–689
Beg AA, Sha WC, Bronson RT, Ghosh S and Baltimore D (1995) Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature 376: 167–170
Li Q, Van Antwerp D, Mercurio F, Lee KF and Verma IM (1999) Severe liver degeneration in mice lacking the IκB kinase 2 gene. Science 284: 321–325
Rudolph D, Yeh WC, Wakeham A, Rudolph B, Nallainathan D, Potter J, Elia AJ and Mak TW (2000) Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev. 14: 854–862
Alcamo E, Mizgerd JP, Horwitz BH, Bronson R, Beg AA, Scott M, Doerschuk CM, Hynes RO and Baltimore D (2001) Targeted mutation of TNF receptor I rescues the RelA-deficient mouse and reveals a critical role for NF-κB in leukocyte recruitment. J. Immunol. 167: 1592–1600
Doi TS, Marino MW, Takahashi T, Yoshida T, Sakakura T, Old LJ and Obata Y (1999) Absence of tumor necrosis factor rescues RelA-deficient mice from embryonic lethality. Proc. Natl. Acad. Sci. USA 96: 2994–2999
Maeda S, Chang L, Li ZW, Luo JL, Leffert H and Karin M (2003) IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα. Immunity 19: 725–737
Chaisson ML, Brooling JT, Ladiges W, Tsai S and Fausto N (2002) Hepatocyte-specific inhibition of NF-κB leads to apoptosis after TNF treatment, but not after partial hepatectomy. J. Clin. Invest. 110: 193–202
Aggarwal BB (2003) Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol. 3: 745–756
Bharti AC and Aggarwal BB (2004) Ranking the role of RANK ligand in apoptosis. Apoptosis 9: 677–690
Nakashima T, Wada T and Penninger JM (2003) RANKL and RANK as novel therapeutic targets for arthritis. Curr. Opin. Rheumatol. 15: 280–287
Mattson MP and Camandola S (2001) NF-κB in neuronal plasticity and neurodegenerative disorders. J. Clin. Invest. 107: 247–254
Bell S, Degitz K, Quirling M, Jilg N, Page S and Brand K (2003) Involvement of NF-κB signalling in skin physiology and disease. Cell Signal. 15: 1–7
Baldwin AS (2001) Control of oncogenesis and cancer therapy resistance by the transcription factor NF-κB. J. Clin. Invest. 107: 241–246
Gilmore TD (2003) The Re1/NF-κB/IκB signal transduction pathway and cancer. Cancer Treat. Res. 115: 241–265
Shishodia S and Aggarwal BB (2004) NF-κB: a friend or a foe in cancer? Biochem. Pharmacol. 68: 1071–1081
Karin M and Greten FR (2005) NF-κB: linking inflammation and immunity to cancer development and progression. Nat. Rev. Immunol. 5: 749–759
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF and Karin M (2004) IKKβ links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118: 285–296
Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E and Ben-Neriah Y (2004) NF-κB functions as a tumour promoter in inflammation-associated cancer. Nature 431: 461–466
Luo JL, Maeda S, Hsu LC, Yagita H and Karin M (2004) Inhibition of NF-κB in cancer cells converts inflammation-induced tumor growth mediated by TNFα to TRAIL-mediated tumor regression. Cancer Cell 6: 297–305
Isaacson PG and Du MQ (2004) MALT lymphoma: from morphology to molecules. Nat. Rev. Cancer 4: 644–653
Ravi R and Bedi A (2004) NF-κB in cancer: a friend turned foe. Drug Resist. Update 7: 53–67
Orlowski RZ and Baldwin Jr AS (2002) NF-κB as a therapeutic target in cancer. Trends Mol. Med. 8: 385–389
Liu H and Pope RM (2003) The role of apoptosis in rheumatoid arthritis. Curr. Opin. Pharmacol. 3: 317–322
Smahi A, Courtois G, Rabia SH, Doffinger R, Bodemer C, Munnich A, Casanova JL and Israel A (2002) The NF-κB signalling pathway in human diseases: from incontinentia pigmenti to ectodermal dysplasias and immune-deficiency syndromes. Hum. Mol. Genet. 11: 2371–2375
Aradhya S and Nelson DL (2001) NF-κB signaling and human disease. Curr. Opin. Genet. Dev. 11: 300–306
Wajant H, Pfizenmaier K and Scheurich P (2003) Tumor necrosis factor signaling. Cell Death Differ. 10: 45–65
Papa S, Zazzeroni F, Pham CG, Bubici C and Franzoso G (2004) Linking JNK signaling to NF-κB: a key to survival. J. Cell Sci. 117: 5197–5208
Micheau O and Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114: 181–190
Schneider-Brachert W, Tchikov V, Neumeyer J, Jakob M, Winoto-Morbach S, Held-Feindt J, Heinrich M, Merkel O, Ehrenschwender M, Adam D, Mentlein R, Kabelitz D and Schutze S (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21: 415–428
Jaattela M and Tschopp J (2003) Caspase-independent cell death in T lymphocytes. Nat. Immunol. 4: 416–423
Edinger AL and Thompson CB (2004) Death by design: apoptosis, necrosis and autophagy. Curr. Opin. Cell Biol. 16: 663–669
Leist M and Jaattela M (2001) Four deaths and a funeral: from caspases to alternative mechanisms. Nat. Rev. Mol. Cell. Biol. 2: 589–598
Danial NN and Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219
Abraham MC and Shaham S (2004) Death without caspases, caspases without death. Trends Cell Biol. 14: 184–193
Launay S, Hermine O, Fontenay M, Kroemer G, Solary E and Garrido C (2005) Vital functions for lethal caspases. Oncogene 24: 5137–5148
Kroemer G and Martin SJ (2005) Caspase-independent cell death. Nat. Med. 11: 725–730
Liu N, Raja SM, Zazzeroni F, Metkar SS, Shah R, Zhang M, Wang Y, Bromme D, Russin WA, Lee JC, Peter ME, Froelich CJ, Franzoso G and Ashton Rickardt PG (2003) NF-κB protects from the lysosomal pathway of cell death. EMBO J. 22: 5313–5322
De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R and Franzoso G (2001) Induction of gadd45β by NF-κB downregulates pro-apoptotic JNK signalling. Nature 414: 308–313
Tang G, Minemoto Y, Dibling B, Purcell NH, Li Z, Karin M and Lin A (2001) Inhibition of JNK activation through NF-κB target genes. Nature 414: 313–317
Javelaud D and Besancon F (2001) NF-κB activation results in rapid inactivation of JNK in TNFα-treated Ewing sarcoma cells: a mechanism for the anti-apoptotic effect of NF-κB. Oncogene 32: 4365–4372
Karin M and Gallagher E (2005) From JNK to pay dirt: Jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life 57: 283–295
Davis RJ (2000) Signal transduction by the JNK group of MAP kinases. Cell 103: 239–252
Chang L and Karin M (2001) Mammalian MAP kinase signalling cascades. Nature 410: 37–40
Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA and Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870–874
Dong C, Davis RJ and Flavell RA (2002) MAP kinases in the immune response. Annu. Rev. Immunol. 20: 55–72
Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P and Flavell RA (1997) Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the jnk3 gene. Nature 389: 865–870
Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ and Flavell RA (1998) Defective T cell differentiation in the absence of JNK1. Science 282: 2092–2095
Sasaki T, Wada T, Kishimoto H, Irie-Sasaki J, Matsumoto G, Goto T, Yao Z, Wakeham A, Mak TW, Suzuki A, Cho SK, Zuniga-Pflucker JC, Oliveira-dos-Santos AJ, Katada T, Nishina H and Penninger JM (2001) The stress kinase mitogen-activated protein kinase kinase (MKK)7 is a negative regulator of antigen receptor and growth factor receptor-induced proliferation in hematopoietic cells. J. Exp. Med. 194: 757–768
Liu ZG, Hsu H, Goeddel DV and Karin M (1996) Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-κB activation prevents cell death. Cell 87: 565–576
Natoli G, Costanzo A, Ianni A, Templeton DJ, Woodgett JR, Balsano C and Levrero M (1997) Activation of SAPK/JNK by TNF receptor 1 through a noncytotoxic TRAF2-dependent pathway. Science 275: 200–203
Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z and Kolesnick RN (1996) Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis. Nature 380: 75–79
Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar-Sagi D and Davis RJ (2002) The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell. Biol. 22: 4929–4942
Ventura JJ, Cogswell P, Flavell RA, Baldwin Jr AS and Davis RJ (2004) JNK potentiates TNF-stimulated necrosis by increasing the production of cytotoxic reactive oxygen species. Genes Dev. 18: 2905–2915
Kamata H, Honda S, Maeda S, Chang L, Hirata H and Karin M (2005) Reactive oxygen species promote TNFα-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120: 649–661
Deng Y, Ren X, Yang L, Lin Y and Wu X (2003) A JNK-dependent pathway is required for TNFα-induced apoptosis. Cell 115: 61–70
Maeda S, Chang L, Li ZW, Luo JL, Leffert H and Karin M (2003) IKKβ is required for prevention of apoptosis mediated by cell-bound but not by circulating TNFα. Immunity 19: 725–737
Park JM, Brady H, Ruocco MG, Sun H, Williams D, Lee SJ, Kato Jr T, Richards N, Chan K, Mercurio F, Karin M and Wasserman SA (2004) Targeting of TAK1 by the NF-κB protein Relish regulates the JNK-mediated immune response in Drosophila. Genes Dev. 18: 584–594
Hoffmann JA and Reichhart JM (2002) Drosophila innate immunity: an evolutionary perspective. Nat. Immunol. 3: 121–126
Boutros M, Agaisse H and Perrimon N (2002) Sequential activation of signaling pathways during innate immune responses in Drosophila. Dev. Cell 3: 711–722
Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T and Miura M (2002) Eiger a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21: 3009–3018
Moreno E, Yan M and Basler K (2002) Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12: 1263–1268
Sakon S, Xue X, Takekawa M, Sasazuki T, Okazaki T, Kojima Y, Piao JH, Yagita H, Okumura K, Doi T and Nakano H (2003) NF-κB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. EMBO J. 22: 3898–3909
Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, Jayawardena S, De Smaele E, Cong R, Beaumont C, Torti FM, Torti SV and Franzoso G (2004) Ferritin heavy chain upregulation by NF-κB inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell 119: 529–542
Matsuzawa A and Ichijo H (2005) Stress-responsive protein kinases in redox-regulated apoptosis signaling. Antioxid. Redox Signal. 7: 472–481
Curtin JF, Donovan M and Cotter TG (2002) Regulation and measurement of oxidative stress in apoptosis. J. Immunol. Methods 265: 49–72
Schoonbroodt S and Piette J (2000) Oxidative stress interference with the NF-κB activation pathways. Biochem. Pharmacol. 60: 1075–1083
Garg AK and Aggarwal BB (2002) Reactive oxygen intermediates in TNF signaling. Mol. Immunol. 39: 509–517
Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and Ichijo H (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2: 222–228
Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ and Shah AM. (2002) Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-α. Circ. Res. 90: 143–150
Lin Y, Choksi S, Shen HM, Yang QF, Hur GM, Kim YS, Tran JH, Nedospasov SA and Liu ZG (2004) Tumor necrosis factor-induced nonapoptotic cell death requires receptor-interacting protein-mediated cellular reactive oxygen species accumulation. J. Biol. Chem. 279: 10822–10828
Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B and Tschopp J (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1: 489–495
Liu CY, Takemasa A, Liles WC, Goodman RB, Jonas M, Rosen H, Chi E, Winn RK, Harlan JM and Chuang PI (2003) Broad-spectrum caspase inhibition paradoxically augments cell death in TNFα -stimulated neutrophils. Blood 101: 295–304
Vercammen D, Brouckaert G, Denecker G, Van de Craen M, Declercq W, Fiers W and Vandenabeele P (1998) Dual signaling of the Fas receptor: initiation of both apoptotic and necrotic cell death pathways. J. Exp. Med. 188: 919–930
Zong WX, Ditsworth D, Bauer DE, Wang ZQ and Thompson CB (2004) Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 18: 1272–1282
Hammerman PS, Fox CJ and Thompson CB (2004) Beginnings of a signal-transduction pathway for bioenergetic control of cell survival. Trends Biochem. Sci. 29: 586–592
Hettmann T, DiDonato J, Karin M and Leiden JM (1999) An essential role for NF-κB in promoting double positive thymocyte apoptosis. J. Exp. Med. 189: 145–158
Bian X, McAllister-Lucas LM, Shao F, Schumacher KR, Feng Z, Porter AG, Castle VP and Opipari Jr AW (2001) NF-κB activation mediates doxorubicin-induced cell death in N-type neuroblastoma cells. J. Biol. Chem. 276: 48921–48929
Ivanov VN and Ronai Z (2000) p38 protects human melanoma cells from UV-induced apoptosis through down-regulation of NF-κB activity and Fas expression. Oncogene 19: 3003–3012
Campbell KJ, Rocha S and Perkins ND (2004) Active repression of antiapoptotic gene expression by RelA(p65) NF-κB. Mol. Cell 13: 853–865
Tergaonkar V, Pando M, Vafa O, Wahl G and Verma I (2002) p53 stabilization is decreased upon NF-κB activation: a role for NF-κB in acquisition of resistance to chemotherapy. Cancer Cell 1: 493–503
Lamb JA, Ventura JJ, Hess P, Flavell RA and Davis RJ (2003) JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell 11: 1479–1489
Reuther-Madrid JY, Kashatus D, Chen S, Li X, Westwick J, Davis RJ, Earp HS, Wang C-Y and Baldwin Jr AS (2002) The p65/RelA subunit of NF-κB suppresses the sustained, antiapoptotic activity of Jun kinase induced by tumor necrosis factor. Mol. Cell. Biol. 22: 8175–8183
Zhang JY, Green CL, Tao S and Khavari PA (2004) NF-κB RelA opposes epidermal proliferation driven by TNFR1 and JNK. Genes Dev. 18: 17–22
Liebermann DA and Hoffman B (2002) Myeloid differentiation (MyD) primary response genes in hematopoiesis. Oncogene 21: 3391–3402
Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, De Smaele E, Tang WJ, D'Adamio L and Franzoso G (2004) Gadd45β mediates the NF-κB suppression of JNK signalling by targeting MKK7/JNKK2. Nat. Cell Biol. 2: 146–153
Tournier C, Dong C, Turner TK, Jones SN, Flavell RA and Davis RJ (2001) MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 15: 1419–1426
Takekawa M and Saito H (1998) A family of stress-inducible GADD45-like proteins mediate activation of the stress-responsive MTK1/MEKK4 MAP3K. Cell 95: 521–530
Amanullah A, Azam N, Balliet A, Hollander C, Hoffman B, Fornace A and Liebermann D (2003) Cell signalling: cell survival and a Gadd45-factor deficiency. Nature 424: 741
Zazzeroni F, Papa S, De Smaele E and Franzoso G (2003) Cell signalling: cell survival and a Gadd45-factor deficiency. Nature 424: 742
Ijiri K, Zerbini LF, Peng H, Correa RG, Lu B, Walsh N, Zhao Y, Taniguchi N, Huang XL, Otu H, Wang H, Fei Wang J, Komiya S, Ducy P, Rahman MU, Flavell RA, Libermann TA and Goldring MB (2005) A novel role for Gadd45β as a mediator of MMP-13 gene expression during chondrocyte terminal differentiation. J. Biol. Chem. 280: 38544–38555
Gupta M, Gupta SK, Balliet AG, Hollander MC, Fornace AJ, Hoffman B and Liebermann DA (2005) Hematopoietic cells from Gadd45α- and Gadd45β-deficient mice are sensitized to genotoxic-stress-induced apoptosis. Oncogene 24: 7170–7179
Lu B, Ferrandino AF and Flavell RA (2004) Gadd45β is important for perpetuating cognate and inflammatory signals in T cells. Nat. Immunol. 5: 38–44
Chi H, Lu B, Takekawa M, Davis RJ and Flavell RA (2004) GADD45β/GADD45γ and MEKK4 comprise a genetic pathway mediating STAT4-independent IFNγ production in T cells. EMBO J. 23: 1576–1586
Salvesen GS and Duckett CS (2002) IAP proteins: blocking the road to death's door. Nat. Rev. Mol. Cell. Biol. 3: 401–410
Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR and Lipp J (1998) Nuclear factor (NF)-κB-regulated X-chromosome-linked IAP gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 188: 211–216
Conte D, Liston P, Wong JV, Wight KE and Korneluk RG (2001) Thymocyte-targeted overexpression of xiap transgene disrupts T lymphoid apoptosis and maturation. Proc. Natl. Acad. Sci. USA 98: 5049–5054
Sanna MG, da Silva Correia J, Ducrey O, Lee J, Nomoto K, Schrantz N, Deveraux QL and Ulevitch RJ (2002) IAP suppression of apoptosis involves distinct mechanisms: the TAK1/JNK1 signaling cascade and caspase inhibition. Mol. Cell. Biol. 22: 1754–1766
Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP and Ma A (2000) Failure to regulate TNF-induced NF-κB and cell death responses in A20-deficient mice. Science 289: 2350–2354
Boone DL, Lee EG, Libby S, Gibson PJ, Chien M, Chan F, Madonia M, Burkett PR and Ma A (2002) Recent advances in understanding NF-κB regulation. Inflamm. Bowel Dis. 8: 201–212
Wertz IE, O'Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, Wu P, Wiesmann C, Baker R, Boone DL, Ma A, Koonin EV and Dixit VM (2004) De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature 430: 694–699
He KL and Ting AT (2002) A20 inhibits tumor necrosis factor (TNF)α-induced apoptosis by disrupting recruitment of TRADD and RIP to the TNF receptor 1 complex in Jurkat T cells. Mol. Cell. Biol. 22: 6034–6045
Torti FM and Torti SV (2002) Regulation of ferritin genes and protein. Blood 99: 3505–3516
Arosio P and Levi S (2002) Ferritin, iron homeostasis, and oxidative damage. Free Radic. Biol. Med. 33: 457–463
Vulcano M, Meiss RP and Isturiz MA (2000) Deferoxamine reduces tissue injury and lethality in LPS-treated mice. Int. J. Immunopharmacol. 2: 635–644
Xie C, Zhang N, Zhou H, Li J, Li Q, Zarubin T, Lin SC and Han J (2005) Distinct roles of basal steady-state and induced H-ferritin in tumor necrosis factor-induced death in L929 cells. Mol. Cell. Biol. 25: 6673–6681
Hentze MW, Muckenthaler MU and Andrews NC (2004) Balancing acts: molecular control of mammalian iron metabolism. Cell 117: 285–297
Benhar M, Engelberg D and Levitzki A (2002) ROS, stress-activated kinases and stress signaling in cancer. EMBO Rep. 3: 420–425
Kakhlon O, Gruenbaum Y and Cabantchik ZI (2001) Repression of ferritin expression increases the labile iron pool, oxidative stress, and short-term growth of human erythroleukemia cells. Blood 97: 2863–2871
Russo J, Yang X, Hu YF, Bove BA, Huang Y, Silva ID, Tahin Q, Wu Y, Higgy N, Zekri A and Russo IH (1998) Biological and molecular basis of human breast cancer. Front. Biosci. 3: D944–D960
Delhalle S, Deregowski V, Benoit V, Merville MP and Bours V (2002) NF-κB-dependent MnSOD expression protects adenocarcinoma cells from TNFα-induced apoptosis. Oncogene 21: 3917–3924
Bernard D, Quatannens B, Begue A, Vandenbunder B and Abbadie C (2001) Antiproliferative and antiapoptotic effects of crel may occur within the same cells via the up-regulation of manganese superoxide dismutase. Cancer Res. 61: 2656–2664
Huang P, Feng L, Oldham EA, Keating MJ and Plunkett W (2000) Superoxide dismutase as a target for the selective killing of cancer cells. Nature 407: 390–395
Sasazuki T, Okazaki T, Tada K, Sakon-Komazawa S, Katano M, Tanaka M, Yagita H, Okumura K, Tominaga N, Hayashizaki Y, Okazaki Y and Nakano H (2004) Genome wide analysis of TNF-inducible genes reveals that antioxidant enzymes are induced by TNF and responsible for elimination of ROS. Mol. Immunol. 41: 547–551
Maeda S, Kamata H, Luo JL, Leffert H and Karin M (2005) IKKβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 121: 977–990
Yamada Y, Kirillova I, Peschon JJ and Fausto N. (1997) Initiation of liver growth by tumor necrosis factor: deficient liver regeneration in mice lacking type I tumor necrosis factor receptor. Proc. Natl. Acad. Sci. USA 94: 1441–1446
Schwabe RF, Bradham CA, Uehara T, Hatano E, Bennett BL, Schoonhoven R and Brenner DA (2003) c-Jun-N-terminal kinase drives cyclin D1 expression and proliferation during liver regeneration. Hepatology 37: 824–832
Kennedy NJ and Davis RJ (2003) Role of JNK in tumor development. Cell Cycle 2: 199–201
Lafarge S, Sylvain V, Ferrara M and Bignon YJ (2001) Inhibition of BRCA1 leads to increased chemoresistance to microtubule-interfering agents, an effect that involves the JNK pathway. Oncogene 20: 6597–6606
Frelin C, Imbert V, Griessinger E, Peyron AC, Rochet N, Philip P, Dageville C, Sirvent A, Hummelsberger M, Berard E, Dreano M, Sirvent N and Peyron JF (2005) Targeting NF-κB activation via pharmacologic inhibition of IKK2-induced apoptosis of human acute myeloid leukemia cells. Blood 105: 804–811
Ziegelbauer K, Gantner F, Lukacs NW, Berlin A, Fuchikami K, Niki T, Sakai K, Inbe H, Takeshita K, Ishimori M, Komura H, Murata T, Lowinger T and Bacon KB (2005) A selective novel low-molecular-weight inhibitor of IκB kinase-beta (IKKβ) prevents pulmonary inflammation and shows broad anti-inflammatory activity. Br. J. Pharmacol. 145: 178–192
Dajee M, Lazarov M, Zhang JY, Cai T, Green CL, Russell AJ, Marinkovich MP, Tao S, Lin Q, Kubo Y and Khavari PA (2003) NF-κB blockade and oncogenic Ras trigger invasive human epidermal neoplasia. Nature 421: 639–643
van Hogerlinden M, Auer G and Toftgard R (2002) Inhibition of Rel/nuclear factor-κB signaling in skin results in defective DNA damage-induced cell cycle arrest and H-ras- and p53-independent tumor development. Oncogene 21: 4969–4977
Acknowledgements
We thank L Sherman for helping with manuscript preparation. This research was supported in part by NIH grants R01-CA84040 and R01-CA098583.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by G Kroemer
Rights and permissions
About this article
Cite this article
Papa, S., Bubici, C., Zazzeroni, F. et al. The NF-κB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease. Cell Death Differ 13, 712–729 (2006). https://doi.org/10.1038/sj.cdd.4401865
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401865
Keywords
This article is cited by
-
Ayurvedic formulations Guduchi and Madhuyashti triggers JNK signaling mediated immune response and adversely affects Huntington phenotype
BMC Complementary Medicine and Therapies (2022)
-
Somatostatin analogues in treatment-refractory meningioma: a systematic review with meta-analysis of individual patient data
Neurosurgical Review (2022)
-
Comprehensive high-throughput meta-analysis of differentially expressed microRNAs in transcriptomic datasets reveals significant disruption of MAPK/JNK signal transduction pathway in Adult T-cell leukemia/lymphoma
Infectious Agents and Cancer (2021)
-
Burn injury induces elevated inflammatory traffic: the role of NF-κB
Inflammation Research (2021)
-
Life, death, and autophagy in cancer: NF-κB turns up everywhere
Cell Death & Disease (2020)
