
Abstract
Friedreich's ataxia (FRDA), the most common autosomal recessively inherited ataxia, is due to a homozygous GAA triplet repeat expansion in the first intron of the FRDA gene in about 96% of patients. Approximately 4% of FRDA patients are compound heterozygotes with a GAA repeat expansion in one allele and a point mutation in the coding region of the second allele. To reinvestigate the mutation spectrum, we searched for mutations including exon deletions in six patients heterozygous for the GAA repeat expansion and found two unknown missense mutations, p.Asn146Lys and p.Leu186Arg, in trans to the expanded FRDA allele. Interestingly, we detected a heterozygous 2776 bp deletion including exon 5a in one of our patients. This deletion removes 50 of the 210 residues of the frataxin. Furthermore, since no FRDA case with two-point mutations is known, we screened eight patients with FRDA phenotype but GAA alleles within the normal range but did not reveal a mutation within the FRDA gene. In addition, DNA polymorphisms have been found in four out of 100 control individuals in this study.
Similar content being viewed by others


Introduction
Friedreich's ataxia (FRDA; MIM 229300), a neurodegenerative disease with an estimated prevalence of 2–4:100 000 in several European populations, is characterized by a progressive ataxia of gait and limbs with early onset usually before the age of 25 years, absence of tendon reflexes, loss of position and vibratory sense, and dysarthria.
The majority of FRDA patients is homozygous for GAA repeat expansions within the first intron of the gene X25.1 In normal alleles, the repeat varies in size between 6 and 36 GAA,2 whereas in mutated alleles the repeat length ranges from 120 to 1700.3 In patients heterozygous for the expanded allele (2–4%), nucleotide substitutions, small deletions and insertions are found. A total of 33 mutations registered in The Human Gene Mutation Database, Cardiff (http://www.hgmd.org) include 17 missense and two nonsense substitutions, four splicing mutations, six small deletions, two small insertions, one indel and one repeat variation.
To date, neither a case with deletion of the complete gene or single exons nor a patient with two-point mutations of the FRDA gene has been described. Therefore, we investigated six ataxia patients with heterozygosity for the repeat expansion and eight patients with FRDA phenotype but normal GAA alleles for exon deletions and point mutations by SSCP analyses and quantitative duplex PCR. Furthermore, we looked for DNA variations in 100 anonymous control individuals.
Subjects and methods
Subjects
DNA analyses were performed for six patients with symptoms of FRDA and heterozygosity for the GAA repeat expansion as well as for eight ataxia patients without expanded FRDA alleles. In addition, 100 samples of anonymous healthy individuals were analyzed.
Mutation analysis
Total genomic DNA was isolated from proteinase K/SDS digest of blood samples. The GAA repeat lengths were determined by PCR as described.2 For amplification of the coding sequence and SSCP analyses,4 exon-specific oligonucleotides (Table 1a) were used. Selected PCR products were sequenced directly using the dideoxy chain termination method on double-stranded DNA templates in the presence of IRD800-labeled exon-specific primers on a Licor 4200 automated sequencer. Results were compared to GenBank sequence U43747 (27 April 1996) of the FRDA gene and numbered with +1 at the A of the ATG start codon.
For gene dosage analysis, a quantitative duplex PCR assay on the LightCycler (Roche Diagnostics) using the fluorescence resonance energy transfer technique was performed for each exon, separately. The β-globin gene was coamplified as an internal standard. The used primer sequences and exon-specific light-emitting single-stranded probes (TIB MolBiol) are shown in Table 1b. For the amplification in a volume of 10 μl, we used 1 μl Hybridization FastStart Mix (Roche), 2 mM MgCl2, 0.2 μ M of the respective hybridization probes, 0.5 μ M of each primer and about 10 ng genomic DNA. PCR conditions were 10 min at 95°C, followed by 35 cycles 10 s at 95°C, 10 s at 55°C and 10 s at 72°C. Fluorescence was measured during the 55°C step. Product concentration was calculated as described5 and results reproduced three-fold.
To analyze the exon 5a deletion, long-range PCR using the primers DEL5a (Table 1a) was performed with the Expand Long Template PCR System (Roche) applying the following PCR conditions: initial 94°C for 5 min/10 cycles: 94°C for 10 s, 63°C for 30 s; 68°C for 8 min/20 cycles: 94°C for 15 s, 63°C for 30 s, 68°C for 8 min+20 s extension/68°C for 10 min, 4°C for ∞. PCR products were cloned into the pCR-XL-TOPO cloning vector (Invitrogen) and sequenced on both strands in the presence of IRD800-labeled M13 (forward and reverse) primers on a Licor 4200 automated sequencer. Location and size of the deletion was determined by comparing the cloned sequence with the genomic sequence AL162730.26 (version 31 January 2002).
Results and discussion
We performed molecular analyses for six ataxia patients with heterozygosity for the GAA repeat expansion, eight patients with FRDA phenotype but GAA alleles within the normal range and 100 healthy controls. The search for nucleotide substitutions, insertions and deletions revealed a deletion of exon 5a in one patient as well as two different substitutions, not seen in control persons, in two patients heterozygous for the repeat expansion. For none of the eight patients clinically affected by FRDA but without detectable GAA repeat expansion in the genomic DNA from lymphocytes, a mutation could be found. In three control individuals, a heterozygous G>A variation in the 5′ untranslated region at position −7 has been found. In one person, the mutation c.258C>A resulting in the amino-acid substitution p.His86Gln was detected. Both DNA variation are not described. Therefore, it is not possible to differentiate in these cases between mutations and polymorphisms.
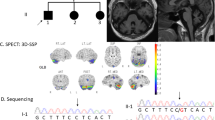
For one female patient (P1), genomic quantitative analysis pointed to a heterozygous deletion of exon 5a (three runs with an average quotient of FRDA exon 5 to β-globin=0.56, standard deviation 0.02), whereas exon 5b is present on both alleles. Performing long-range PCR, we obtained one normal allele with the expected length of 4 kb and a smaller fragment of 1.3 kb length (Figure 1). Subsequent sequencing of the small PCR product showed a 2776 bp deletion between position 120032 and 122808 (g.120032_122808del) of the genomic clone AL162730.26. The deletion (Figure 1) covers 1315 bp of intron 4 (intron length 7576 bp in the wild-type gene), the complete exon 5a (490 bp) plus 971 bp genomic downstream sequence including a potential polyadenylation signal. The 3′ breakpoint is located directly within the AluI recognition sequence of a repetitive element according to the NCBI AluI repeat database. The loss of 50 amino acids (of 210) at the carboxy-terminus would result in a truncated frataxin and/or in the expression of an isoform resulting from exon 5b. The repeat expansion could be traced back to the maternal allele, the deletion to the paternal one. The compound heterozygosity of repeat expansion and deletion explain the FRDA phenotype in the patient.

Deletion analysis. The comparison of PCR products of the patient (2) and her parents (1, 3) revealed heterozygosity for a deletion in the patient and her father. Molecular weight marker M: Boehringer X. The schematic drawing shows the location of the primers P1 and P2 (FA-Del5F+R) flanking exon 5a. White box=coding sequence; hatched box=3′UTR. The deletion (upper part) affects the complete exon 5a including upstream and downstream sequences.
In a second patient (P2), we found a point mutation in exon 4 of the FRDA gene in addition to the GAA repeat expansion. The transversion C>G at position 438 is resulting in the amino-acid exchange p.Asn146Lys. Investigation of the parental FRDA genes revealed the GAA repeat expansion in the mother and the point mutation in the father.
In patient 3 (P3), SSCP and sequence analyses revealed the nucleotide substitution c.557T>G (exon 5a) resulting in p.Leu186Arg within frataxin. This point mutation was inherited by the patient's mother and the repeat expansion by the father.
Both amino acids, exchanged by the mutations, are conserved in mouse and yeast.6 The FRDA phenotype in both patients in addition to the compound heterozygosity and the evolutionary conservation of the affected amino acids underlines the disease-causing potential of these mutations.
The detection of private mutations (summarized in Table 2), in three of six individuals with a single expanded GAA repeat, was highly important for the diagnosis and counseling in the three families. For example, the molecular genetic findings were relevant for the treatment of the severe cardiomyopathy of patient P3 with idebenone. Therefore, the introduction of appropriate standards in molecular genetic analyses including gene dosage measurements seems reasonable to us in these cases.
None of the eight patients with symptoms of FRDA but missing repeat expansion could be asserted by molecular findings. Patients with two-point mutations in the FRDA gene will be highly rare. Assuming that 2% of the FRDA alleles are point mutations in a population with a carrier frequency of one in 100, the probability to find two unrelated point mutations is one in 100 million individuals. Thus, in one of 2500 FRDA patients this constellation could be found.7 However, it can not be excluded that mutations could not be seen by the applied SSCP conditions or occurs in gene regions not presented in the PCR products, for example, the promoter or exon sequences included in the PCR primers. The detection of these rare cases may be complicated additionally by the fact that the clinical phenotype of this group of patients is not really known. Possibly, persons with two missense mutations will not develop symptoms typical for FRDA or will present with a late age of onset. Furthermore, a prenatal lethality is not excluded.
References
Campuzano V, Montermini L, Molto MD et al: Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996; 271: 1423–1427.
Montermini L, Andermann E, Labuda M et al: The Friedreich ataxia GAA triplet repeat: premutation and normal alleles. Hum Mol Genet 1997; 6: 1261–1266.
Cossee M, Schmitt M, Campuzano V et al: Evolution of the Friedreich's ataxia trinucleotide repeat expansion: founder effect and premutations. Proc Natl Acad Sci USA 1997; 94: 7452–7457.
Zühlke CH, Laccone F, Cossee M, Kohlschütter A, Koenig M, Schwinger E : Mutation of the start codon in the FRDA1 gene: linkage analysis of three pedigrees with the ATG to ATT transversion points to a unique common ancestor. Hum Genet 1998; 103: 102–105.
Hedrich K, Kann M, Lanthaler AJ et al: The importance of gene dosage studies: mutational analysis of the parkin gene in early-onset Parkinsonism. Hum Mol Genet 2001; 10: 1649–1656.
Koutnikova H, Campuzano V, Foury F, Dolle P, Cazzalini O, Koenig M : Studies of human, mouse and yeast homologues indicate a mitochondrial function of frataxin. Nat Genet 1997; 16: 345–351.
Pandolfo M, Montermini L : Prenatal diagnosis of Friedreich ataxia. Prenatal Diagn 1998; 18: 831–833.
Acknowledgements
We thank U Gehlken and J Atici for excellent technical help. We thank all patients for providing blood samples for scientific research as well as the German Heredo-Ataxia Society (DHAG), whose cooperation and support is essential in our work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zühlke, C., Dalski, A., Habeck, M. et al. Extension of the mutation spectrum in Friedreich's ataxia: detection of an exon deletion and novel missense mutations. Eur J Hum Genet 12, 979–982 (2004). https://doi.org/10.1038/sj.ejhg.5201257
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201257
Keywords
This article is cited by
-
DNA methylation in Friedreich ataxia silences expression of frataxin isoform E
Scientific Reports (2022)
-
Friedreich ataxia is not only a GAA repeats expansion disorder: implications for molecular testing and counselling
Journal of Applied Genetics (2016)
-
Friedreich ataxia in Norway – an epidemiological, molecular and clinical study
Orphanet Journal of Rare Diseases (2015)
-
Modeling of Friedreich ataxia-related iron overloading cardiomyopathy using patient-specific-induced pluripotent stem cells
Pflügers Archiv - European Journal of Physiology (2014)
-
Klinik und Genetik der rezessiven Ataxien
Der Nervenarzt (2011)
