
Abstract
Prion diseases have stimulated intense scientific scrutiny since it was proposed that the infectious agent was devoid of nucleic acid. Despite this finding, genetics has played a key role in understanding the pathobiology and clinical aspects of prion disease through the effects of a series of polymorphisms and mutations in the prion protein gene (PRNP). The advent of variant Creutzfeldt–Jakob disease has confirmed one of the most powerful human genetic susceptibility factors, as all tested patients have an identical genotype at polymorphic codon 129 of PRNP. This review will also consider the accrued reports of inherited prion disease and attempt a genotype–phenotype correlation. The prospects for detection of novel genetic susceptibility factors using mouse models and human genetic association studies will be explored.
Similar content being viewed by others

Main
Prion diseases are a group of human and animal neurodegenerative conditions that have in common a key role for the prion protein in their pathogenesis. Traditionally, human prion diseases are clinically classified as Creutzfeldt–Jakob disease (CJD), Gerstmann–Straussler–Scheinker syndrome (GSS), fatal familial insomnia (FFI) and kuru. They may also be classified by aetiology as acquired (transmitted between animals or humans), inherited or sporadic (unknown cause). The primacy of a single protein in a disease with these diverse mechanisms is unique in biology.
There is now overwhelming evidence to support the ‘protein-only’ hypothesis of prion disease. The infectious agent of prion diseases is critically comprised of abnormal isoforms of a protein (PrP) encoded by PRNP. A disease-associated isoform of PrP acts as a template to promote the conversion of normal PrP to the pathological state.1 The normal prion protein is widely expressed and bound to the cell-surface by a glycosylphosphatidylinositol anchor, its function is unknown. PrP has an unstructured N-terminal domain of around 100 amino acids, and a structured C-terminal domain of similar size, which includes a single disulphide bond and two glycosylation sites. It is hypothesised that a range of abnormal PrP conformations and glycosylation states provides the basis for multiple prion ‘strains’ with consistent clinicopathological correlates (eg Bruce et al2). The genetic determinants of prion strain-type are beyond the scope of this review.
Human and mouse genetics have made major contributions to prion disease research. Perhaps most prominent among these was the linkage to chromosome 20 and mutation discovery in PRNP in families with dominantly inherited neurodegenerative diseases.3, 4 The fact that heterogeneous diseases caused by mutation of PRNP were known to be transmissible to laboratory animals obviated the need to the search for a cryptic infectious organism. A great deal is now known about the inherited prion diseases (IPD) as molecular genetic diagnosis has been available for over a decade. An explanation for the heterogeneity of this category has the potential to provide useful insights into the pathogenesis of the group as a whole.
PRNP comprises two exons (three in mice) with the entire open reading frame contained within the larger second exon. The two domains of PrP are distinct in terms of the corresponding genetic variation of PRNP (Figure 1). Codons 51–91 of the N-terminal domain encode a 5-mer repeat region consisting of a nonapeptide followed by four identical octapeptides. Alterations in the number of repeats are found as polymorphisms and pathogenic mutations, but there are no point mutations or common single-nucleotide polymorphisms (SNP) in this region. Point mutation causing IPD and a series of SNPs are found in the C-terminal domain. A common coding polymorphism at codon 129 of PRNP between methionine and valine (c.385A>G) has a critical role in susceptibility and modification of prion disease.

Definite or suspected pathogenic mutations are shown above this representation of the prion protein gene. Neutral or prion disease susceptibility/modifying polymorphisms are shown below. Nucleotide changes for those pathogenic mutations not discussed in the text are as follows: P105L (c.314C>T), G114V (c.341G>T), A117V (c.350C>T), G131V (c.392G>T), Y145X (c.435T>G), R148H (c.443G>A), Q160X (c.478C>T), V180I (c.538G>A), T183A (c.547A>G), H187R (c.560A>G), T188A (c.562A>G), T188 K (c.563C>A), T188R (c.563C>G), E196 K (c.586G>A), F198S (c.593T>C), D202N (c.604G>A), V203I (c.607G>A), R208H (c.623G>A), V210I (c.628G>A), E211Q (c.631G>C), Q212P (c.635A>G), Q217R (c.650A>G), M232R (c.695T>G) and M232T (c.695T>C). Nucleotide changes for synonymous changes not discussed in the text are as follows: P68P (c.204T>C), A117A (c.351A>G), G124G (c.372C>G), V161V (c.483G>A), H177 H (c.531C>T), Q212Q (c.636G>A), R228R (c.684A>G), S230S (c.690G>A).
In the last decade, prion diseases have provoked particular public and scientific scrutiny because of the epidemic of bovine spongiform encephalopathy in the UK and other European countries and the subsequent demonstration of its transmission to the human population as a new phenotype of acquired prion disease, variant Creutzfeldt-Jakob disease (vCJD).5, 6 Acquired prion diseases are known to have long silent incubation times and so uncertainty remains about the eventual size of the outbreak of vCJD.7 With justification, there is a strong suspicion that genetic susceptibility loci will be of importance.8
Inherited prion disease
All familial concurrence of prion disease is accounted for by mutation of PRNP, which in total are responsible for 10–15% of the incidence of prion disease.9, 10 There are three types of pathogenic PRNP mutation: point mutations leading to an amino-acid substitution or premature stop codon, and insertion of additional octapeptide repeats (OPRI) (Figure 1). Over 30 different mutations have been described: some are typically associated with particular clinical categories of prion disease; others are associated with a spectrum of clinical phenotypes, often with striking phenotypic variability. Despite the rarity of PRNP mutation, the protracted clinical course of some IPDs leads to a high population prevalence relative to sporadic and acquired prion diseases.
Historically, variation in clinical phenotype of IPD has been encapsulated by three clinical categories: GSS (Gerstmann, 1936, reviewed by Hainfellner et al11), FFI12, and familial Creutzfeldt–Jakob disease (fCJD) (Jakob, 1923, described by Brown et al13). The description of these syndromes predates molecular genetic diagnosis. The core features of these categories include slow progression of ataxia followed by later onset dementia in GSS; refractory insomnia, hallucinations, dysautonomia and motor signs in FFI; and rapidly progressive dementia, with myoclonus and pseudoperiodic discharges on electroencephalogram in fCJD. The clinical categories of IPD may be seen as extremes of phenotype, in reality the syndromes overlap considerably. With the development of genetic diagnosis, the usefulness of the clinical categories has waned.
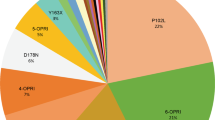
A number of surveillance reports and screening studies provide useful data about the epidemiology and importance of IPD. Mutation screening of dementia has shown that prion disease is a frequent cause of inherited early onset dementia. Finckh et al,14 for example, tested four genes (APP, PSEN1, PSEN2 and PRNP) in 36 patients with familial early onset dementia and found PRNP accounted for 4/12 mutation positive cases. A general epidemiological account of IPD is problematic, however, for a number of reasons. Firstly, there is considerable geographical variation in the incidence of IPD related to the population genetic effects of drift and migration on ancestral mutations. Secondly, an accurate incidence of IPD as a proportion of the totality of prion disease is difficult because of the poor overall ascertainment of prion disease. An autopsy study found that 40% of cases of neuropathological prion disease were undiagnosed while alive.15 Indeed, for some IPD, notably OPRI, the neuropathological features are subtle and might be overlooked without PrP immunocytochemistry (eg Case VII).16 These issues are compounded by the phenotypic scope of IPD, which overlaps those of the common causes of dementia in old age. The referral of cases to national CJD surveillance units is likely to be biased to the phenotype of sporadic CJD rather than Alzheimer's disease. Despite these issues, a number of units have reviewed the population incidence of IPD. In Germany, Windl found 40 cases of IPD from 578 suspect prion disease cases referred to a surveillance unit.10 In total, 13/40 cases were found to have D178N, 8/40 had E200K, 7/40 had P102L, 6/40 had V210I (c.628G>A) and 5/40 had OPRI. In France, however, Laplanche did not detect any D178N cases out of 57, but 8/57 had E200K.17 For Italian IPD, Pocchiari et al18 found 6/38 with D178N, 2/38 with P102L, and 30/38 with either E200K or V210I. In Finland, 12 familial cases out of 44 prion disease patients all had the D178N mutation.19 Goldfarb et al20 found that 45/54 worldwide families with IPD had the E200K mutation, 6/54 had D178N. In the UK, a single type of 6-OPRI mutation is the most frequently detected PRNP mutation. It remains to be seen if the large populations of South and East Asia will have a similar epidemiology of IPD, but if a generality is to be made in the absence of data from these regions, the most common worldwide PRNP mutations are E200K, D178N, P102L, and OPRI. These mutations illustrate the complexity of inherited prion disease well and will be explored further below.
Of the case reports considered in this review, the four common PRNP pathogenic mutations listed above comprised 350/492. Of the remaining 142 mutations shown in Figure 1, A117V, F198S, V210I are of particular interest in that they have been described in large pedigrees or multiple families worldwide.21, 22, 23, 24 A mutation at codon 145 generating a premature STOP codon is notable for a prominent vascular deposition of PrP and marked tau-protein histopathology.25, 26 Some of the other mutations occur in single cases or small families and their pathogenicity remains likely but unproven. In particular for genetic counselling, a lack of clarity regarding pathogenicity and penetrance is problematic.
Data collected from the 492 published cases are summarised in Figures 2 and 3. Three mutations are notable for their early age of onset, OPRI with insertion of more than five repeats, G114V27 and H187R.28 These mutations are associated with early neuropsychiatric symptoms and/or premorbid personality problems. As the three mutations are distributed across PRNP domains and different in type, it is possible that this aspect of clinical phenotype is consequent upon early age of onset of prion disease rather than a specific mutation related phenomenon. Further support for an association between age of onset and clinical phenotype is evidenced by a strong negative correlation between age of onset and duration of disease in the IPD group as a whole (duration vs age, pearson correlation −0.48, P<0.001, n=264). One possible explanation of this observation is deterioration in the ability of the ageing brain to degrade the putative toxic component of prion disease.29 A cluster of mutations towards the C-terminal end of PrP, together with the small OPRI mutations, share a late median age of onset and shorter duration of disease, mimicking sporadic CJD. It is unknown whether this is phenomenon specifically related to these mutations, or a more general finding related to their lower pathogenicity, later age of onset and the resulting impact of age on phenotype.

(a) Age of onset and (b) duration of disease distribution for 492 reported cases of inherited prion disease. In (b) the annualised number of cases with a duration of disease of 11–15 years is shown as 15, 16–20 years as 20 and 21–30+ years as 30.

Age of onset (no fill) and duration of disease (grey fill) for different PRNP mutations. Error bars show two times standard error of the mean.
The phenotypic variability of inherited prion disease is striking when compared with other neurodegenerative diseases. A multitude of possible explanations are only beginning to be characterised. Known or suspected genetic factors include the polymorphic codon 129 genotype of the mutant and wild-type alleles, the haplotype background of the mutation and unlinked genetic susceptibility loci. At the protein level, mutated PrPs appears to be able to fold into a number of different pathogenic conformers (eg Piccardo et al30 and our own observations). This diversity may be partly constrained by PrP primary structure, known as the conformation selection hypothesis.8 Diversity of PrP conformation within a single pedigree may account for phenotypic variability if different pathological conformers have differing toxicity or neuropathological targeting (Piccardo et al,30 Parchi et al,31 and our own observations). A related issue is the possibility of a variable contribution to the disease process from the product of the wild-type PRNP allele (Chen et al32 and our own observations).
E200K (c.598G>A)
The typical clinical presentation is a rapidly progressive dementia with myoclonus and pyramidal, cerebellar or extrapyramidal signs. Median age of onset of 112 reported cases (including some summarised data) was 58 with a median duration of 7 months.20, 33, 34, 35, 36, 37, 38, 39 Although the age of onset is slightly younger than that for sporadic CJD, Kahana found no unique clinical features that distinguish E200K patients from those with sporadic CJD.40 The neuropathology is also similar to sporadic CJD, with an absence of PrP plaques. However, in E200K patients with 129M homozygosity, a peculiar perpendicular stripe-like PrP deposit has been described in the molecular layer of the cerebellum.41
E200K is the most common cause of IPD worldwide. As mutation occurs by deamination of a CpG dinucleotide, it is not surprising that haplotype ancestry has shown multiple occurrences in human history.20, 42, 43 Multiple ancestral mutations have led to distinct geographical clusters with a high incidence of disease, most notably of Libyan Jews, which had prompted investigators to make an epidemiological link with ingestion of sheep's brain. At least four separate mutational events are responsible for the global distribution of the mutation.42 Clustering has led to the occurrence of a small number of individuals homozygous for the E200K mutation. These patients have a slightly earlier age of onset at 50, but overall, the homozygous and heterozygous phenotypes are similar, confirming the true dominance of this PRNP mutation.44
Expressivity of E200K is highly variable, manifesting in a wide range of age of onset of disease. Examination and genetic testing of unaffected relatives detect asymptomatic mutation carriers in old age, implying that penetrance is incomplete. The codon 129 polymorphism may determine these to a limited extent.39 The E219K polymorphism (c.655G>A) may also modify phenotype.43 Atypical clinical presentations include those with peripheral neuropathy,37, 45 supranuclear gaze palsy46 and with sleep disturbance,36 the pathogenesis of these atypical presentations is not well understood.
Octapeptide repeat insertion (OPRI, typically p.Q75_P76ins32_72)
Insertion of more than three additional octapeptide repeats in the N-terminal region of PrP causes inherited prion disease. Variability of clinical phenotype is again a major theme: the median age of onset of 109 reported OPRI patients (>3 repeats) was 35 (range 21–82) and duration of disease 7 years (range 3 months–21 years).47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62 The prototypic example is the insertion of an additional six extra octapeptide repeats. This mutation has some historical importance as it was the first described PRNP mutation, in a small UK family.3 Genealogical work by Poulter and Collinge63 in 1992 demonstrated a common ancestor of this small family and a larger pedigree with over 50 affected individuals. In 2005, this pedigree comprised over 80 affected individuals and 100 at-risk of disease affording a detailed analysis of phenotype and its determinants.
Clinical and pathological features of the UK pedigree are again highly variable. Cortical dementia, often with apraxia, is the core feature with additional neurological signs including cerebellar ataxia, pyramidal, extrapyramidal, myoclonus, chorea, seizures in declining order of frequency. Age of onset ranges from the third to the sixth decade and duration of disease ranges from an aggressive condition mistaken for sporadic CJD to a slowly progressive neurodegenerative disease over more than two decades. The polymorphism at PRNP codon 129 accounts for a proportion of this variability. Insert carriers associated with heterozygosity at codon 129 have a delayed age of onset by around a decade compared with patients homozygous at codon 129.63
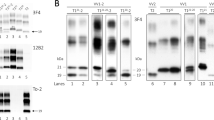
The neuropathology of OPRI mutation has been studied by Vital et al64 and King et al.65 The degree of spongiosis and astrocytosis seen was highly variable between cases. Obvious plaques of PrP deposition in the cerebellum were only seen in those patients with 8- or 9-OPRI mutations. For smaller OPRI mutations, PrP deposition was visualised by immunocytochemistry as elongated deposits in the molecular layer of the cerebellum (Figure 4).

Typical histological features of inherited prion disease 4-OPRI (96 bp), D178N and P102L. Histopathologically, classical GSS is associated with uni- or multicentric PrP plaques in the cerebellar and cerebral cortex with variable spongiform change and neurofibrillary tangles; FFI is associated with anterior and dorsomedial thalamic atrophy with variable extrathalamic spongiosis and gliosis; and classical fCJD is associated with cortical neuronal loss, spongiosis, gliosis and variable PrP deposits. 4-OPRI shows stripe-like deposits of PrPSc in the molecular layer of the cerebellum, D178N shows spongiform change and ‘fluffy’ deposits of PrPSc and P102L shows multiple unicentric plaques of PrPSc deposition. Immunohistochemistry with prion protein antibody ICSM35 is shown. Scale bar is 150 μm.
The existence of a premorbid personality disorder in prion disease was first reported in this pedigree, characterised by criminality, aggression, delinquency and hypersexuality.16 Other clinicians have ascribed psychiatric symptoms in IPD to the early stages of a neurodegenerative disease (eg Rodriguez et al27 and Laplanche et al61). The presence of personality disorders with such an early onset may indicate a role for the normal function of PrP in the healthy brain that is abrogated by certain mutations. The related issue of identifying clinical onset of inherited prion disease will become increasingly important as treatments become available.
A number of reports of prion disease associated with small OPRI (<4 extra repeats) appear to have a distinct phenotype relative to the larger repeat insertions, with a lack of family history, later age of onset and shorter duration. This has been noted by Croes et al,66 finding a negative correlation between age of onset and size of the insertion. Small insertions have been detected incidentally or by population surveys.67 It remains possible that small OPRI are neutral rare polymorphisms that CJD surveillance units will occasionally detect in elderly patients referred with otherwise sporadic CJD.
P102L (c.305C>T)
The P102L mutation, first reported in 1989 in a UK and US family,4 is the archetypal example of the GSS syndrome (Figure 4), which was later described in association with the less frequently occurring F198S, A117V, P105L, G131V, Y145X, H187R and some D178N mutations. Phenotypic variability is once more a major feature to a similar extent as OPRI: median age of onset in 52 reported cases was 50 (range 25–70) and median duration of disease was 4 years (range 5 months–17 years).11, 30, 68, 69, 70, 71 The traditional GSS phenotype of a slowly progressive ataxia with later dementia is commonly found, but families with amyotrophic features72 and patients with a rapid course are seen. Interestingly, in marked contrast to OPRI, there appears to be only limited modifying effect of codon 129 in P102L cases (median age of onset for 12 codon 129 homozygous cases was 53 compared with a median age of 55 for 7 heterozygous cases).
D178N (c.532G>A)
This mutation was first reported in Finnish families with a CJD-like illness.73 Subsequently, a large family case series was reported by Medori et al,74 who described an untreatable insomnia, dysautonomia, and myoclonus. Histopathologically, selective degeneration of the anteroventral and dorsomedial thalamus was seen with cortical pathology and weak PrP immunocytochemical staining (Figure 4). This clinicopathological description is termed FFI. In 1992, Goldfarb et al75 established a haplotypic relationship between codon 129 and 178, whereby the mutation on a 129M chromosome leads to FFI, and the mutation on a 129V chromosome leads to familial CJD. More recent experience questions such a tight association between haplotype and phenotype: some families do not obey the rule,76 there are pedigrees that segregate both the FFI and CJD phenotype,77 and it has become increasingly recognised that autonomic and sleep disturbances may accompany other PRNP mutations, sometimes overtly.36 Cases reported to the CJD surveillance unit in Germany were less clinically distinct than the first reported FFI families in that none were clinically diagnosed as FFI due to an absence of obvious insomnia and no positive family history was obtained in 4/9.78 Pocchiari et al18 noted that of patients presenting with overt sleep disturbance to a CJD unit, 4/9 had sporadic CJD, 1/9 had a V210I mutation, but none had D178N. Of the 72 case reports reviewed here, the median age of onset was 50 (range 20–71) and median duration of disease was 11 months (range 5 months–4 years).
PRNP polymorphisms and prion disease susceptibility
The coding sequences of mammalian prion protein genes are highly conserved, in a similar way to other structured proteins, presumably by deleterious selection against coding mutations. Despite this conservation, a number of human coding polymorphisms have been characterised in different populations that achieve intermediate frequencies.79, 80 First and foremost among these is a polymorphism at PRNP codon 129 between methionine and valine, which has a strong disease susceptibility and phenotype modifying effect.81 There are marked differences in codon 129 allele frequency worldwide: in the UK and Northern European population, the 129M allele frequency is around 0.65 with a slightly increasing cline through Europe into Africa79, 82 and a marked increasing cline through Asia,67, 83, 84 129V is rare in Japan. 129V is the more frequent PRNP allele in only two regions: the Eastern Highlands of Papua New Guinea (PNG) at 0.55, and some Native American populations. A polymorphism at codon 219 between glutamine and lysine (E219K, c.655G>A), found in East Asian populations, also has a profound effect on susceptibility to prion disease.83 Polymorphisms at codons 14279 (G142S, c.424G>A) and 171 (N171S, c.512A>G), most frequent in African populations, have not been tested for disease susceptibility effects. A 1-octapeptide repeat deletion (1-OPRD), of which there are differing genetic types with identical protein products, occurs as an uncommon polymorphism in the European population. There is good evidence that 1-OPRD has no susceptibility effect in sporadic CJD.85
Codon 129 has a susceptibility or modifying effect in all categories of prion disease. Variant CJD demonstrates this effect most dramatically: all genetically tested cases have been homozygous for 129M.86, 87 A single codon 129 heterozygous patient, who had received blood from donor subsequently diagnosed with vCJD, was found to have widespread prion protein deposition in the peripheral lymphoreticular system at autopsy, having died of an unrelated cause.88 It remains to be seen whether individuals of different genotypes will succumb to vCJD, or a similar phenotype of prion disease, in the BSE-exposed UK and other European populations. Thankfully, the annual incidence of vCJD appears to be declining, although it will be some time before it will become clear whether the disease is in permanent decline.7
Outbreaks of acquired prion disease have been caused by human–human transmission of prions by cadaver derived growth hormone injections, the use of dura mater in neurosurgery, corneal transplantation, and intracerebral electrodes. These outbreaks have remained small due to the limited exposed group but patients with a heterozygous genotype at codon 129 are protected. Our largest experience of an acquired human prion disease comes from kuru: a devastating epidemic of prion disease spread by ritualistic cannibalism in the Fore and neighbouring linguistic groups of the Eastern Highlands of PNG. At its peak in the 1950s, kuru was the leading cause of death in young women in the South Fore. Although precise epidemiological data are not available, both methionine and valine homozygous individuals had a mean onset of disease in early adulthood, whereas heterozygous individuals had a delayed onset into middle age.89 Such a high disease incidence, coupled with the effect of heterozygosity to delay disease onset through reproductive age, affords the opportunity for strong selection pressure. Surviving elderly women in the present-day South Fore population, who lived through the epidemic, show marked Hardy–Weinberg disequilibrium at PRNP with an excess of codon 129 heterozygosity, illustrating the population genetic effect of the kuru epidemic.79, 90 The strong evolutionary selection pressure in favour of prion disease resistance alleles in the Fore population provides an opportunity to test for the presence of other resistance loci.
Sporadic CJD also shows a susceptibility effect at both codon 129 and 219. Again, the susceptible individuals are homozygous for codon 129M or 129V. At codon 219, the rarity of the 219K allele precludes inference about heterozygosity; however, no cases of sporadic CJD with either one or two 219K alleles have ever been reported.83
The search for other prion disease susceptibility loci
The laboratory mouse has proved to be a useful genetic model of acquired prion disease. After intracerebral inoculation with prions, mice succumb after an incubation time that may vary from 100 days to a natural lifespan. Many factors influence this time: the route of inoculation (brain shortest, oral longest), dose, prion strain and species the inoculum was derived from, and importantly, the genetic background of the recipient. When these are controlled for, intracerebral inoculation of defined inbred mouse strains in a laboratory environment results in population incubation times with remarkably small variation. It is now well established that different inbred mouse strains have distinct incubation times, a finding first reported by Dickinson and MacKay in 1964.91 Prnp alleles are a major, but not the only, contributor to this phenotypic variability. Haplotypes comprised of alleles at codons 108 and 189 of the mouse prion protein gene confer a short or long incubation time.
The use of prion disease incubation time in mice as a quantitative trait has prompted a series of mapping studies. These utilise a backcross or an F2-intercross of inbred mouse strains with indentical Prnp alleles. Over 20 loci have been mapped on eight mouse chromosomes, although to-date, none of quantitative trait nucleotides (QTN) responsible for these loci have been characterised. Fine mapping of these loci is underway using congenics and alternative genetic strategies.
Human studies have been similarly unsuccessful, suffering from necessarily small sample sizes related to the rarity of prion disease and the lack of technology for a true genome-wide association study. Various candidate regions have been examined, including putative polymorphisms of regulatory regions of PRNP. Strong linkage disequilibrium exists around PRNP including at least 10 kb of upstream sequence leading to two major haplotype groups in the European population containing either 129M or 129V.92 One methionine haplotype was found to be overrepresented in a series of sporadic CJD patients, but the effect was small and this finding has not been replicated. Similarly, a putative association of vCJD and the HLA-DQB7 serogroup has not been replicated in a larger sample.93, 94 The prion-like doppel gene (PRND)95, 96, 97, 98, 99 and APOE genotypes100, 101 have also been tested for association with prion disease, with inconclusive results.
Despite the rarity of human prion disease and the current lack of success in identifying new susceptibility genes, there should still be optimism as the technology for genome-wide association study will very soon be available. The rewards for a detailed understanding of prion disease susceptibility, including the identification of high-risk groups, improving animal models, and rational therapeutics, are potentially significant.
Reference sequences
CCDS Database CCDS13080.1
References
Prusiner SB : Novel proteinaceous infectious particles cause scrapie. Science 1982; 216: 136–144.
Bruce ME, Will RG, Ironside JW et al: Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature 1997; 389: 498–501.
Owen F, Poulter M, Lofthouse R et al: Insertion in prion protein gene in familial Creutzfeldt–Jakob disease. Lancet 1989; 1: 51–52.
Hsiao K, Baker HF, Crow TJ et al: Linkage of a prion protein missense variant to Gerstmann–Straussler syndrome. Nature 1989; 338: 342–345.
Will RG, Ironside JW, Zeidler M et al: A new variant of Creutzfeldt–Jakob disease in the UK. Lancet 1996; 347: 921–925.
Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF : Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature 1996; 383: 685–690.
Clarke P, Ghani AC : Projections of the future course of the primary vCJD epidemic in the UK: includsion of subclinical infection and the possibility of wider genetic susceptibility. J Roy Soc Interf 2005; 1–13.
Collinge J : Variant Creutzfeldt–Jakob disease. Lancet 1999; 354: 317–323.
Masters CL, Gajdusek DC, Gibbs Jr CJ : The familial occurrence of Creutzfeldt–Jakob disease and Alzheimer's disease. Brain 1981; 104: 535–558.
Windl O, Giese A, Schulz-Schaeffer W et al: Molecular genetics of human prion diseases in Germany. Hum Genet 1999; 105: 244–252.
Hainfellner JA, Brantner-Inthaler S, Cervenáková L et al: The original Gerstmann–Straussler–Scheinker family of Austria: divergent clinicopathological phenotypes but constant PrP genotype. Brain Pathol 1995; 5: 201–211.
Lugaresi E, Medori R, Baruzzi PM et al: Fatal familial insomnia and dysautonomia, with selective degeneration of thalamic nuclei. N Engl J Med 1986; 315: 997–1003.
Brown P, Cervenákova L, Boellaard JW, Stavrou D, Goldfarb LG, Gajdusek DC : Identification of a PRNP gene mutation in Jakob's original Creutzfeldt–Jakob disease family. Lancet 1994; 344: 130–131.
Finckh U, Muller-Thomsen T, Mann U et al: High prevalence of pathogenic mutations in patients with early-onset dementia detected by sequence analyses of four different genes. Am J Hum Genet 2000; 66: 110–117.
Bruton CJ, Bruton RK, Gentleman SM, Roberts GW : Diagnosis and incidence of prion (Creutzfeldt–Jakob) disease: a retrospective archival survey with implications for future research. Neurodegeneration 1995; 4: 357–368.
Collinge J, Brown J, Hardy J et al: Inherited prion disease with 144 base pair gene insertion: II: Clinical and pathological features. Brain 1992; 115: 687–710.
Laplanche J-L, Delasnerie-Lauprêtre N, Brandel JP et al: Molecular genetics of prion diseases in France. French Research Group on Epidemiology of Human Spongiform Encephalopathies. Neurology 1994; 44: 2347–2351.
Pocchiari M, Ladogana A, Petraroli R, Cardone F, D’Alessandro M : Recent Italian FFI cases. Brain Pathol 1998; 8: 564–566.
Kovanen J : Clinical characteristics of familial and sporadic Creutzfeldt–Jakob disease in Finland. Acta Neurol Scand 1993; 87: 469–474.
Goldfarb LG, Brown P, Mitrova E et al: Creutzfeldt–Jacob disease associated with the PRNP codon 200Lys mutation: an analysis of 45 families. Eur J Epidemiol 1991; 7: 477–486.
Mallucci GR, Campbell TA, Dickinson A et al: Inherited prion disease with an alanine to valine mutation at codon 117 in the prion protein gene. Brain 1999; 122: 1823–1837.
Tranchant C, Doh-ura K, Warter JM et al: Gerstmann–Sträussler–Scheinker disease in an Alsatian family: clinical and genetic studies. J Neurol Neurosurg Psychiatry 1992; 55: 185–187.
Dlouhy SR, Hsiao K, Farlow MR et al: Linkage of the Indiana kindred of Gerstmann–Sträussler–Scheinker disease to the prion protein gene. Nat Genet 1992; 1: 64–67.
Pocchiari M, Salvatore M, Cutruzzolá F et al: A new point mutation of the prion protein gene in Creutzfeldt–Jakob disease. Ann Neurol 1993; 34: 802–807.
Kitamoto T, Iizuka R, Tateishi J : An amber mutation of prion protein in Gerstmann–Straussler syndrome with mutant PrP plaques. Biochem Biophys Res Commun 1993; 192: 525–531.
Ghetti B, Piccardo P, Spillantini MG et al: Vascular variant of prion protein cerebral amyloidosis with τ-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci USA 1996; 93: 744–748.
Rodriguez MM, Peoc’h K, Haik S et al: A novel mutation (G114V) in the prion protein gene in a family with inherited prion disease. Neurology 2005; 64: 1455–1457.
Hall DA, Leehey MA, Filley CM et al: PRNP H187R mutation associated with neuropsychiatric disorders in childhood and dementia. Neurology 2005; 64: 1304–1306.
Mallucci G, Collinge J : Rational targeting for prion therapeutics. Nat Rev Neurosci 2005; 6: 23–34.
Piccardo P, Dlouhy SR, Lievens PMJ et al: Phenotypic variability of Gerstmann–Straussler–Scheinker disease is associated with prion protein heterogeneity. J Neuropathol Exp Neurol 1998; 57: 979–988.
Parchi P, Chen SG, Brown P et al: Different patterns of truncated prion protein fragments correlate with distinct phenotypes in P102L Gerstmann–Sträussler–Scheinker disease. Proc Natl Acad Sci USA 1998; 95: 8322–8327.
Chen SG, Parchi P, Brown P et al: Allelic origin of the abnormal prion protein isoform in familial prion diseases. Nat Med 1997; 3: 1009–1015.
Bertoni JM, Brown P, Goldfarb LG, Rubenstein R, Gajdusek DC : Familial Creutzfeldt–Jakob disease (codon 200 mutation) with supranuclear palsy. JAMA 1992; 268: 2413–2415.
Inoue I, Kitamoto T, Doh-ura K, Shii H, Goto I, Tateishi J : Japanese family with Creutzfeldt–Jakob disease with codon 200 point mutation of the prion protein gene. Neurology 1994; 44: 299–301.
Collinge J, Palmer MS, Campbell TA, Sidle KCL, Carroll D, Harding AE : Inherited prion disease (PrP lysine 200) in Britain: two case reports. BMJ 1993; 306: 301–302.
Chapman J, Arlazoroff A, Goldfarb LG et al: Fatal insomnia in a case of familial Creutzfeldt–Jakob disease with the codon 200Lys mutation. Neurology 1996; 46: 758–761.
Antoine JC, Laplanche JL, Mosnier JF, Beaudry P, Chatelain J, Michel D : Demyelinating peripheral neuropathy with Creutzfeldt–Jakob disease and mutation at codon 200 of the prion protein gene. Neurology 1996; 46: 1123–1127.
Miyakawa T, Inoue K, Iseki E et al: Japanese Creutzfeldt–Jakob disease patients exhibiting high incidence of the E200K PRNP mutation and located in the basin of a river. Neurol Res 1998; 20: 684–688.
Mitrova E, Belay G : Creutzfeldt–Jakob disease with E200 K mutation in Slovakia: characterization and development. Acta Virol 2002; 46: 31–39.
Kahana E, Zilber N : Do Creutzfeldt–Jakob disease patients of Jewish Libyan origin have unique clinical features? Neurology 1991; 41: 1390–1392.
Jarius C, Kovacs GG, Belay G, Hainfellner JA, Mitrova E, Budka H : Distinctive cerebellar immunoreactivity for the prion protein in familial (E200K) Creutzfeldt–Jakob disease. Acta Neuropathol (Berl) 2003; 105: 449–454.
Lee HS, Sambuughin N, Cervenakova L et al: Ancestral origins and worldwide distribution of the PRNP 200 K mutation causing familial Creutzfeldt–Jakob disease. Am J Hum Genet 1999; 64: 1063–1070.
Seno H, Tashiro H, Ishino H, Inagaki T, Nagasaki M, Morikawa S : New haplotype of familial Creutzfeldt–Jakob disease with a codon 200 mutation and a codon 219 polymorphism of the prion protein gene in a Japanese family. Acta Neuropathol (Berl) 2000; 99: 125–130.
Simon ES, Kahana E, Chapman J et al: Creutzfeldt–Jakob disease profile in patients homozygous for the PRNP E200K mutation. Ann Neurol 2000; 47: 257–260.
Chapman J, Brown P, Goldfarb LG, Arlazoroff A, Gajdusek DC, Korczyn AD : Clinical heterogeneity and unusual presentations of Creutzfeldt–Jakob disease in Jewish patients with the PRNP codon 200 mutation. J Neurol Neurosurg Psychiatry 1993; 56: 1109–1112.
Bertoni JM, Label LS, Sackelleres JC, Hicks SP : Supranuclear gaze palsy in familial Creutzfeldt–Jakob disease. Arch Neurol 1983; 40: 618–622.
Goldfarb LG, Brown P, Little BW et al: A new (two-repeat) octapeptide coding insert mutation in Creutzfeldt–Jakob disease. Neurology 1993; 43: 2392–2394.
Laplanche JL, Delasnerie Laupretre N, Brandel JP, Dussaucy M, Chatelain J, Launay JM : Two novel insertions in the prion protein gene in patients with late-onset dementia. Hum Mol Genet 1995; 4: 1109–1111.
Campbell TA, Palmer MS, Will RG, Gibb WRG, Luthert P, Collinge J : A prion disease with a novel 96-base pair insertional mutation in the prion protein gene. Neurology 1996; 46: 761–766.
Goldfarb LG, Brown P, McCombie WR et al: Transmissible familial Creutzfeldt–Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci USA 1991; 88: 10926–10930.
Skworc KH, Windl O, Schulz-Schaeffer WJ et al: Familial Creutzfeldt–Jakob disease with a novel 120-bp insertion in the prion protein gene. Ann Neurol 1999; 46: 693–700.
Cochran EJ, Bennett DA, Cervenáková L et al: Familial Creutzfeldt–Jakob disease with a five-repeat octapeptide insert mutation. Neurology 1996; 47: 727–733.
Nicholl D, Windl O, De Silva R et al: Inherited Creutzfeldt–Jakob disease in a British family associated with a novel 144 base pair insertion of the prion protein gene. J Neurol Neurosurg Psychiatry 1995; 58: 65–69.
Oda T, Kitamoto T, Tateishi J et al: Prion disease with 144 base pair insertion in a Japanese family line. Acta Neuropathol (Berl) 1995; 90: 80–86.
Capellari S, Vital C, Parchi P et al: Familial prion disease with a novel 144-bp insertion in the prion protein gene in a Basque family. Neurology 1997; 49: 133–141.
Kitamoto T, Doh-ura K, Muramoto T, Miyazono M, Tateishi J : The primary structure of the prion protein influences the distribution of abnormal prion protein in the central nervous system. Am J Pathol 1992; 141: 271.
Brown P, Goldfarb LG, McCombie WR et al: Atypical Creutzfeldt–Jakob disease in an American family with an insert mutation in the PRNP amyloid precursor gene. Neurology 1992; 42: 422–427.
Levy E, Carman MD, Fernandez Madrid IJ et al: Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990; 248: 1124–1126.
Goldfarb LG, Brown P, Vrbovska A et al: An insert mutation in the chromosome 20 amyloid precursor gene in a Gerstmann–Straussler–Scheinker family. J Neurol Sci 1992; 111: 189–194.
Van Gool WA, Hensels GW, Hoogerwaard EM, Wiezer JHA, Wesseling P, Bolhuis PA : Hypokinesia and presenile dementia in a Dutch family with a novel insertion in the prion protein gene. Brain 1995; 118: 1565–1571.
Laplanche JL, El Hachimi KH, Durieux I et al: Prominent psychiatric features and early onset in an inherited prion disease with a new insertional mutation in the prion protein gene. Brain 1999; 122: 2375–2386.
Krasemann S, Zerr I, Weber T et al: Prion disease associated with a novel nine octapeptide repeat insertion in the PRNP gene. Brain Res Mol Brain Res 1995; 34: 173–176.
Poulter M, Baker HF, Frith CD et al: Inherited prion disease with 144 base pair gene insertion: I: genealogical and molecular studies. Brain 1992; 115: 675–685.
Vital C, Gray F, Vital A et al: Prion encephalopathy with insertion of octapeptide repeats: the number of repeats determines the type of cerebellar deposits. Neuropathol Appl Neurobiol 1998; 24: 125–130.
King A, Doey L, Rossor M, Mead S, Collinge J, Lantos P : Phenotypic variability in the brains of a family with a prion disease characterized by a 144-base pair insertion in the prion protein gene. Neuropathol Appl Neurobiol 2003; 29: 98–105.
Croes EA, Theuns J, Houwing-Duistermaat JJ et al: Octapeptide repeat insertions in the prion protein gene and early onset dementia. J Neurol Neurosurg Psychiatry 2004; 75: 1166–1170.
Yu SL, Jin L, Sy MS et al: Polymorphisms of the PRNP gene in Chinese populations and the identification of a novel insertion mutation. Eur J Hum Genet 2004.
Young K, Clark HB, Piccardo P, Dlouhy SR, Ghetti B : Gerstmann–Straussler–Scheinker disease with the PRNP P102L mutation and valine at codon 129. Mol Brain Res 1997; 44: 147–150.
Barbanti P, Fabbrini G, Salvatore M et al: Polymorphism at codon 129 or codon 219 of PRNP and clinical heterogeneity in a previously unreported family with Gerstmann–Straussler–Scheinker disease (PrP-P102L mutation). Neurology 1996; 47: 734–741.
Adam J, Crow TJ, Duchen LW, Scaravilli F, Spokes E : Familial cerebral amyloidosis and spongiform encephalopathy. J Neurol Neurosurg Psychiatry 1982; 45: 37–45.
Tanaka Y, Minematsu K, Moriyasu H et al: A Japanese family with a variant of Gerstmann-Straussler-Scheinker disease. J Neurol Neurosurg Psychiatry 1997; 62: 454–457.
Kretzschmar HA, Kufer P, Riethmuller G, DeArmond SJ, Prusiner SB, Schiffer D : Prion protein mutation at codon 102 in an Italian family with Gerstmann–Straussler–Scheinker syndrome. Neurology 1992; 42: 809–810.
Goldfarb LG, Haltia M, Brown P et al: New mutation in scrapie amyloid precursor gene (at codon 178) in Finnish Creutzfeldt–Jakob kindred. Lancet 1991; 337: 425.
Medori R et al: in Prusiner SB, Collinge J, Powell J, Anderton B (eds): Prion Diseases of Humans and Animals. London: Ellis Horwood, 1992, pp 180–187.
Goldfarb LG, Petersen RB, Tabaton M et al: Fatal familial insomnia and familial Creutzfeldt–Jakob disease: disease phenotype determined by a DNA polymorphism. Science 1992; 258: 806–808.
Brown P, Goldfarb LG, Kovanen J et al: Phenotypic characteristics of familial Creutzfeldt–Jakob disease associated with the codon 178AsnPRNP mutation. Ann Neurol 1992; 31: 282–285.
McLean CA, Storey E, Gardner RJM, Tannenberg AEG, Cervenáková L, Brown P : The D178N (cis-129M) ‘fatal familial insomnia’ mutation associated with diverse clinicopathologic phenotypes in an Australian kindred. Neurology 1997; 49: 552–558.
Zerr I, Giese A, Windl O et al: Phenotypic variability in fatal familial insomnia (D178N-129M) genotype. Neurology 1998; 51: 1398–1405.
Mead S, Stumpf MP, Whitfield J et al: Balancing selection at the prion protein gene consistent with prehistoric Kurulike epidemics. Science 2003; 300: 640–643.
Soldevila M, Calafell F, Andres AM et al: Prion susceptibility and protective alleles exhibit marked geographic differences. Hum Mutat 2003; 22: 104–105.
Palmer MS, Dryden AJ, Hughes JT, Collinge J : Homozygous prion protein genotype predisposes to sporadic Creutzfeldt–Jakob disease. Nature 1991; 352: 340–342.
Lucotte G, Mercier G : The population distribution of the Met allele at the PRNP129 polymorphism (a high risk factor for Creutzfeldt–Jakob disease) in various regions of France and in West Europe. Infect Genet Evol 2005; 5: 141–144.
Shibuya S, Higuchi J, Shin RW, Tateishi J, Kitamoto T : Codon 219 Lys allele of PRNP is not found in sporadic Creutzfeldt–Jakob disease. Ann Neurol 1998; 43: 826–828.
Jeong BH, Nam JH, Lee YJ et al: Polymorphisms of the prion protein gene (PRNP) in a Korean population. J Hum Genet 2004.
Palmer MS, Mahal SP, Campbell TA et al: Deletions in the prion protein gene are not associated with CJD. Hum Mol Gen 1993; 2: 541–544.
Zeidler M, Stewart G, Cousens SN, Estebeiro K, Will RG : Codon 129 genotype and new variant CJD. Lancet 1997; 350: 668.
Hill AF, Butterworth RJ, Joiner S et al: Investigation of variant Creutzfeldt–Jakob disease and other human prion diseases with tonsil biopsy samples. Lancet 1999; 353: 183–189.
Peden AH, Head MW, Ritchie DL, Bell JE, Ironside JW : Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 2004; 364: 527–529.
Cervenakova L, Goldfarb L, Garruto R, Lee HS, Gajdusek CD, Brown P : Phenotype-genotype studies in kuru: implications for new variant Creutzfeldt–Jakob disease. Proc Natl Acad Sci USA 1999; 95: 13239–13241.
Lee HS, Brown P, Cervenáková L et al: Increased susceptibility to Kuru of carriers of the PRNP 129 methionine/methionine genotype. J Infect Dis 2001; 183: 192–196.
Dickinson AG, Mackay JMW : Genetical control of the incubation period in mice of the neurological disease, scrapie. Heredity (Edinburgh) 1964; 19: 279–288.
Mead S, Mahal SP, Beck J et al: Sporadic – but not variant – Creutzfeldt–Jakob disease is associated with polymorphisms upstream of PRNP exon 1. Am J Hum Genet 2001; 69: 1225–1235.
Jackson GS, Beck JA, Navarrete C et al: HLA-DQ7 antigen and resistance to variant CJD. Nature 2001; 414: 269–270.
Pepys MB, Bybee A, Booth DR et al: MHC typing in variant Creutzfeldt–Jakob disease. Lancet 2003; 361: 487–489.
Jeong BH, Kim NH, Kim JI, Carp RI, Kim YS : Polymorphisms at codons 56 and 174 of the prion-like protein gene (PRND) are not associated with sporadic Creutzfeldt–Jakob disease. J Hum Genet 2005.
Peoc’h K, Guerin C, Brandel JP, Launay JM, Laplanche JL : First report of polymorphisms in the prion-like protein gene (PRND): implications for human prion diseases. Neurosci Lett 2000; 286: 144–148.
Mead S, Beck J, Dickinson A, Fisher EMC, Collinge J : Examination of the human prion protein-like gene Doppel for genetic susceptibility to sporadic and variant Creutzfeldt–Jakob disease. Neurosci Lett 2000; 290: 117–120.
Croes EA, Alizadeh BZ, Bertoli-Avella AM et al: Polymorphisms in the prion protein gene and in the doppel gene increase susceptibility for Creutzfeldt–Jakob disease. Eur J Hum Genet 2004.
Jeong BH, Kim NH, Choi EK et al: Polymorphism at 3′ UTR +28 of the prion-like protein gene is associated with sporadic Creutzfeldt–Jakob disease. Eur J Hum Genet 2005.
Chapman J, Cervenakova L, Petersen RB et al: APOE in non-Alzheimer amyloidoses: transmissible spongiform encephalopathies. Neurology 1998; 51: 548–553.
Van Everbroeck B, Croes EA, Pals P et al: Influence of the prion protein and the apolipoprotein E genotype on the Creutzfeldt–Jakob Disease phenotype. Neurosci Lett 2001; 313: 69–72.
Acknowledgements
Ray Young assisted with figure design. John Beck prepared Figure 1. Jacqueline Linehan and Professor Sebastian Brandner prepared Figure 3. The author is funded by the Medical Research Council, UK. Professor John Collinge is Head of the Department of Neurodegenerative disease and Director MRC Prion Unit.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is cited by
-
Sporadic Creutzfeldt–Jakob Disease in the young (50 and below): 10-year review of United Kingdom surveillance
Journal of Neurology (2023)
-
Cell biology of prion strains in vivo and in vitro
Cell and Tissue Research (2023)
-
The importance of ongoing international surveillance for Creutzfeldt–Jakob disease
Nature Reviews Neurology (2021)
-
Bank vole prion protein extends the use of RT-QuIC assays to detect prions in a range of inherited prion diseases
Scientific Reports (2021)
-
Environmental and host factors that contribute to prion strain evolution
Acta Neuropathologica (2021)
