
Abstract
The discovery of B-cell lymphoma-2 (BCL-2) over 20 years ago revealed a new paradigm in cancer biology: the development and persistence of cancer can be driven by molecular roadblocks along the natural pathway to cell death. The subsequent identification of an expansive family of BCL-2 proteins provoked an intensive investigation of the interplay among these critical regulators of cell death. What emerged was a compelling tale of guardians and executioners, each participating in a molecular choreography that dictates cell fate. Ten years into the BCL-2 era, structural details defined how certain BCL-2 family proteins interact, and molecular targeting of the BCL-2 family has since become a pharmacological quest. Although many facets of BCL-2 family death signaling remain a mechanistic mystery, small molecules and peptides that effectively target BCL-2 are eliminating the roadblock to cell death, raising hopes for a medical breakthrough in cancer and other diseases of deregulated apoptosis.
Similar content being viewed by others

Introduction
First identified at the chromosomal breakpoint of t(14;18)(q32;q21) lymphomas, B-cell lymphoma-2 (BCL-2) is the founding member of a family of proteins that regulate cell death.1, 2, 3 Gene rearrangement places BCL-2 under the transcriptional control of the immunoglobulin heavy chain locus, resulting in high-level BCL-2 expression and pathologic cell survival.4, 5 The oncogenic activity of BCL-2 derives from its ability to block cell death following a wide variety of stimuli.6, 7, 8 Transgenic mice bearing a BCL-2-Ig minigene initially displayed a polyclonal follicular lymphoproliferation that selectively expanded a small resting IgM/IgD B-cell population.4, 9 These recirculating B cells accumulated because of an extended survival rather than increased proliferation. Despite a fourfold increase in resting B cell counts, BCL-2-Ig mice were initially quite healthy. However, over time these transgenics progressed from an indolent follicular hyperplasia to a diffuse large cell, and often immunoblastic, lymphoma.10 The long latency period and progression from polyclonal hyperplasia to monoclonal high-grade malignancy implicated secondary genetic abnormalities in BCL-2-driven lymphomagenesis. Indeed, approximately half of the high-grade tumors possessed a c-myc translocation involving an immunoglobulin heavy-chain locus.10 These tumor cells complemented an inherent survival advantage (bcl-2) with a gene that promotes proliferation (c-myc). By preventing the apoptotic demise of activated lymphocytes, BCL-2 enabled the acquisition of additional genetic aberrations and the emergence of monoclonal neoplasms. Doubly transgenic mice engineered to overexpress both BCL-2 and c-myc displayed synergistic tumorigenesis.11 When leukemic mice with doubly deregulated BCL-2 and c-myc were conditionally induced to cease BCL-2 expression, tumor regression was observed, confirming a role for BCL-2 in tumor maintenance.12 Thus, the discovery of BCL-2 established the new paradigm in cancer biology that prolonging cell survival by evasion of apoptosis can both initiate and sustain cancer.
The BCL-2 family has expanded significantly and now includes both pro- and antiapoptotic proteins, which form a complex network of checks and balances that regulate cell fate.13, 14 Disrupting the balance imposed by the BCL-2 family can lead to a host of human conditions that are characterized by excessive cellular demise, such as in neurodegenerative disease15 or relentless cellular survival, such as in cancer16 (Figure 1). BCL-2 proteins are defined both by their structure17 and function (Figure 2). The survival proteins such as BCL-2 and BCL-XL share three to four conserved BCL-2 homology (BH1-4) domains, and are thus termed ‘multidomain antiapoptotic’ members. The executioner proteins such as BAX and BAK share three conserved domains (BH1-3) and are known as ‘multidomain proapoptotic’ proteins. A subgroup of proapoptotic proteins only displays conservation in the third BH domain. These ‘BH3-only’ members function as death sentinels that are situated throughout the cell, poised to transmit signals of cellular injury through multidomain members. A variety of physiological death signals, as well as pathological cellular insults, trigger the genetically programmed pathway of apoptosis13, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40 (Figure 3). Depending upon the nature of apoptotic stimuli and the cellular context, a BH3-only protein's death signal will either be neutralized by antiapoptotic proteins or delivered, directly or indirectly, to the mitochondrial executioners BAX and BAK. When activated, these proapoptotic multidomain members induce permeabilization of the outer mitochondrial membrane, enabling released mitochondrial factors to activate caspases, which irreversibly execute the death program.

Apoptosis maintains tissue homeostasis by balancing cellular life and death. Deregulated apoptotic pathways disrupt the balance, resulting in diseases of premature cell loss or unrelenting cell survival

BCL-2 family proteins are structurally defined by their BCL-2 homology domains (BH domains) and functionally categorized by their ability to inhibit or activate cell death. Human BCL-2 family proteins are drawn to scale based upon polypeptide length and aligned by their BH3 domains

Cytotoxic signals activate the apoptotic program through diverse pathways, recruiting distinct BH3-only members to engage downstream multidomain BCL-2 family proteins
The network of interactions among BCL-2 family members is complex and remains a focus of intensive investigation. Diverse cellular signaling pathways engage the apoptotic program by launching particular BH3-only proteins41, 42 (Figure 3). For example, activation of death receptors Fas and tumor necrosis factor receptor 1 triggers caspase-8-induced cleavage of BH3-only protein BID.43, 44 The amino terminus of truncated BID (tBID) becomes myristolated, which facilitates its mitochondrial targeting and subsequent transmission of a plasma membrane death signal to multidomain BCL-2 family members at the mitochondria.45 In the DNA-damage response, nuclear p53 induces gene transcription of BH3-only PUMA,46 which in turn displaces cytoplasmic p53 from BCL-XL, enabling direct protein interaction-based activation of BAX by p53.47, 48 Just as distinct signaling pathways employ specific BH3-only proteins, BH3-only proteins display sequence-dependent specificity for their target multidomain antiapoptotic proteins.12, 49 The ability of antiapoptotic proteins to form heterodimers with multidomain proapoptotic proteins suggests that neutralizing competition plays an important role in their suppression of cell death.50, 51, 52, 53, 54, 55, 56 BH3-only proteins can promote apoptosis by antagonizing antiapoptotic proteins, and thereby relieve antiapoptotic inhibition of BAX/BAK. Such BH3-only proteins (e.g. BAD) have been termed ‘sensitizers’57 or ‘derepressors’.58 Alternatively, select BH3-only proteins such as BID and BIM may also trigger BAX/BAK directly,22, 58, 59, 60, 61, 62 and are therefore termed ‘activators’.57 In addition to these internal family-based interactions, BCL-2 proteins associate with a host of other cellular proteins31, 47, 63, 64, 65, 66, 67, 68, 69, 70, 71 and have emerging roles in diverse physiologic pathways including glucose metabolism20 and the DNA-damage response.24, 40
BCL-2 Family Form and Function
A pivotal milestone in the apoptosis field was achieved in 1996 when the first X-ray and nuclear magnetic resonance (NMR) structure of a BCL-2 family protein was reported72 (Figure 4a). The architecture of BCL-XL consists of eight α-helices, two of which form a central hydrophobic core reminiscent of the membrane insertion domains of pore-forming Diphtheria toxin and colicins.72 This structural analogy led to experimental confirmation that BCL-2 family members can mediate pore formation in liposomal and mitochondrial systems,73, 74, 75, 76 an activity that is dependent upon core helices 5 and 6.76, 77, 78 Another critical architectural feature of BCL-XL was identified on its protein surface, a hydrophobic groove formed at the apex by the confluence of BH1, 2, and 3 domains and at the base by α-helices 3 and 4. The structure of a BAK BH3 peptide in complex with BCL-XL revealed that the hydrophobic groove was indeed the contact site for proapoptotic binding53 (Figure 4b). Thus, the protein interaction that accounts for BCL-2 family member homo- and heteroassociations,50, 54, 56 and believed to regulate pore formation, was explicitly defined. The subsequent NMR structures of BCL-2,79 a BAD BH3 peptide/BCL-XL complex,80 BCL-w81, 82, 83 and MCL-184 highlighted the three-dimensional theme of an antiapoptotic hydrophobic groove fashioned to engage the BH3 death helix of proapoptotic members (Figure 4b). Discrete differences in the amino-acid composition among antiapoptotic grooves and BH3 ligands dictate the specificity of apoptotic-binding partners.49, 57, 79, 80, 84 With an essential rule of engagement structurally defined, the mechanics of BCL-2 family interactions came into focus.

The structures of antiapoptotic BCL-2 family members (a) and their BH3 peptide complexes (b) revealed a multidomain hydrophobic groove that serves as the critical binding interface for α-helical BH3 domains. Hydrophobic residues are indicated in green and BH3-binding grooves bracketed in black. Despite their opposing functions, pro- and antiapoptotic multidomain members share overall structural similarities, including two hydrophobic core α-helices surrounded by 6–7 amphipathic α-helices, a flexible N-terminal loop, a surface hydrophobic groove, and a C-terminal transmembrane domain (a, c). BH3-only BID contains an extended amino terminus that contains the cleavage site for caspase-8, which triggers BID activation upon death receptor engagement (c). BAX and BCL-w contain a structurally defined C-terminal ninth helix (α9) that may regulate access to the BH3-binding groove (c, d). A comparison of BCL-w structures with and without BID BH3 peptide binding demonstrates how α9 (red) overlies the binding pocket until displaced by the BH3 helix (blue), which targets the hydrophobic groove (d)
On the proapoptotic side, NMR structures of BH3-only BID85, 86 and multidomain proapoptotic BAX52 disclosed striking architectural similarities between the proponents and opponents of cell death (Figure 4c). BID and BAX likewise possess two central core helices that are surrounded by 6 or 7 amphipathic helices, respectively. The amino-terminal portions of BID and BAX contain unstructured loops (Figure 4c), as do select antiapoptotic proteins such as BCL-2 and BCL-XL. This loop region has distinctive lengths and primary sequences among BID, BAX, and the antiapoptotics BCL-2 and BCL-XL, and is believed to regulate their apoptotic functions. For example, phosphorylation within the loop region of BCL-2 differentially modulates its antiapoptotic activity depending upon the cellular context,87, 88 and caspase-mediated cleavage at the loop can actually transform BCL-2 into a proapoptotic protein.64 Caspase-8-mediated cleavage of BID within the unstructured loop results in exposure of the BH3 helix of tBID,85, 86 which is targeted to the mitochondria for apoptosis induction.43 The amino terminus of BAX has been implicated in BH3 ligand binding,59 intracellular localization,89, 90 and negative regulation.91, 92 Calpain-mediated cleavage of BAX just prior to its relatively short unstructured loop generates a truncated form with enhanced apoptogenic activity,93, 94, 95 which may reflect its functional conversion to a BH3-only-type protein.96
Multidomain pro- and antiapoptotic proteins contain C-terminal transmembrane domains that insert into the mitochondrial outer membrane. BH3-only BID lacks this C-terminal domain and interacts with lipid membrane with its helices parallel to the surface rather than by transmembrane insertion.97, 98 Whereas the constructs for the BCL-XL, BCL-2, and MCL-1 structures lack the C-terminal transmembrane region, proapoptotic BAX and antiapoptotic BCL-w have a structurally defined ninth α-helix (α9) at the C-terminus52, 82, 83 (Figure 4c and d). These α9 helices fold back into the hydrophobic groove in an orientation opposite to that delineated for the interactions of BAK and BAD BH3 peptides with BCL-XL. The C-terminal helix effectively blocks both access to the groove and exposure of the BH3 domain and C-terminal hydrophobic residues (Figure 4d). Indeed, binding of BH3 peptides to the hydrophobic groove of BCL-w is impaired by α-9, as reflected by decreased BH3 peptide affinity for full-length versus C-terminally truncated BCL-w.82, 83 The distinctive C-terminal structure likely accounts for the cytosolic disposition of BAX and BCL-w by optimizing solubility until triggered to undergo a conformational change, which releases α9 for membrane insertion.52, 82, 83, 99 By obstructing the protein interaction site, α9 may also contribute to maintaining BAX and BCL-w in the monomeric form. Of note, α9 of BCL-w is less hydrophobic and more mobile than that of BAX by NMR, suggesting that the activation criteria for BAX α9 disengagement and resultant BAX translocation are stringent by design. Consistent with the need for tight structural control over BAX activation, numerous proteins in addition to BCL-2 family antiapoptotics have been identified that bind and inhibit BAX or BAK.63, 64, 66, 67, 69
The published structures of BCL-XL, BCL-2, BID, BAX, BCL-w, MCL-1, and several of their complexes with BH3 peptides have provided tremendous insights into the functional roles of BCL-2 family members and the protein interactions that enable their apoptotic activities. The explicit mechanics of how BCL-2 family members regulate mitochondrial pore formation, and how select BH3-only members may engage proapoptotic multidomain proteins, remain active areas of investigation. Structural studies that evaluate BCL-2 family activities and interactions in the lipid environment continue to provide new details regarding the complexity of BCL-2 family conformational changes that occur during mitochondrial apoptosis induction.97, 98, 100, 101, 102 Most importantly, structural delineation of the helical folds responsible for forming both a multidomain groove and its BH3 ligand established a rational means for targeting apoptosis by chemical design.
Getting into the Groove
The fields of chemical genetics and developmental therapeutics share the mission of identifying small molecules that directly and specifically alter protein function so that physiologic activities can be investigated and manipulated on a conditional basis in real-time. The objective of generating small molecules to selectively target apoptotic protein interactions and specifically manipulate their corresponding pathways in vivo has been challenging due to the size and complexity of the intracellular protein-binding interface. However, virtual and small molecule screens, in addition to peptidomimetic, secondary structure reinforcement, and NMR-based strategies, have yielded a diverse group of compounds that target the BCL-2 family hydrophobic groove (Figures 5 and 6, Table 1). The development of these compounds as biological tools and clinical candidates promises to deepen our understanding of BCL-2 family biology and deliver a new era of treatments to patients suffering from oncologic, neurodegenerative, autoimmune, and a host of other diseases characterized by an imbalance between cell survival and death.

Virtual and biochemical small molecule screening, NMR-based methodologies, and parallel synthesis have yielded a heterogeneous group of antiapoptotic groove binders

Strategies to reinforce BH3 peptide α-helicity or chemically simulate key projections of the BH3 helix on a synthetic scaffold have generated peptidic and peptidomimetic compounds for BCL-2 family targeting
The Hunt for Small Molecules
One of the earlier strategies that successfully identified BCL-2 inhibitors involved computer-based screening of small molecule databases for structures that matched likely binding sites on the surface of BCL-2. This approach yielded HA14–1 (IC50=9 μM for competing BAK BH3/BCL-2 interaction),103 several micromolar affinity hits from the National Cancer Institute 3D database including Compound 6,104 and YC137 (IC50=1.3 μM for competing BID BH3/BCL-2 interaction)105 (Figure 5a–c). Antimycin A3 emerged as a BCL-2 groove binder (Kd=0.82 μM) upon screening mitochondrial respiration inhibitors for proapoptotic activity in hepatocyte cell lines with graded expression of BCL-XL106, 107 (Figure 5d). A BAK BH3/BCL-XL competitive binding assay-based screen identified two small molecule inhibitors, BH3I-1 and BH3I-2, with Kis in the 2–16 μM range108 (Figure 5e–f). Similar binding assay strategies were used to screen natural product libraries, identifying chelerythrine (IC50=1.5 μM, BAK BH3/BCL-XL competition),109 gossypol (IC50=0.5 μM, BAD BH3/BCL-XL competition),110, 111 and purpurogallin (IC50=2.2 μM, BAD BH3/BCL-XL competition)110 as BCL-XL groove binders (Figure 5g–j). Natural polyphenols found in extracts of green tea (e.g. epigallocatechingallate) and black tea (e.g. theaflavanin) compete with BAD BH3 for BCL-XL and BCL-2 binding with Kis in the 120–1230 nanomolar range112 (Figure 5k and l). A polypyrrole derivative identified from a natural compound library screen was developed to yield GX15-070, which binds to BCL-XL, BCL-w, and MCL-1 in the 500 nM range113, 114 (Figure 5m).
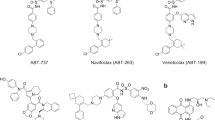
A major hurdle of small molecule library screening approaches, whether virtual or biochemical, is the chemical optimization required to achieve high-potency target-binding activity from hits that are typically in the micromolar range. An alternative strategy developed by Fesik and co-workers115 circumvents this shortcoming of library screening by chemically linking ligands that bind to adjacent sites within a target interface, effectively converting relatively low affinity interactors into conjoined high-affinity compounds. The method is called structure–activity relationships (SAR) by NMR, reflecting that compound optimization is derived from SAR determined by NMR. One of the seminal fruits of this strategy is ABT-737, a small molecule that binds to BCL-2, BCL-XL, and BCL-w at subnanomolar affinity and demonstrates potent antitumor activity in vitro and in vivo116 (Figure 5n). The SAR by NMR approach has recently been combined with parallel synthesis to generate optimized BCL-XL inhibitors117, 118 (Figure 5o and p).
Rebuilding the BH3 Helix
The nanomolar-binding affinities and native selectivities of BH3 peptides for their antiapoptotic targets prompted the development of derivatized peptides for BCL-2 family targeting. In order to overcome cell impermeability, cell penetrating peptides were synthesized by tagging the BH3 domain with moieties that facilitate uptake, including Antennapedia119, 120, 121 and poly-D-arginine57 amino-acid sequences, and fatty acids such as decanoic acid.122 Despite their cell permeability, several drawbacks of these derivatized peptides include loss of α-helical structure, protease sensitivity, low cellular potency, and in certain circumstances apoptosis induction independent of BCL-2 targeting.119
Short peptides taken out of context from a protein typically lack native three-dimensional structure, and therefore, may fail to exhibit biologic functionality at physiologic doses. As the α-helix participates in a wide variety of intermolecular biological recognition events, a major focus of modern organic chemistry is the development of synthetic strategies to mimic or stabilize the architecture of biologically active structures for both basic research and medicinal purposes (Figure 6). Schepartz and co-workers123, 124 developed a protein grafting strategy in which bioactive α-helical residues, such as BAK BH3 peptide, are inserted into a stable protein scaffold, and then subjected to rounds of phage display to evolve high-affinity ligands with preferences for BCL-2 or BCL-XL (Figure 6a). In lieu of the natural amide backbone of α-helical peptides, synthetic scaffolds have been developed that effectively present critical amino acid residues to the antiapoptotic groove. For example, constructing compounds using terphenyl,125, 126 terpyridine,127 or terephthalamide128 scaffolds that project essential BH3 motifs yielded groove binders with micromolar and nanomolar Kds (Figure 6b). Gellman and co-workers129 developed BAK BH3 ‘foldamers’ that are peptidic compounds built from oligomers with defined folding propensities. Chimeric peptides that juxtapose native BAK BH3 sequence with alternating α- and β-peptide foldamer sequence can also present critical groove-binding motifs, resulting in compounds with nanomolar-binding affinities for BCL-XL(Figure 6c).
An important strategic breakthrough in stabilizing natural α-helices derived from installing a covalent bond between amino acids in an attempt to ‘lock’ the peptide structure into place.130, 131, 132, 133, 134 The incorporation of lactam bridges into the BAK BH3 peptide increased α-helical content from 14% to as high as 78%135 (Figure 6d). However, stabilization methods that incorporate polar or labile crosslinks may not address peptide shortcomings of instability and cell impermeability in vivo. Grubbs and co-workers133 generated a covalent crosslink between O-allyl serine residues on adjacent turns of an α-helix using ruthenium-catalyzed ring closing metathesis (RCM), which employs a metal catalyst to form a covalent bond between non-natural amino acid residues containing terminal double bonds or ‘olefins’. This novel chemical approach was successful in generating a covalent hydrocarbon crosslink, but little to no enhancement of peptide α-helicity was observed. Subsequently, Verdine and co-workers134 developed an alternate ‘olefin metathesis’-based approach, which employed α,α-disubstituted non-natural amino acids containing alkyl tethers (Figure 6e). By experimenting with alternative placement of these non-natural amino acids along the α-helix, in addition to varying stereochemistry and alkyl tether length, the chemical features required to dramatically stabilize a model helical peptide using an all-hydrocarbon chain crosslink were defined.
Stabilizing the helical form of biologically active peptides is expected to favor target binding by facilitating structural preorganization.136 Furthermore, helix formation buries the polar amide backbone, which should increase resistance to proteolytic cleavage and decrease the barrier to cell penetration. In our first biological application of this chemical approach, we developed and tested Stabilized Alpha-Helices of Bcl-2 domains, or ‘SaHBs’, modeled after BID BH3.137 An all-hydrocarbon staple inserted into the BID BH3 peptide sequence successfully (1) restored and stabilized α-helical structure, (2) enhanced peptide half-life, (3) improved binding potency, and (4) conferred cellular permeability such that the genetic pathway of apoptosis could be reactivated in cancer cells in vitro and in vivo. Thus, synthetic approaches such as ‘hydrocarbon stapling’ that reinforce native peptide sequences offer an alternative strategy for studying and manipulating protein interactions, and provide prototypes for novel therapeutics designed to target aberrant signaling pathways in human disease. The appeal of this strategy includes retaining the complexity, potency, and specificity of natural bioactive peptide sequences, the short timeframe for compound development, and the potential for broad applicability in targeting cell surface, intracellular, and organelle-based protein interactions.
Neutralizing the Proapoptotics
Small molecules that target apoptotic protein interfaces other than the multidomain antiapoptotic groove have also been identified. Using the strategy named SAR by interligand nuclear Overhauser effect (ILOEs), of Reed and co-workers138 designed a panel of 4-phenylsulfanyl-phenylamine compounds that bind a deep groove on the surface of BID with micromolar affinity. The molecules inhibited tBID-induced mitochondrial SMAC release, caspase-3 activation, and cell death in the 20–100 μM range. 3,6-Dibromocarbazole piperazine derivatives of 2-propanol were developed as the first small molecule modulators of BAX-induced mitochondrial and liposomal release activity.139 Subsequently, a small molecule screen using a BAX-induced liposomal release assay identified two additional BAX channel blockers.140 The Bax channel inhibitors Bci1 and Bci2 prevented cytochrome c release from mitochondria and protected cells from apoptosis in vitro at micromolar dosing. Bcis were neuroprotective in an in vivo model of transient brain ischemia. Peptides derived from humanin63 and Ku70141 proteins also interact with BAX and inhibit its activation. Thus, in addition to targeting the antiapoptotic groove to stimulate cell death, small molecule and peptide-based approaches to inhibiting proapoptotic proteins may lead to the development of cytoprotective therapeutics.
Toward a Therapeutic Reality
From a 20-year multidisciplinary dissection of BCL-2 family interactions and pathways has emerged the promise of novel therapeutics to treat human disease. More than a dozen small molecules and peptidic compounds are currently in preclinical and clinical development for targeting the structurally defined multidomain antiapoptotic groove113, 142 (Table 1). Preclinical development of small molecule and peptide inhibitors of proapoptotic BCL-2 family proteins is also underway. A BCL-2 antisense therapeutic,143, 144, 145, 146 Genasense, is in Phase III clinical testing. For the clinicians and scientists who discovered BCL-2 and for those who have subsequently dedicated their lives' work to elucidating and targeting the BCL-2 family of proteins, successful translation of these efforts into the clinic in the form of FDA-approved medicines will be a crowning achievement.
Conflict of Interest
LDW is an advisor to Renegade Therapeutics, an emerging biopharmaceutical company developing next-generation targeted therapeutics.
Abbreviations
- BCL-2:
-
B-cell lymphoma-2
- BH:
-
BCL-2 homology
- NMR:
-
nuclear magnetic resonance
- SAR:
-
structure-activity relationship
- RCM:
-
ring-closing metathesis
- SAHB:
-
stabilized alpha-helices of BCL-2 domains
- ILOE:
-
interligand nuclear Overhauser effect
References
Tsujimoto Y, Gorham J, Cossman J, Jaffe E and Croce CM (1985) The t(14;18) chromosome translocations involved in B-cell neoplasms result from mistakes in VDJ joining. Science 229: 1390–1393.
Cleary ML and Sklar J (1985) Nucleotide sequence of a t(14;18) chromosomal breakpoint in follicular lymphoma and demonstration of a breakpoint-cluster region near a transcriptionally active locus on chromosome 18. Proc. Natl. Acad. Sci. USA 82: 7439–7443.
Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL and Korsmeyer SJ (1985) Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18. Cell 41: 899–906.
McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP and Korsmeyer SJ (1989) bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell 57: 79–88.
Seto M, Jaeger U, Hockett RD, Graninger W, Bennett S, Goldman P and Korsmeyer SJ (1988) Alternative promoters and exons, somatic mutation and deregulation of the Bcl-2-Ig fusion gene in lymphoma. EMBO J. 7: 123–131.
Vaux DL, Cory S and Adams JM (1988) Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 335: 440–442.
Nunez G, Seto M, Seremetis S, Ferrero D, Grignani F, Korsmeyer SJ and Dalla-Favera R (1989) Growth- and tumor-promoting effects of deregulated BCL2 in human B-lymphoblastoid cells. Proc. Natl. Acad. Sci. USA 86: 4589–4593.
Hockenbery D, Nunez G, Milliman C, Schreiber RD and Korsmeyer SJ (1990) Bcl-2 is an inner mitochondrial membrane protein that blocks programmed cell death. Nature 348: 334–336.
McDonnell TJ, Nunez G, Platt FM, Hockenberry D, London L, McKearn JP and Korsmeyer SJ (1990) Deregulated Bcl-2-immunoglobulin transgene expands a resting but responsive immunoglobulin M and D-expressing B-cell population. Mol. Cell Biol. 10: 1901–1907.
McDonnell TJ and Korsmeyer SJ (1991) Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14; 18). Nature 349: 254–256.
Strasser A, Harris AW, Bath ML and Cory S (1990) Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348: 331–333.
Letai A, Sorcinelli MD, Beard C and Korsmeyer SJ (2004) Antiapoptotic BCL-2 is required for maintenance of a model leukemia. Cancer Cell 6: 241–249.
Cory S and Adams JM (2002) The Bcl2 family: regulators of the cellular life-or-death switch. Nat. Rev. Cancer 2: 647–656.
Danial NN and Korsmeyer SJ (2004) Cell death: critical control points. Cell 116: 205–219.
Raff MC, Whitmore AV and Finn JT (2002) Axonal self-destruction and neurodegeneration. Science 296: 868–871.
Baliga BC and Kumar S (2002) Role of Bcl-2 family of proteins in malignancy. Hematol. Oncol. 20: 63–74.
Petros AM, Olejniczak ET and Fesik SW (2004) Structural biology of the Bcl-2 family of proteins. Biochim. Biophys. Acta. 1644: 83–94.
Barry M, Heibein JA, Pinkoski MJ, Lee SF, Moyer RW, Green DR and Bleackley RC (2000) Granzyme B short-circuits the need for caspase 8 activity during granule-mediated cytotoxic T-lymphocyte killing by directly cleaving Bid. Mol. Cell Biol. 20: 3781–3794.
Bruick RK (2000) Expression of the gene encoding the proapoptotic Nip3 protein is induced by hypoxia. Proc. Natl. Acad. Sci. USA 97: 9082–9087.
Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP and Korsmeyer SJ (2003) BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424: 952–956.
Fukazawa H, Noguchi K, Masumi A, Murakami Y and Uehara Y (2004) BimEL is an important determinant for induction of anoikis sensitivity by mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitors. Mol. Cancer Ther. 3: 1281–1288.
Harada H, Quearry B, Ruiz-Vela A and Korsmeyer SJ (2004) Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc. Natl. Acad. Sci. USA 101: 15313–15317.
Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL and Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328.
Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, Haimovich G, Lerenthal Y, Marcellus RC and Gross A (2005) Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 122: 593–603.
Kim JY, Ahn HJ, Ryu JH, Suk K and Park JH (2004) BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J. Exp. Med. 199: 113–124.
Kuribara R, Honda H, Matsui H, Shinjyo T, Inukai T, Sugita K, Nakazawa S, Hirai H, Ozawa K and Inaba T (2004) Roles of Bim in apoptosis of normal and Bcr-Abl-expressing hematopoietic progenitors. Mol. Cell Biol. 24: 6172–6183.
Lei K and Davis RJ (2003) JNK phosphorylation of Bim-related members of the Bcl2 family induces Bax-dependent apoptosis. Proc. Natl. Acad. Sci. USA 100: 2432–2437.
Li J, Lee B and Lee AS (2006) Endoplasmic Reticulum Stress-induced Apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J. Biol. Chem. 281: 7260–7270.
Mathai JP, Germain M and Shore GC (2005) BH3-only BIK regulates BAX, BAK-dependent release of Ca2+ from endoplasmic reticulum stores and mitochondrial apoptosis during stress-induced cell death. J. Biol. Chem. 280: 23829–23836.
Nikrad M, Johnson T, Puthalalath H, Coultas L, Adams J and Kraft AS (2005) The proteasome inhibitor bortezomib sensitizes cells to killing by death receptor ligand TRAIL via BH3-only proteins Bik and Bim. Mol. Cancer Ther. 4: 443–449.
Puthalakath H, Villunger A, O'Reilly LA, Beaumont JG, Coultas L, Cheney RE, Huang DC and Strasser A (2001) Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science 293: 1829–1832.
Qin JZ, Ziffra J, Stennett L, Bodner B, Bonish BK, Chaturvedi V, Bennett F, Pollock PM, Trent JM, Hendrix MJ, Rizzo P, Miele L and Nickoloff BJ (2005) Proteasome inhibitors trigger NOXA-mediated apoptosis in melanoma and myeloma cells. Cancer Res. 65: 6282–6293.
Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME and Korsmeyer SJ (2003) Bad-deficient mice develop diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 100: 9324–9329.
Reginato MJ, Mills KR, Paulus JK, Lynch DK, Sgroi DC, Debnath J, Muthuswamy SK and Brugge JS (2003) Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 5: 733–740.
Ruefli AA, Ausserlechner MJ, Bernhard D, Sutton VR, Tainton KM, Kofler R, Smyth MJ and Johnstone RW (2001) The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc. Natl. Acad. Sci. USA 98: 10833–10838.
Valentijn AJ and Gilmore AP (2004) Translocation of full-length Bid to mitochondria during anoikis. J. Biol. Chem. 279: 32848–32857.
Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausserlechner MJ, Adams JM and Strasser A (2003) p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 302: 1036–1038.
Yu J, Wang Z, Kinzler KW, Vogelstein B and Zhang L (2003) PUMA mediates the apoptotic response to p53 in colorectal cancer cells. Proc. Natl. Acad. Sci. USA 100: 1931–1936.
Zhang Y, Adachi M, Kawamura R and Imai K (2006) Bmf is a possible mediator in histone deacetylase inhibitors FK228 and CBHA-induced apoptosis. Cell Death Differ. 13: 129–140.
Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A and Korsmeyer SJ (2005) A role for proapoptotic BID in the DNA-damage response. Cell 122: 579–591.
Willis SN and Adams JM (2005) Life in the balance: how BH3-only proteins induce apoptosis. Curr. Opin. Cell Biol. 17: 617–625.
Huang DC and Strasser A (2000) BH3-Only proteins-essential initiators of apoptotic cell death. Cell 103: 839–842.
Li H, Zhu H, Xu CJ and Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501.
Luo X, Budihardjo I, Zou H, Slaughter C and Wang X (1998) Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 94: 481–490.
Zha J, Weiler S, Oh KJ, Wei MC and Korsmeyer SJ (2000) Posttranslational N-myristoylation of BID as a molecular switch for targeting mitochondria and apoptosis. Science 290: 1761–1765.
Nakano K and Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 7: 683–694.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M and Green DR (2004) Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 303: 1010–1014.
Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD and Green DR (2005) PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science 309: 1732–1735.
Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM and Huang DC (2005) Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol. Cell. 17: 393–403.
Diaz JL, Oltersdorf T, Horne W, McConnell M, Wilson G, Weeks S, Garcia T and Fritz LC (1997) A common binding site mediates heterodimerization and homodimerization of Bcl-2 family members. J. Biol. Chem. 272: 11350–11355.
Gross A, Jockel J, Wei MC and Korsmeyer SJ (1998) Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17: 3878–3885.
Suzuki M, Youle RJ and Tjandra N (2000) Structure of Bax: coregulation of dimer formation and intracellular localization. Cell 103: 645–654.
Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB and Fesik SW (1997) Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275: 983–986.
Sedlak TW, Oltvai ZN, Yang E, Wang K, Boise LH, Thompson CB and Korsmeyer SJ (1995) Multiple Bcl-2 family members demonstrate selective dimerizations with Bax. Proc. Natl. Acad. Sci. USA 92: 7834–7838.
Oltvai ZN, Milliman CL and Korsmeyer SJ (1993) Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell 74: 609–619.
Wang K, Gross A, Waksman G and Korsmeyer SJ (1998) Mutagenesis of the BH3 domain of BAX identifies residues critical for dimerization and killing. Mol. Cell Biol. 18: 6083–6089.
Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S and Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192.
Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR and Newmeyer DD (2005) BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol. Cell. 17: 525–535.
Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P, Meflah K, Vallette FM and Juin P (2004) The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol. Cell. 16: 807–818.
Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR and Newmeyer DD (2002) Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 111: 331–342.
Terrones O, Antonsson B, Yamaguchi H, Wang HG, Liu J, Lee RM, Herrmann A and Basanez G (2004) Lipidic pore formation by the concerted action of proapoptotic BAX and tBID. J. Biol. Chem. 279: 30081–30091.
Marani M, Tenev T, Hancock D, Downward J and Lemoine NR (2002) Identification of novel isoforms of the BH3 domain protein Bim which directly activate Bax to trigger apoptosis. Mol. Cell Biol. 22: 3577–3589.
Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC and Reed JC (2003) Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature 423: 456–461.
Cheng EH, Sheiko TV, Fisher JK, Craigen WJ and Korsmeyer SJ (2003) VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science 301: 513–517.
Ohtsuka T, Ryu H, Minamishima YA, Macip S, Sagara J, Nakayama KI, Aaronson SA and Lee SW (2004) ASC is a Bax adaptor and regulates the p53-Bax mitochondrial apoptosis pathway. Nat. Cell Biol. 6: 121–128.
Sawada M, Sun W, Hayes P, Leskov K, Boothman DA and Matsuyama S (2003) Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat. Cell Biol. 5: 320–329.
Xu Q and Reed JC (1998) Bax inhibitor-1, a mammalian apoptosis suppressor identified by functional screening in yeast. Mol. Cell. 1: 337–346.
Leu JI, Dumont P, Hafey M, Murphy ME and George DL (2004) Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell Biol. 6: 443–450.
Zhang H, Kim JK, Edwards CA, Xu Z, Taichman R and Wang CY (2005) Clusterin inhibits apoptosis by interacting with activated Bax. Nat. Cell Biol. 7: 909–915.
Puthalakath H, Huang DC, O'Reilly LA, King SM and Strasser A (1999) The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex. Mol. Cell. 3: 287–296.
Zha J, Harada H, Yang E, Jockel J and Korsmeyer SJ (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87: 619–628.
Muchmore SW, Sattler M, Liang H, Meadows RP, Harlan JE, Yoon HS, Nettesheim D, Chang BS, Thompson CB, Wong SL, Ng SL and Fesik SW (1996) X-ray and NMR structure of human Bcl-xL, an inhibitor of programmed cell death. Nature 381: 335–341.
Saito M, Korsmeyer SJ and Schlesinger PH (2000) BAX-dependent transport of cytochrome c reconstituted in pure liposomes. Nat. Cell Biol. 2: 553–555.
Narita M, Shimizu S, Ito T, Chittenden T, Lutz RJ, Matsuda H and Tsujimoto Y (1998) Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl. Acad. Sci. USA 95: 14681–14686.
Nouraini S, Six E, Matsuyama S, Krajewski S and Reed JC (2000) The putative pore-forming domain of Bax regulates mitochondrial localization and interaction with Bcl-X(L). Mol. Cell Biol. 20: 1604–1615.
Schendel SL, Xie Z, Montal MO, Matsuyama S, Montal M and Reed JC (1997) Channel formation by antiapoptotic protein Bcl-2. Proc. Natl. Acad. Sci. USA 94: 5113–5118.
Heimlich G, McKinnon AD, Bernardo K, Brdiczka D, Reed JC, Kain R, Kronke M and Jurgensmeier JM (2004) Bax-induced cytochrome c release from mitochondria depends on alpha-helices-5 and -6. Biochem. J. 378: 247–255.
Garcia-Saez AJ, Coraiola M, Serra MD, Mingarro I, Muller P and Salgado J (2006) Peptides corresponding to helices 5 and 6 of Bax can independently form large lipid pores. FEBS. J. 273: 971–981.
Petros AM, Medek A, Nettesheim DG, Kim DH, Yoon HS, Swift K, Matayoshi ED, Oltersdorf T and Fesik SW (2001) Solution structure of the antiapoptotic protein bcl-2. Proc. Natl. Acad. Sci. USA 98: 3012–3017.
Petros AM, Nettesheim DG, Wang Y, Olejniczak ET, Meadows RP, Mack J, Swift K, Matayoshi ED, Zhang H, Thompson CB and Fesik SW (2000) Rationale for Bcl-xL/Bad peptide complex formation from structure, mutagenesis, and biophysical studies. Protein Sci. 9: 2528–2534.
Denisov AY, Chen G, Sprules T, Moldoveanu T, Beauparlant P and Gehring K (2006) Structural model of the BCL-w-BID peptide complex and its interactions with phospholipid micelles. Biochemistry 45: 2250–2256.
Denisov AY, Madiraju MS, Chen G, Khadir A, Beauparlant P, Attardo G, Shore GC and Gehring K (2003) Solution Structure of Human BCL-w: modulation of ligand binding by the C-terminal helix. J. Biol. Chem. 278: 21124–21128.
Hinds MG, Lackmann M, Skea GL, Harrison PJ, Huang DC and Day CL (2003) The structure of Bcl-w reveals a role for the C-terminal residues in modulating biological activity. EMBO J. 22: 1497–1507.
Day CL, Chen L, Richardson SJ, Harrison PJ, Huang DC and Hinds MG (2005) Solution structure of prosurvival Mcl-1 and characterization of its binding by proapoptotic BH3-only ligands. J. Biol. Chem. 280: 4738–4744.
McDonnell JM, Fushman D, Milliman CL, Korsmeyer SJ and Cowburn D (1999) Solution structure of the proapoptotic molecule BID: a structural basis for apoptotic agonists and antagonists. Cell 96: 625–634.
Chou JJ, Li H, Salvesen GS, Yuan J and Wagner G (1999) Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 96: 615–624.
Ito T, Deng X, Carr B and May WS (1997) Bcl-2 phosphorylation required for anti-apoptosis function. J. Biol. Chem. 272: 11671–11673.
Srivastava RK, Mi QS, Hardwick JM and Longo DL (1999) Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc. Natl. Acad. Sci. USA 96: 3775–3780.
Cartron PF, Priault M, Oliver L, Meflah K, Manon S and Vallette FM (2003) The N-terminal end of Bax contains a mitochondrial-targeting signal. J. Biol. Chem. 278: 11633–11641.
Cartron PF, Moreau C, Oliver L, Mayat E, Meflah K and Vallette FM (2002) Involvement of the N-terminus of Bax in its intracellular localization and function. FEBS Lett. 512: 95–100.
Goping IS, Gross A, Lavoie JN, Nguyen M, Jemmerson R, Roth K, Korsmeyer SJ and Shore GC (1998) Regulated targeting of BAX to mitochondria. J. Cell Biol. 143: 207–215.
Parikh N, Sade H, Kurian L and Sarin A (2004) The Bax N terminus is required for negative regulation by the mitogen-activated protein kinase kinase and Akt signaling pathways in T cells. J. Immunol. 173: 6220–6227.
Gao G and Dou QP (2000) N-terminal cleavage of bax by calpain generates a potent proapoptotic 18-kDa fragment that promotes bcl-2-independent cytochrome c release and apoptotic cell death. J. Cell. Biochem. 80: 53–72.
Wood DE, Thomas A, Devi LA, Berman Y, Beavis RC, Reed JC and Newcomb EW (1998) Bax cleavage is mediated by calpain during drug-induced apoptosis. Oncogene 17: 1069–1078.
Wood DE and Newcomb EW (2000) Cleavage of Bax enhances its cell death function. Exp. Cell Res. 256: 375–382.
Cartron PF, Oliver L, Juin P, Meflah K and Vallette FM (2004) The p18 truncated form of Bax behaves like a Bcl-2 homology domain 3-only protein. J. Biol. Chem. 279: 11503–11512.
Gong XM, Choi J, Franzin CM, Zhai D, Reed JC and Marassi FM (2004) Conformation of membrane-associated proapoptotic tBid. J. Biol. Chem. 279: 28954–28960.
Oh KJ, Barbuto S, Meyer N, Kim RS, Collier RJ and Korsmeyer SJ (2005) Conformational changes in BID, a pro-apoptotic BCL-2 family member, upon membrane binding. A site-directed spin labeling study. J. Biol. Chem. 280: 753–767.
Wilson-Annan J, O'Reilly LA, Crawford SA, Hausmann G, Beaumont JG, Parma LP, Chen L, Lackmann M, Lithgow T, Hinds MG, Day CL, Adams JM and Huang DC (2003) Proapoptotic BH3-only proteins trigger membrane integration of prosurvival Bcl-w and neutralize its activity. J. Cell Biol. 162: 877–887.
Denisov AY, Chen G, Sprules T, Moldoveanu T, Beauparlant P and Gehring K (2006) Structural Model of the BCL-w-BID Peptide Complex and Its Interactions with Phospholipid Micelles. Biochemistry 45: 2250–2256.
Losonczi JA, Olejniczak ET, Betz SF, Harlan JE, Mack J and Fesik SW (2000) NMR studies of the anti-apoptotic protein Bcl-xL in micelles. Biochemistry 39: 11024–11033.
Yethon JA, Epand RF, Leber B, Epand RM and Andrews DW (2003) Interaction with a membrane surface triggers a reversible conformational change in Bax normally associated with induction of apoptosis. J. Biol. Chem. 278: 48935–48941.
Wang JL, Liu D, Zhang ZJ, Shan S, Han X, Srinivasula SM, Croce CM, Alnemri ES and Huang Z (2000) Structure-based discovery of an organic compound that binds Bcl-2 protein and induces apoptosis of tumor cells. Proc. Natl. Acad. Sci. USA 97: 7124–7129.
Enyedy IJ, Ling Y, Nacro K, Tomita Y, Wu X, Cao Y, Guo R, Li B, Zhu X, Huang Y, Long YQ, Roller PP, Yang D and Wang S (2001) Discovery of small-molecule inhibitors of Bcl-2 through structure-based computer screening. J. Med. Chem. 44: 4313–4324.
Real PJ, Cao Y, Wang R, Nikolovska-Coleska Z, Sanz-Ortiz J, Wang S and Fernandez-Luna JL (2004) Breast cancer cells can evade apoptosis-mediated selective killing by a novel small molecule inhibitor of Bcl-2. Cancer Res. 64: 7947–7953.
Kim KM, Giedt CD, Basanez G, O'Neill JW, Hill JJ, Han YH, Tzung SP, Zimmerberg J, Hockenbery DM and Zhang KY (2001) Biophysical characterization of recombinant human Bcl-2 and its interactions with an inhibitory ligand, antimycin A. Biochemistry 40: 4911–4922.
Tzung SP, Kim KM, Basanez G, Giedt CD, Simon J, Zimmerberg J, Zhang KY and Hockenbery DM (2001) Antimycin A mimics a cell-death-inducing Bcl-2 homology domain 3. Nat. Cell Biol. 3: 183–191.
Degterev A, Lugovskoy A, Cardone M, Mulley B, Wagner G, Mitchison T and Yuan J (2001) Identification of small-molecule inhibitors of interaction between the BH3 domain and Bcl-xL. Nat. Cell Biol. 3: 173–182.
Chan SL, Lee MC, Tan KO, Yang LK, Lee AS, Flotow H, Fu NY, Butler MS, Soejarto DD, Buss AD and Yu VC (2003) Identification of chelerythrine as an inhibitor of BclXL function. J. Biol. Chem. 278: 20453–20456.
Kitada S, Leone M, Sareth S, Zhai D, Reed JC and Pellecchia M (2003) Discovery, characterization, and structure-activity relationships studies of proapoptotic polyphenols targeting B-cell lymphocyte/leukemia-2 proteins. J. Med. Chem. 46: 4259–4264.
Becattini B, Kitada S, Leone M, Monosov E, Chandler S, Zhai D, Kipps TJ, Reed JC and Pellecchia M (2004) Rational design and real time, in-cell detection of the proapoptotic activity of a novel compound targeting Bcl-X(L). Chem. Biol. 11: 389–395.
Leone M, Zhai D, Sareth S, Kitada S, Reed JC and Pellecchia M (2003) Cancer prevention by tea polyphenols is linked to their direct inhibition of antiapoptotic Bcl-2-family proteins. Cancer Res. 63: 8118–8121.
Reed JC and Pellecchia M (2005) Apoptosis-based therapies for hematologic malignancies. Blood 106: 408–418.
Shore GC and Viallet J (2005) Modulating the bcl-2 family of apoptosis suppressors for potential therapeutic benefit in cancer. Hematology (Am. Soc. Hematol. Educ. Program) 2005: 226–230.
Shuker SB, Hajduk PJ, Meadows RP and Fesik SW (1996) Discovering high-affinity ligands for proteins: SAR by NMR. Science 274: 1531–1534.
Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW and Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435: 677–681.
Petros AM, Dinges J, Augeri DJ, Baumeister SA, Betebenner DA, Bures MG, Elmore SW, Hajduk PJ, Joseph MK, Landis SK, Nettesheim DG, Rosenberg SH, Shen W, Thomas S, Wang X, Zanze I, Zhang H and Fesik SW (2006) Discovery of a potent inhibitor of the antiapoptotic protein Bcl-xL from NMR and parallel synthesis. J. Med. Chem. 49: 656–663.
Wendt MD, Shen W, Kunzer A, McClellan WJ, Bruncko M, Oost TK, Ding H, Joseph MK, Zhang H, Nimmer PM, Ng SC, Shoemaker AR, Petros AM, Oleksijew A, Marsh K, Bauch J, Oltersdorf T, Belli BA, Martineau D, Fesik SW, Rosenberg SH and Elmore SW (2006) Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopotentiation activity in vitro and in vivo. J. Med. Chem. 49: 1165–1181.
Schimmer AD, Hedley DW, Chow S, Pham NA, Chakrabartty A, Bouchard D, Mak TW, Trus MR and Minden MD (2001) The BH3 domain of BAD fused to the Antennapedia peptide induces apoptosis via its alpha helical structure and independent of Bcl-2. Cell Death Differ. 8: 725–733.
Vieira HL, Haouzi D, El Hamel C, Jacotot E, Belzacq AS, Brenner C and Kroemer G (2000) Permeabilization of the mitochondrial inner membrane during apoptosis: impact of the adenine nucleotide translocator. Cell Death Differ. 7: 1146–1154.
Shangary S, Oliver CL, Tillman TS, Cascio M and Johnson DE (2004) Sequence and helicity requirements for the proapoptotic activity of Bax BH3 peptides. Mol. Cancer Ther. 3: 1343–1354.
Wang JL, Zhang ZJ, Choksi S, Shan S, Lu Z, Croce CM, Alnemri ES, Korngold R and Huang Z (2000) Cell permeable Bcl-2 binding peptides: a chemical approach to apoptosis induction in tumor cells. Cancer Res. 60: 1498–1502.
Gemperli AC, Rutledge SE, Maranda A and Schepartz A (2005) Paralog-selective ligands for bcl-2 proteins. J. Am. Chem. Soc. 127: 1596–1597.
Chin JW and Schepartz A (2001) Design and Evolution of a Miniature Bcl-2 Binding Protein We thank the HHMI Biopolymer/Keck Foundation Biotechnology Resource Laboratory (Yale University School of Medicine, New Haven, CT) for oligonucleotide and peptide synthesis and amino acid analysis and Professor Jennifer Doudna (Yale University) for use of a Perseptive Voyager-DE (MALDI-TOF) mass spectrometer. We are grateful also to Dr. Junying Yuan and Dr. Alexi Degterev (Harvard Medical School) for a generous gift of Bcl-X(L)-His(6) and Stacey E. Rutledge for helpful comments. This work was supported by the National Institutes of Health. Angew. Chem. Int. Ed. Engl. 40: 3806–3809.
Kutzki O, Park HS, Ernst JT, Orner BP, Yin H and Hamilton AD (2002) Development of a potent Bcl-x(L) antagonist based on alpha-helix mimicry. J. Am. Chem. Soc. 124: 11838–11839.
Yin H, Lee GI, Park HS, Payne GA, Rodriguez JM, Sebti SM and Hamilton AD (2005) Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew. Chem. Int. Ed. Engl. 44: 2704–2707.
Davis JM, Truong A and Hamilton AD (2005) Synthesis of a 2, 3′;6′, 3″-terpyridine scaffold as an alpha-helix mimetic. Org. Lett. 7: 5405–5408.
Yin H, Lee GI, Sedey KA, Rodriguez JM, Wang HG, Sebti SM and Hamilton AD (2005) Terephthalamide derivatives as mimetics of helical peptides: disruption of the Bcl-x(L)/Bak interaction. J. Am. Chem. Soc. 127: 5463–5468.
Sadowsky JD, Schmitt MA, Lee HS, Umezawa N, Wang S, Tomita Y and Gellman SH (2005) Chimeric (alpha/beta + alpha)-peptide ligands for the BH3-recognition cleft of Bcl-XL: critical role of the molecular scaffold in protein surface recognition. J. Am. Chem. Soc. 127: 11966–11968.
Phelan JC, Skelton NJ, Braisted AC and McDowell RS (1997) A general method for constraining short peptides to an a-helical conformation. J. Am. Chem. Soc. 119: 455–460.
Bracken C, Gulyas J, Taylor JW and Baum J (1994) Synthesis and nuclear magnetic resonance structure determination of an alpha-helical, bicyclic, lactam-bridged hexapeptide. J. Am. Chem. Soc. 116: 6431–6432.
Jackson DY, King DS, Chmielewski J, Singh S and Schultz PG (1991) General approach to the synthesis of short a-helical peptides. J. Am. Chem. Soc. 113: 9391–9392.
Blackwell HE and Grubbs RH (1998) Highly Efficient Synthesis of Covalently Cross-Linked Peptide Helices by Ring-Closing Metathesis. Angew. Chem. Int. Ed. 37: 3281–3284.
Schafmeister C, Po J and Verdine G (2000) An all-hydrocarbon cross-linking system for enhancing the helicity and metabolic stability of peptides. J. Am. Chem. Soc. 122: 5891–5892.
Yang B, Liu D and Huang Z (2004) Synthesis and helical structure of lactam bridged BH3 peptides derived from pro-apoptotic Bcl-2 family proteins. Bioorg. Med. Chem. Lett. 14: 1403–1406.
Andrews MJI and Tabor AB (1999) Forming stable helical peptides using natural and artificial amino acids. Tetrahedron 55: 11711–11743.
Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright R, Wagner G, Verdine GL and Korsmeyer SJ (2004) Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305: 1466–1470.
Becattini B, Sareth S, Zhai D, Crowell KJ, Leone M, Reed JC and Pellecchia M (2004) Targeting apoptosis via chemical design: inhibition of bid-induced cell death by small organic molecules. Chem. Biol. 11: 1107–1117.
Bombrun A, Gerber P, Casi G, Terradillos O, Antonsson B and Halazy S (2003) 3, 6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J. Med. Chem. 46: 4365–4368.
Hetz C, Vitte PA, Bombrun A, Rostovtseva TK, Montessuit S, Hiver A, Schwarz MK, Church DJ, Korsmeyer SJ, Martinou JC and Antonsson B (2005) Bax channel inhibitors prevent mitochondrion-mediated apoptosis and protect neurons in a model of global brain ischemia. J. Biol. Chem. 280: 42960–42970.
Sawada M, Hayes P and Matsuyama S (2003) Cytoprotective membrane-permeable peptides designed from the Bax-binding domain of Ku70. Nat. Cell Biol. 5: 352–357.
Fesik SW (2005) Promoting apoptosis as a strategy for cancer drug discovery. Nat. Rev. Cancer 5: 876–885.
Badros AZ, Goloubeva O, Rapoport AP, Ratterree B, Gahres N, Meisenberg B, Takebe N, Heyman M, Zwiebel J, Streicher H, Gocke CD, Tomic D, Flaws JA, Zhang B and Fenton RG (2005) Phase II study of G3139, a Bcl-2 antisense oligonucleotide, in combination with dexamethasone and thalidomide in relapsed multiple myeloma patients. J. Clin. Oncol. 23: 4089–4099.
Cotter FE, Johnson P, Hall P, Pocock C, al Mahdi N, Cowell JK and Morgan G (1994) Antisense oligonucleotides suppress B-cell lymphoma growth in a SCID-hu mouse model. Oncogene 9: 3049–3055.
Kitada S, Miyashita T, Tanaka S and Reed JC (1993) Investigations of antisense oligonucleotides targeted against bcl-2 RNAs. Antisense Res. Dev. 3: 157–169.
Reed JC, Cuddy M, Haldar S, Croce C, Nowell P, Makover D and Bradley K (1990) BCL2-mediated tumorigenicity of a human T-lymphoid cell line: synergy with MYC and inhibition by BCL2 antisense. Proc. Natl. Acad. Sci. USA 87: 3660–3664.
Acknowledgements
This work is dedicated to the memory of our beloved mentor and role model Stanley J Korsmeyer, who is so deeply missed. His guidance, integrity, and laser-focused commitment to his research and his trainees continue to inspire our daily work. I thank E Smith for invaluable editorial and computer graphics assistance. LDW is supported by NHLBI grant K08HL074049, a Burroughs Wellcome Career Award in the Biomedical Sciences, a Culpeper Scholarship in Medical Science, and a V Scholar Award.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by C Borner
Rights and permissions
About this article
Cite this article
Walensky, L. BCL-2 in the crosshairs: tipping the balance of life and death. Cell Death Differ 13, 1339–1350 (2006). https://doi.org/10.1038/sj.cdd.4401992
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.cdd.4401992
Keywords
This article is cited by
-
Mechanism and significance of apoptosis of the immortalized human oral mucosal epithelial cells established by Lentivirus-mediated hTERT
Molecular Biology Reports (2020)
-
CKT0353, a novel microtubule targeting agent, overcomes paclitaxel induced resistance in cancer cells
Investigational New Drugs (2020)
-
Melatonin Alleviates Intracerebral Hemorrhage-Induced Secondary Brain Injury in Rats via Suppressing Apoptosis, Inflammation, Oxidative Stress, DNA Damage, and Mitochondria Injury
Translational Stroke Research (2018)
-
Select Bcl-2 antagonism restores chemotherapy sensitivity in high-risk neuroblastoma
BMC Cancer (2016)
-
Time depended Bcl-2 inhibition might be useful for a targeted drug therapy
Cancer Cell International (2015)
