
Abstract
Congenital cutis laxa is a genetically heterogeneous condition presenting with loose and redundant skin folds, decreased elasticity of the skin, connective tissue involvement and a highly variable spectrum of associated features. The most common forms are inherited in an autosomal recessive or dominant fashion. Fibulin 5 and elastin mutations were detected in a limited number of patients, but in most cases the etiology is not known. Based on a previous observation of an abnormal transferrin isoelectric focusing pattern in a patient with cutis laxa indicating an N-glycosylation defect, we performed a screening for disorders of protein glycosylation in unrelated children with cutis laxa syndrome, including a recently developed test for defective O-glycosylation. Here, we describe five patients from consanguineous marriages with a cutis laxa syndrome with skeletal and joint involvement, developmental delay and neurological findings. Three of these five children have an inborn error of glycan biosynthesis affecting the synthesis of both N- and O-linked glycans. Two patients had normal glycosylation patterns. All known causes of secondary glycosylation disorders were excluded in the children. No mutations were found in the FBLN5 gene. In conclusion, we have identified a new combined glycosylation defect with a distinct clinical phenotype. Our results suggest that a combined defect of glycosylation might be a causative factor in congenital cutis laxa. This is the first report where abnormal N- and O-linked glycosylation is implicated in the etiology of cutis laxa syndrome.
Similar content being viewed by others

Introduction
Cutis laxa syndrome is a rare, genetically heterogeneous condition1, 2, 3, 4 presenting in the early months of life with loose and redundant skin folds mostly on the trunk and the extremities, and decreased elasticity of the skin.1, 5, 6 A highly variable spectrum of associated features7, 8, 9, 10, 11 has been reported. The skin histology characteristically demonstrates abnormal elastic fiber structure with a significantly decreased amount of elastin.5, 6, 12 The most common forms are inherited in an autosomal recessive or dominant fashion (MIM 219200, MIM 219100, MIM 123700, MIM 150240).
Two distinct clinical types of autosomal recessive cutis laxa (ARCL) have been described in detail. In ARCL type I (MIM 219100), congenital cutis laxa presents with a generalized disorder of the elastic tissue with diaphragmatic or other hernias, diverticula of the gastrointestinal or urinary tract and infantile emphysema. The disease is often fatal within the first year. In this severe form, missense mutations have been detected in the fibulin-5 gene.1, 3 The phenotypic features observed in a fibulin-5 knockout mouse model are similar to those of the human ARCL type I.12
In patients with ARCL type II (MIM 219200), the cutis laxa is combined with widely patent anterior fontanel, a variety of malformations, retarded growth and abnormal development.7, 8, 11, 13, 14 Central nervous system anomalies and ophthalmologic findings are rare findings in ARCL type II.9, 10 So far, the genetic etiology of ARCL type II is not known.
Congenital defects of glycosylation (CDG) are a group of phenotypically diverse, multisystemic disorders caused by various enzyme defects of the N-glycan synthesis. The generally used screening method for CDG is transferrin isoelectric focusing (TIEF).15 In some of these patients, the underlying enzyme defect is not yet known. During a systemic screening program for CDG in patients with multisystem disorders without a known etiology, we observed an abnormal TIEF pattern, indicating an N-glycosylation defect (classified as CDG type IIx) in a patient with cutis laxa. Based on this observation, we performed a metabolic evaluation for disorders of protein glycosylation (including N-glycan and O-linked glycan biosynthesis) in further unrelated patients with cutis laxa and developmental delay.
Apolipoprotein C-III (apoC-III) isoelectric focusing (IEF) has recently been described as a new screening method for ‘core 1 mucin type O-glycans’.16 Defects of the O-glycan biosynthesis may also lead to clinically very distinct disorders. Muscle–eye–brain disease and Walker–Warburg syndrome17 have been identified as O-mannosylation disorders. The progeroid type of Ehlers Danlos18 and multiple exostoses syndrome19 are due to biosynthesis defects in O-xylosyl-proteoglycans. The gene underlying familial tumoral calcinosis encodes a glycosyltransferase responsible for initiating mucin-type O-glycosylation.20
Here, we present five patients with a cutis laxa syndrome associated with skeletal and joint involvement, muscle hypotonia and developmental delay. Some of these patients had epilepsy and congenital brain malformations as well. Three out of the five children were found to have a defect affecting the biosynthesis of both the N- and the O-linked glycans.
Case reports
The clinical and laboratory findings of five patients with cutis laxa syndrome are summarized in Table 1. Here we describe in detail the phenotype of the three patients (patients 1–3) with an inborn error of glycosylation.
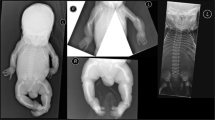
Patient 1: Patient 1 (Figure 1a) was born as the second child of healthy parents of Turkish origin, who were first cousins. The third offspring had nonimmune fetal hydrops and died at the 20th week of the pregnancy. Our patient presented at term with a birth weight of 2730 g (−2SD), a birth length of 47 cm (−2SD), microcephaly (HC: 31.0 cm, −3.0SD), large fontanel (5.5 × 7 cm), brachycephaly, flat face, midfacial hypoplasia, anteverted nares, long philtrum, neonatal teeth (left incisor), down-slanting palpebral fissures, strabismus, blue sclera, generalized cutis laxa and single umbilical artery. Additionally, hyperflexible joints, muscle hypotonia, cryptorchidism and swallowing difficulties were reported. Cranial and abdominal ultrasound, EEG and a cranial MRI were normal. VEP and BAEP were both abnormal showing decreased conduction velocities. A skeletal survey showed luxated hypoplastic femur heads with hip dysplasia. Chromosome analysis, Hb, Htc, MCV, MCH, MCHC, cell counts, serum creatinine, BUN, uric acid, electrolytes, calcium, phosphorus, ALP, ASAT, ALAT, γGT, LDH, CK, cholesterol, TG, serum proteins, immunoglobulins, lactic acid, glucose, blood gas (ASTRUP), coagulation studies (APT, APTT, factor VII, VIII, IX, X, XI, XII, protein C and S), alpha1-antitrypsin, endocrine evaluation (TSH, free T4, thyroxine binding globulin (TBG), ACTH, GH, cortisol), markers for a possible infection and initial metabolic studies (serum amino-acid profile, acyl-carnitine profile, homocysteine, studies on cholesterol synthesis, phytanic acid, lysosomal enzymes, urine organic acid pattern, urine amino acids, purines/pyrimidines) were all normal. The skin histology showed a normal epidermis, homogeneous staining of the collagen bundles, with somewhat shorter collagen fibers, and the elastic fiber staining detected a rarefaction of elastic fibers, which were severely fragmented. The electromicroscopy showed a decreased elastic material. Intact elastic fibers could not be observed.

Family tree of the three patients (patients 1–3) with a combined glycosylation disorder.
The fontanel was closed at the age of 3 years (length: 101 cm, +1SD; weight: 18 kg, 0SD; HC: 46.5 cm, −2.5SD). At the age of 10 years, the patient (Figure 2a) had a normal growth with microcephaly (length: 146 cm, +1SD; weight: 31 kg, −1SD; HC: 49 cm, −2.5SD). He wore hearing aids, showed a developmental delay (IQ 69) and followed special education. He had mild hypotonia and a minimal hyperlaxity of the joints (the hip luxation was successfully operated). He showed a gradual improvement of the skin disease. He continued to have a very dry skin, but at 10 years of age the cutis laxa was localized only in the abdominal region (Figure 2d).

Patient 1: facial features with down-slanting palpebral fissures, midfacial hypoplasia, long philtrum (a) and mild abdominal cutis laxa (d) at the age of 10 years. Patient 2: characteristic facial features with high forehead, down-slanting palpebral fissures, midfacial hypoplasia (b) and gluteal fat-pads (e) at the age of 7 years. Patient 3: facial features with down-slanting palpebral fissures, midfacial hypoplasia and facial cutis laxa at the age of 3 years (c), and cutis laxa on the extremities at the age of 3.5 years (f).
Patient 2: Patient 2 (Figure 1b) was born premature at 36 weeks of gestation as the first child of healthy parents of European origin, who were second cousins. She presented with a birth weight of 2250 g (−2SD), length of 44 cm (−2SD), microcephaly (HC: 29 cm, −2.5SD), large fontanel (5 × 6 cm), brachycephaly, flat face, midfacial hypoplasia, anteverted nares, long philtrum, down-slanting palpebral fissures, strabismus, a generalized, severe cutis laxa, dry skin, sparse hair, pes adductus, hyperflexible joints, muscle hypotonia and feeding difficulties. Abdominal ultrasound and echocardiography were normal. From the age of 5 months (Figure 3a), she received anticonvulsive medication due to a West syndrome and abnormal EEG. A cranial MRI at the age of 2.5 years showed bilateral pachygyria (Figure 4a). VEP and BAEP were normal. A skeletal survey showed a large, open fontanel, narrow pelvis and generalized osteopenia. She had recurrent urinary infections and a gastro-esophageal reflux. Chromosome analysis and blood and urine analysis including initial metabolic analysis (see Patient 1) were all normal. The skin histology showed a normal epidermis, increased subcutaneous fat and homogeneous staining of the collagen bundles; the Weigert elastic fiber technique detected a rarefaction of elastic fibers, which were short and fragmented. Intact elastic fibers could not be observed. The Western blotting for collagens and procollagens was normal. Electromicroscopy showed decreased elastic material.

Evolving phenotype in patient 2 showing an improvement of the skin symptoms at the age of 5 months (a), 18 months (b), 3.5 years (c) and 7 years (d).

Cranial MRI picture of patient 2 with bilateral pachygyria (a) and patient 3 with mild partial fronto-temporal pachygyria in the sagittal and vertical view (b).
The patient had no significant feeding problems at the age of 3 years (Figure 3c) and showed a normal growth with a microcephaly (HC: 45.5 cm, −2.5SD). Her fontanel was closed at the age of 2.5 years. At the age of 7.5 years, our patient (Figures 2b and 3d) was severely retarded, but able to communicate and very social. Her growth was age appropriate (length: 122.5 cm, −1SD; weight: 22.5 kg, −1SD) with a microcephaly (HC: 47, −2.5SD) and psychomotor developmental delay (IQ 52). She walked clumsy but without help. She had a significant improvement of the cutis laxa since birth with a mostly abdominal localization (Figure 3b–d), a very dry skin and gluteal fad-pads (Figure 2e).
Patient 3: Patient 3 (Figure 1c) was born as the first child of healthy parents of Turkish origin, who were first cousins. The patient presented at term with a birth weight of 3605 g (+1SD), length of 53 cm (+1SD), microcephaly (31.5, −2.5SD), large fontanel (4.5 × 5 cm), flat face, midfacial hypoplasia, anteverted nares, down-slanting palpebral fissures, strabismus, generalized cutis laxa, clubfeet, bilateral clinodactyly of the IVth fingers, hyperflexible joints and muscle hypotonia. She had recurrent urinary tract infections. A mild bilateral hydronephrosis was found by an abdominal ultrasound. Further studies detected bilateral pyelo-urethral stenosis. The echocardiography was normal. From as early as the second day of life, she suffered from tonic clonic seizures, her EEG was abnormal and she was started on anticonvulsive therapy. A cranial MRI showed a partial, bilateral fronto-temporal pachygyria (Figure 4b). She was diagnosed with a myopia of 5.5 diopters. VEP showed decreased conduction velocities. BAEP was normal. A skeletal survey showed a large, open fontanel, and hypoplastic iliac wings and acetabulae. Chromosome analysis and blood and urine analysis including initial metabolic analysis (see Patient 1) were all normal. The skin histology showed a slight hyperkeratosis; the Gleason staining detected shorter and thin collagen fibers and the elastic fibers were short, scattered and severely fragmented. Electromicroscopy showed a severe decrease in the amount of elastic fibers. The fibers present were all abnormal in structure.
The patient at the age of 3 years had no feeding problems, showed a normal growth and microcephaly (length: 98 cm, +0.5SD; weight: 16 kg, +1SD; HC: 46.0 cm, −2SD). Her fontanel was closed at the age of 2 years. She had a developmental delay and speech delay; she started to walk at 15 months of age but could not speak till the age of 22 months, and she had muscle hypotonia, significant hyperlaxity of the joints and cutis laxa. At the age of 4 years (Figure 2c), she still had generalized cutis laxa (Figure 2f), hyperelastic joints, hypotonia and a developmental delay (IQ 59).
Patient 4: The clinical phenotype of patient 4 (see Table 1) has been described in detail earlier.9
Patient 5: Patient 5 had a mild intrauterine growth retardation, severe congenital generalized cutis laxa with joint hyperlaxity, psychomotor retardation, hypotonia, epilepsy, transient feeding problems and normal growth after the first year of life. He had no microcephaly or brain malformations (see Table 1).
Methods
Patient selection
Metabolic screening for inborn errors of protein glycosylation was performed in five patients with cutis laxa syndrome, consecutively diagnosed in the neonatal period. These unrelated patients were all born from consanguineous marriages. The clinical features are summarized in Table 1.
Sample preparation for isoelectric focusing of apoC-III, transferrin and TBG
After centrifugation for cell separation, plasma and/or serum samples were stored without delay at −80°C until analysis. Samples used for IEF of apoC-III were diluted 10-fold with saline. Samples used for TIEF were saturated with a 10 mM ferric citrate and 0.5 mM sodium hydrogen carbonate solution (2:1) in a ratio of 10:3 for 30 min. This saturated plasma sample is diluted five times with water.
IEF of apoC-III
ApoC-III is a mucin type core I O-glycosylated plasma protein. In control samples, equal amounts of disialo and monosialo isoforms are present with only a very small amount of asialo-apoC-III.
IEF of apoC-III was carried out as described by Wopereis et al.16 Plasma was separated on a hydrated dry IEF gel (pH 3.5–5.0 and 8 M urea) on a PhastSystem. After IEF, a Western blot was performed. The apoC-III isoforms were specified by adding rabbit anti-human apoC-III antibody and visualized by electrochemiluminescence. The relative amounts of apoC-III isoforms were determined using densitometry.
IEF of transferrin
Transferrin is an N-glycosylated protein. Multiple isoforms occur in plasma, with the tetrasialo isoform being the most abundant. Transferrin isofocusing is used as a screening method for the detection of abnormally high amount of asialo, monosialo, disialo and trisialo isoforms in N-glycan biosynthesis defects.
TIEF was carried out essentially as described.21 Iron-saturated plasma or serum was separated on a hydrated immobiline gel (pH 4–7) on a Ultraphore system (Amersham Pharmacia Biotech). After IEF, the isoforms were visualized by adding rabbit anti-human transferrin antibodies (Dako, Glostrup, Denmark) and stained with Coomassie blue. The relative amounts of isoforms were scanned using an Image master Labscan, Ver. 3.00 (Amersham Pharmacia Biotech) and quantified using Image master 1D gel analysis, Ver. 4.10, software (Amersham Pharmacia Biotech).
IEF of TBG
Pure plasma was applied to a Phastgel (pH 4–6.5) (Amersham Pharmacia Biotech) and analyzed using the PhastSystem. After addition of rabbit TBG antibody (Dako), the TBG pattern was detected by silver staining.
Mutation analysis of FBLN5
Genomic DNA was isolated from cultured dermal fibroblasts of patients 1 and 2 and from leukocytes of patient 3. Mutation analysis of the FBLN5 gene was performed as described by Loeys et al.3
Results
Transferrin and TBG isofocusing
Plasma samples of patients 1–3 showed reproducibly an abnormal isofocusing pattern with a decrease of the tetrasialotransferrin isoforms but increase of hypoglycosylated transferrin isoforms (tri-, di- and monosialylated isoform) (Figure 5a). TBG isoforms were also abnormal, confirming a generalized defect in N-glycosylation (data not shown). All known causes of secondary glycosylation disorders were excluded in the three children. These three patients were classified as having an unidentified CDG type II, suggesting a defect in glycan biosynthesis in the Golgi.

IEF results: abnormal transferrin pattern with decreased tetrasialotransferrin and increased hypoglycosylated tri-, di- and monosialylated transferrin isoforms (a) and abnormal apoC-III pattern with increased levels of apoC-III1, decreased levels of apoC-III2 and normal apoC-III0 in patients 1–3 (b).
The IEF patterns in patients 4 and 5 were repeatedly normal.
ApoC-III isofocusing
Plasma and/or serum samples of patients 1–3 showed an abnormal isofocusing profile for apoC-III (Figure 5b) in duplicate over a period of a year. All three patients had increased apoC-III1, decreased apoC-III2 and normal apoC-III0 (patient 1: apoC-III1: 73%, controls: 33–67%; apoC-III2: 16%, controls: 27–60%; apoC-III0: 11%, controls: 0–12%; patient 2: apoC-III1: 76%, controls: 33–67%; apoC-III2: 14%, controls: 27–60%; apoC-III0: 10%, controls: 0–12%; patient 3: apoC-III1: 86%, controls: 33–67%; apoC-III2: 8%, controls: 27–60%; apoC-III0: 6%, controls: 0–12%).
The apoC-III IEF patterns in patients 4 and 5 were normal.
Mutation analysis of FBLN5
No mutations were found in the FBLN5 gene in the cutis laxa patients with abnormal glycosylation profiles.
Discussion
We performed a metabolic evaluation for glycosylation disorders in five unrelated patients with cutis laxa syndrome. We found three patients out of the five with the clinical presentation of cutis laxa syndrome and developmental delay who were diagnosed with an inborn error of metabolism affecting N- and O-glycan synthesis. All known causes of secondary CDG forms were excluded by appropriate measures.
The patients with the combined glycosylation defect presented with a phenotype similar to that of ARCL type II (generalized cutis laxa, mental retardation, microcephaly, late closure of the fontanel, joint laxity). However, some features, like the bilateral urethral-pelvic stenosis and vesico-urethral reflux in patient 3, support the presence of urethral wall involvement, which is more characteristic for ARCL type I.
The presence of seizures, found in two of these three patients with glycan abnormalities and in one with normal IEF patterns, is not characteristic in cutis laxa syndrome, but common in CDG syndrome.15 Structural abnormalities of the brain occur frequently in CDGS as well. The finding of pachygyria as in patients 2 and 3 is a rare observation in cutis laxa patients. However, pachygyria have been reported in children with O-glycan biosynthesis defects16 supporting the importance of glycosylation in normal brain development.
There was an obvious difference observed in the severity of the cutis laxa at birth (with the most severe phenotype in patient 2), but all of the five children showed a significant improvement of the skin symptoms at an older age. Based on some of the phenotypical overlap observed in these children with a glycosylation defect and in patients with known fibulin mutations, our findings further support the presence of genetic heterogeneity in cutis laxa syndrome.
Cutis laxa syndrome is a congenital disorder with major structural and functional defects of the elastic fiber system. Elastin is a nonglycosylated extracellular matrix (ECM) protein found in vasculature, lung, skin and connective tissue. Crosslinked elastin can be found in association with several microfibril-associated glycoproteins like fibrillin-1, fibrillin-2, fibronectin or fibulin-1 and fibulin-5 suggesting that these proteins contribute to elastic fiber assembly, structure and function.22, 23, 24 In the ECM, the final structure and adequate function of collagen and elastin fibers are dependent on the perfect function and anchoring of these essential matrix glycoprotein molecules.
Fibulins are widely expressed secretory glycoproteins with a complex structure including N- and O-glycosylated sites and are present in the basement membranes and the stroma of most tissues.12, 24, 25, 26 Their roles have been implicated in organogenesis and vasculogenesis.22, 25 In fibuline knockout mice, fragmented elastin can be detected in different tissues without an increase of elastase activity, indicating a defective development of elastic fibers, similar to that in cutis laxa syndromes.12 In some patients with ARCL type I and in a large family with a dominant form of cutis laxa, mutations have been detected in the fibulin 5 gene.1, 2, 3
An inborn error of metabolism leading to a combined defect of N- and O-glycosylation might alter the structure and therefore the function of several ECM glycoproteins, like fibulin 5. This may cause developmental defects already in the early embryonic period and lead to abnormal structure and malfunction of different tissues and organs. In the three patients described here with a cutis laxa syndrome, the combined N- and O-linked glycosylation defect might explain the presence of the characteristic phenotype without a fibulin 5 gene mutation, and the presence of features overlapping with other syndromes with N- or O-glycosylation defects. However, in the absence of an evident enzyme deficiency, the primary defect remains speculative.27
Recently Wu et al28 described patients with a lethal form of CDG syndrome and a defect in the COG7 subunit of the COG complex (conserved oligomeric Golgi complex). Disruption of this complex caused by COG7 deficiency leads to abnormal localization of enzymes and transporters resulting in severe hypoglycosylation of glycoproteins. The defect also affects both N- and O-glycosylation. These patients had a multisystem disease with neurological involvement, and wrinkled skin. The partial phenotypic overlap with the three cutis laxa cases further delineates the importance of O-linked protein glycosylation in the normal skin structure and brain development. The subunits of COG complex and other proteins, associated with the cytoplasmic surface of the Golgi apparatus, and involved in intracellular trafficking, are among the candidate proteins for the primary defect in our cutis laxa patients.
Our findings support the presence of a new cutis laxa syndrome, based on an inborn error of glycan biosynthesis. We suggest that all patients with cutis laxa syndrome, especially with microcephaly, late closure of the fontanels and associated features of the central nervous system, should be screened for glycosylation defects.
References
de Schepper S, Loeys B, de Paepe A, Lambert J, Naeyaert JM : Cutis laxa of the autosomal recessive type in a consanguineous family. Eur J Dermatol 2003; 13: 529–533.
Markova D, Zou Y, Ringpfeil F et al: Genetic heterogeneity of cutis laxa: a heterozygous tandem duplication within the fibulin-5 (FBLN5) gene. Am J Hum Genet 2003; 72: 998–1004.
Loeys B, Van Maldergem L, Mortier G et al: Homozygosity for a missense mutation in fibulin-5 (FBLN5) results in a severe form of cutis laxa. Hum Mol Genet 2002; 11: 2113–2118.
Zhang MC, He L, Giro M, Yong SL, Tiller GE, Davidson JM : Cutis laxa arising from frameshift mutations in exon 30 of the elastin gene (ELN). J Biol Chem 1998; 74: 981–986.
Andiran N, Sarikayalar F, Saraclar M, Caglar M : Autosomal recessive form of congenital cutis laxa: more than the clinical appearance. Pediatr Dermatol 2002; 19: 412–414.
Hashimoto K, Kanzaki T : Cutis laxa. Ultrastructural and biochemical studies. Arch Dermatol 1975; 111: 861–873.
Reisner SH, Seelenfreund M, Ben-Bassat M : Cutis laxa associated with severe intrauterine growth retardation and congenital dislocation of the hip. Acta Paediatr Scand 1971; 60: 357–360.
Beighton P : The dominant and recessive forms of cutis laxa. J Med Genet 1972; 9: 216–221.
Litzman J, Buckova H, Ventruba J, Holcikova A, Mikyska P, Lokaj J : A concurrent occurrence of cutis laxa, Dandy–Walker syndrome and immunodeficiency in a girl. Acta Paediatr 2003; 92: 861–864.
Biver A, De Rijcke S, Toppet V, Ledoux-Corbusier M, Van Maldergem L : Congenital cutis laxa with ligamentous laxity and delayed development, Dandy–Walker malformation and minor heart and osseous defects. Clin Genet 1994; 45: 318–322.
Van Maldergem L, Ogur G, Yuksel M : Facial anomalies in congenital cutis laxa with retarded growth and skeletal dysplasia. Am J Med Genet 1989; 32: 265.
Nakamura T, Lozano PR, Ikeda Y et al: Fibulin-5/DANCE is essential for elastogenesis in vivo. Nature 2002; 415: 171–175.
Kermane A, Tachfouti S, Lezrek M, Mohcine Z : Cutis laxa syndrome. Bull Soc Belge Ophtalmol 2004; 292: 5–8.
Tuysuz B, Arapoglu M, Ilikkan B, Demirkesen C, Perk Y : Congenital cutis laxa syndrome: type II autosomal recessive inheritance. Turk J Pediatr 2003; 45: 265–268.
Grünewald S, Matthijs G, Jaeken J : Congenital disorders of glycosylation: a review. Pediatr Res 2002; 52: 618–624.
Wopereis S, Grunewald S, Morava E et al: Apolipoprotein C-III isofocusing in the diagnosis of genetic defects in O-glycan biosynthesis. Clin Chem 2003; 49: 1839–1845.
Beltran-Valero de Bernabe D, Currier S, Steinbrecher A et al: Mutations in the O-mannosyltransferase gene POMT1 give rise to the severe neuronal migration disorder Walker–Warburg syndrome. Am J Hum Genet 2002; 71: 1033–1043.
Okajima T, Fukumoto S, Furukawa K, Urano T, Furukawa K : Molecular basis for the progeroid variant of Ehlers–Danlos syndrome. J Biol Chem 1999; 274: 2841–2844.
Wuyts W, Van Hul W : Molecular basis of multiple exostoses: mutations in the EXT1 and EXT2 genes. Hum Mutat 2000; 15: 220–227.
Topaz O, Shurman DL, Bergman R et al: Mutations in GALNT3, encoding a protein involved in O-linked glycosylation, cause familial tumoral calcinosis. Nat Genet 2004; 36: 579–581.
Van Eijk HG, van Noort WL : The analysis of human serum transferrins with the PhastSystem quantitation of microheterogeneity. Electrophoresis 1992; 13: 354–358.
Visconti RP, Barth JL, Keeley FW, Little CD : Codistribution analysis of elastin and related fibrillar proteins in early vertebrate development. Matrix Biol 2003; 22: 109–121.
Yanagisawa H, Davis EC, Starcher BC et al: Fibulin-5 is an elastin-binding protein essential for elastic fiber development in vivo. Nature 2002; 415: 168–171.
Argraves WS, Greene LM, Cooley MA, Gallagher WM : Fibulins: physiological and disease perspectives. EMBO Rep 2003; 4: 1127–1131.
Timpl R, Sasaki T, Kostka G, Chu ML : Fibulins: a versatile family of extracellular matrix proteins. Nat Rev Mol Cell Biol 2003; 4: 479–489.
Hansen JE, Lund O, Engelbrecht J, Bohr H, Nielsen JO, Hansen JE : Prediction of O-glycosylation of mammalian proteins: specificity patterns of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferase. Biochem J 1995; 308: 801–813.
Wopereis S, Morava E, Grunewald S et al: A combined defect in the biosynthesis of N- and O-glycans in patients with cutis laxa and neurological involvement: biochemical characteristics. Bioch Biophys Acta 2005; in press.
Wu X, Steet RA, Bohorov O et al: Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat Med 2004; 10: 518–523.
Acknowledgements
We thank Karin Huijben for the technical assistance and Dr Litzman for providing a serum sample of patient 4 for this study. The authors were supported by EUROGLYCANET 512131.
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Rights and permissions
About this article
Cite this article
Morava, E., Wopereis, S., Coucke, P. et al. Defective protein glycosylation in patients with cutis laxa syndrome. Eur J Hum Genet 13, 414–421 (2005). https://doi.org/10.1038/sj.ejhg.5201361
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201361
Keywords
This article is cited by
-
Genetik der kortikalen Fehlbildungen
Medizinische Genetik (2018)
-
Clinical glycomics for the diagnosis of congenital disorders of glycosylation
Journal of Inherited Metabolic Disease (2018)
-
Polymicrogyria and myoclonic epilepsy in autosomal recessive cutis laxa type 2A
neurogenetics (2016)
-
Mass spectrometry of transferrin and apolipoprotein C-III for diagnosis and screening of congenital disorder of glycosylation
Glycoconjugate Journal (2016)
-
Glycosylation disorders of membrane trafficking
Glycoconjugate Journal (2013)
