
Abstract
Hereditary nonpolyposis colorectal cancer (HNPCC) is an autosomal dominant syndrome caused by germline mutations of the mismatch repair (MMR) genes. Only a few studies have taken into account the selection of families tested for these mutations in estimating colorectal cancer (CRC) risk in carriers. They found much lower estimates of CRC risks than previous ones, but these estimates lacked precision despite the large number of families. The aim of this study was to evaluate the efficiency of the ‘genotype restricted likelihood’ (GRL) method that provides unbiased estimates of risks whatever the ascertainment process of families, and to estimate CRC and endometrial cancer risk for carriers of the MMR genes. Efficiency of the GRL method was evaluated using simulations. Risks were estimated from a sample of 36 families diagnosed with HNPCC and carrying a mutation of MSH2 or MLH1, ascertained through a cancer family clinic in Lyon (France). The efficiency of the GRL method was found to be strongly dependent on the proportion of family members tested. By age 70 years, CRC risk was estimated at 47% (95% confidence interval: 12–98%) for men and 33% (95% confidence interval: 24–54%) for women. The endometrial cancer risk was only 14% (confidence interval: 6–20%). As methods allowing for the selection of families lack efficiency, large-scale family studies should be undertaken and data should be pooled to provide reliable and precise estimates of risks for an optimal familial management.
Similar content being viewed by others


Introduction
Hereditary nonpolyposis colorectal cancer (HNPCC) is an autosomal dominant syndrome that predisposes carriers to colorectal and endometrial cancer and cancers of other organs.1 Mutations of the mismatch repair (MMR) genes (essentially MLH1, MSH2 and MSH6) have been shown to be responsible for a majority of families with this syndrome. Mutations are usually identified in families that fulfil the so-called Amsterdam criteria.2, 3 These criteria include having three close relatives with an HNPCC-associated cancer (of the colon, rectum, endometrium, small bowel, ureter or renal pelvis). If a mutation is identified in one family member (index case), genetic testing is offered to relatives. If they are found to be carriers, they may undergo intensive surveillance, which considerably improves the prognosis of the disease.
Most studies estimate the risk of colorectal cancer in families with HNPCC syndrome selected according to Amsterdam criteria without correcting for selection bias.4, 5, 6, 7, 8, 9 Estimates in these studies range from 0.68 to 0.82, but these values have been shown to be substantially overestimated.10 Only a few studies have taken the ascertainment process into account,11, 12, 13 and their estimates are lower than those of the other studies. Penetrance values for endometrial cancer range from 0.4 to 0.6.4, 6, 8, 11 Because the criteria used to select families did not include this tumour, these values should be unbiased.
We proposed an ascertainment-adjusted method for estimating the age-specific cumulative risk (penetrance) of a given disease associated with deleterious mutations in families in which these mutations have been identified.14 This likelihood, called the ‘genotype-restricted likelihood’ (GRL), provides unbiased penetrance estimates, regardless of the criteria used to select the families and without modelling the ascertainment process. It also corrects for the bias that is introduced by selection according to genotype and which is inherent in this selection because genotypes are available in relatives only if a mutation is detected in the index case.
In the most recent study of Quehenberger et al,13 endometrial and colorectal cancer risks were estimated for carriers of the MLH1 and MSH2 gene by using a maximum likelihood method that corrected for ascertainment by conditioning on all observed phenotypes, as in the GRL method. They confirmed that previous estimates of colorectal cancer risks were largely overestimated, as colorectal cancer risks by age 70 years were 26.7% for men and 22.4% for women. Despite the large number of families (84), the confidence intervals were quite large, suggesting a lack of efficiency of the method. Indeed, the retrospective likelihoods based on modelling genotypes as a function of given phenotypes are affected by a lack of efficiency.15 This issue might be particularly crucial in case of missing genotypes, that is, the most usual situation. Using such methods, another question is whether or not to include parts of the pedigree in which the phenotypes of relatives are known but their genotypes are not available.
In this paper, we studied the efficiency of the GRL method according to the proportion of relatives tested in the families and to the amount of family information available for the analysis. We also evaluated this method in a sample of 36 families diagnosed with HNPCC.
Methods
Genotype restricted likelihood
The GRL is a function of observed genotypes (Gen), given observed phenotypes (Phen), and ascertainment (Asc) of families. It can be written as

Let g denote the genotype of noncarriers of the mutated allele and G that of carriers, Geni is the genotype of individual i, P(Geni) is the corresponding probability, and P(Pheni/Geni) is the probability of individual i phenotype given his/her genotype. Thus, the contribution of a given family f with s members can be written as (see Carayol and Bonaïti-Pellié14 for a complete demonstration):

where Γ corresponds to the set of genotypic configurations compatible with the genotypes of the individuals tested, ΩC, to the set of genotypic configurations compatible with the selection criteria (ie, the index case carries the mutation), and Geni,v and Geni,w, to the genotypes of individual i in genotypic configuration v and w, respectively. The product on j is taken over all individuals whose parents' status is unknown (grandparents and spouses) and the product on {l,m,n} over all parent–offspring triplets.
For an individual i with genotype Geni, P(Geni) is expressed as a function of the frequency of the mutated allele in the general population for a founder, assuming Hardy–Weinberg proportions. Otherwise, this probability depends on parental genotypes, assuming Mendelian transmission.
Finally, let  (t) be the penetrance function at age t (cumulative risk by age t). If individual i is unaffected at age ti, the contribution of i to the likelihood is
(t) be the penetrance function at age t (cumulative risk by age t). If individual i is unaffected at age ti, the contribution of i to the likelihood is

that is, the probability that individual i is still unaffected at age ti (survival probability).
If individual i is affected at age ti, the contribution of i to the likelihood is

that is, the probability of being affected at age ti included in the 1-year interval [ti;ti+1[.
For the age-dependent penetrance function according to Geni, we chose a Weibull model with parameters  (scale parameter) and
(scale parameter) and  (shape parameter). This model is widely used in parametric survival analysis because of its ability to adjust to observed data.
(shape parameter). This model is widely used in parametric survival analysis because of its ability to adjust to observed data.
To take into account the possibility that some carriers will never develop the disease, we introduced a third parameter,  , corresponding to the fraction of individuals who will never be affected.16, 17 Finally, the penetrance function may be written as
, corresponding to the fraction of individuals who will never be affected.16, 17 Finally, the penetrance function may be written as

Simulation of family data
We used simulations to study the efficiency of the GRL in cases where some family members had unknown genotypes. As in a previous paper,14 samples of three-generation families with at least two affected members were simulated, with various penetrance values. The simulated pedigrees had a fixed structure: a couple of ancestors with four offsprings and their spouses, each with four offsprings. We simulated the genotypes of family members according to Mendel's laws for subjects whose parents were in the pedigree, ignoring the possibility of de novo mutation and according to the frequency of the mutated allele for founders. To obtain samples of sufficient size with at least one carrier individual (the index case), without simulating too many families, this frequency was set at 0.10. Phenotypes were simulated according to the age-dependent penetrance function, with the Weibull model. For noncarriers (Gen=g), the parameter κg was set at 0 and parameters λg and ag at values corresponding to a cumulative risk of 0.02 by age 80. For carriers (Gen=G), we considered two different risk values, the first one corresponding to a cumulative risk of 0.2 (called ‘low true penetrance’) by age 80 and the second one to 0.5 (called ‘high true penetrance’) by the same age. We did not consider any gender differences in risks.
The families were selected if at least two members were affected. To keep sample fluctuations to a minimum, sample size was fixed to 10 000 families after selection.
The loss (or gain) of efficiency was investigated by computing asymptotic relative efficiencies (AREs) of penetrance estimates, that is, the inverse of the ratio of the variance estimate in a given situation to the variance obtained in a reference situation. To evaluate the variance of the penetrance, we simulated, in each situation, 1000 replicates of the family sample and computed the variance of the estimate by age 70.
The efficiency of the GRL according to the proportion of genotyped individuals in families was studied by comparing the variance of the cumulative hazard functions calculated with varying proportions of genotyped individuals (25, 50 and 75%) to the variance computed when all genotypes are known. We also considered the most extreme situation, where only two genotypes are known (the index case and one relative). Note that if only the index case is genotyped, Γ and Ωc are identical, and the likelihood is a constant.
To study the information provided by family branches with no genotypic data, we selected families in which the index case's nuclear family included an affected relative tested for the mutation, and members of the secondary nuclear families of the third generation were not tested. We then compared the variance of the cumulative hazard function in four different situations, according to whether the sample included for each family (Figure 1): (1) only the ancestors and members of the index case's nuclear family (pedigree A); (2) pedigree A + members of secondary nuclear families with at least one affected (ie, family types B and C); (3) pedigree A + members of secondary nuclear families with at least two affected (family type C); and (4) all family members.

Pedigree structure with (a) index case's ancestors and nuclear family and (b,c) secondary nuclear families (index case marked with an arrow).
Parameters of the penetrance function were estimated by maximising the likelihood of simulated samples. We wrote a program that includes the maximisation procedure GEMINI as a subroutine18 and provides maximum likelihood estimates of the parameters λG and aG for carriers. Because κG was set at 0 in the simulation process, we did not estimate this parameter. We assumed that the penetrance was known for noncarriers and the three parameters were set at the same values as in the family simulation process.
HNPCC families
The index cases investigated in this study are patients referred by their physicians or self-referred for genetic counselling at the Centre Leon Bérard in Lyon (France) from January 1994 to January 2004. MMR testing was offered when they fulfilled the Amsterdam criteria I, which include only colorectal tumours,2 or II, which include extracolonic tumours associated with the syndrome,3 or even less stringent criteria, when one of the classic criteria was missing. All the individuals included in this study signed an informed consent for genetic testing. As this study did not involve any additional intervention, it was exempt under French law from ethical review board approval. Blood samples were subjected to germline mutation screening of MLH1 (NM_000249 for cDNA and NC_000003 for genomic DNA) and MSH2 (NM_000251 for cDNA and NC_000002 for genomic DNA) genes using genomic DNA sequencing.19 Of the 161 index cases meeting one of the selection criteria, 42 were found to carry a deleterious mutation of MLH1 or MSH2. Five families were not informative because none of the index case's relatives underwent mutation testing, and were therefore excluded. Another family was excluded because numerous consanguineous loops made the program unfeasible. Among the 36 mutated informative families, 22 index cases (61.1%) carried a mutation of MLH1 and 14 (38.9%) a mutation of MSH2. Genetic testing identified 129 mutation carriers (51 men and 78 women) and 59 noncarriers. Clinical information was available for 1185 family members (577 men and 608 women), 216 of whom were affected by colorectal cancer (97 men and 94 women). Age at diagnosis ranged from 20 to 89 years in men (mean: 44 years) and 18 to 82 years in women (mean: 45 years). Endometrial cancer was reported in 30 women. The youngest woman was diagnosed at age 32 years and the oldest one at age 88. Other tumours associated with the syndrome were observed (of the ovary, urinary tract, stomach and small intestine), but there were too few cases to allow estimation of penetrance. The cancer diagnosis was confirmed by medical and pathological reports in the great majority of affected relatives (85%).
The GRL method was used to estimate the parameters of the penetrance function. For each family member, the age t was taken as the age at last news or age of death if unaffected and the age at first diagnosis of colorectal cancer or endometrial cancer if affected. We assumed a frequency of 10−3 for the mutated allele and a de novo mutation frequency of 10−5, after verifying that estimates of penetrance were not sensitive to errors in these values. Parameters for noncarriers were fixed at values that fit their incidence in the French population.20 Maximum likelihood was used to estimate the three parameters of the penetrance function: λG, aG and κG. Analyses were conducted separately for men and women.
Confidence intervals were calculated with the bootstrap method. One thousand samples were constructed by resampling the 36 HNPCC families, and the penetrance function was estimated for each new sample. We used the 2.5 and 97.5 percentiles of the distribution of estimated penetrance at different ages to determine the corresponding lower and upper bounds of the confidence interval of the risk for each cancer.
Results
Efficiency
As shown in Table 1, efficiency decreased with the percentage of relatives tested, whatever the penetrance value. This reduction was particularly marked when only one family member besides the index case was tested in which case efficiency fell to 7%.
Whatever the penetrance value, the information provided by family branches without genotypic data did not increase efficiency of penetrance estimate with an ARE of about 1.00 in all cases. This clearly indicates that the inclusion of family branches in the analysis provides no significant information when genotypes are not available.
We could check that, whatever the proportion of missing genotypes and the family branches included in the analysis, penetrance estimates using the GRL were unbiased.
Estimation of cancer risk in HNPCC
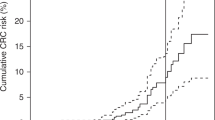
Figure 2 summarizes the penetrance functions of colorectal cancer estimated with the GRL from the 36 HNPCC families. Penetrance was negligible before 30 years. Although some cases of colorectal cancer were diagnosed before this age, most were index cases, which do not contribute to the likelihood. Penetrance was found to be higher in men than in women, with estimates of 0.47 and 0.34, respectively, at 70 years. Confidence intervals were rather large: 0.12–0.98 for men and 0.24–0.54 for women.

Penetrance function and confidence intervals at 30, 50 and 70 years of colorectal cancer risk in MSH2 and MLH1 mutation carriers for men (solid line) and women (dotted line), estimated from the 36 HNPCC families.
Estimated penetrance for endometrial cancer was very low before 40 years, because only three women developed this tumour at an earlier age, two of them being index cases (Figure 3). The cumulative risk at 70 years was estimated to be 0.14, with a confidence interval of 0.06–0.20.

Penetrance function and confidence intervals at 30, 50 and 70 years of endometrial cancer risk in MSH2 and MLH1 mutation carriers, estimated from the 36 HNPCC families.
Discussion
The results reported here show that efficiency may be problematic when only a few individuals are tested. The proportion of relatives undergoing genetic testing in families with such a genetic mutation and associated disease appears quite low, despite the benefits of molecular screening and endoscopic surveillance. For example, in 32 Italian families with germline mutations of MSH2, MLH1 or MSH6, only 34% of the first-degree relatives of affected individuals underwent genetic testing.21 In this study, only 24% of the 292 first-degree relatives of the 36 index cases were tested. This proportion may increase in the future, as families come to understand better the benefits of genetic testing.
We applied the GRL to estimate the risks of colorectal and endometrial cancer in families with HNPCC syndrome selected by familial criteria and identification of a MSH2 or MLH1 gene mutation. Lifetime penetrance of colorectal cancer was estimated at 47% for men and 33% for women. These risks were considerably lower than the first estimates reported in the literature, and were consistent with the values determined by studies taking into account the ascertainment bias.11, 12, 13 Dunlop et al11 selected subjects as a function of age at diagnosis of the index case (at or below 35 years of age) and presence of microsatellite instability (MSI) in the patient's tumour; MSI is characteristic of tumours due to MMR mutations, that is, independent of family history. They obtained risk estimates of 52% for colorectal cancer (CRC), and 42 % for uterine cancer by the age of 70 years. Parc et al12 analysed data from families of patients referred to a cancer family clinic and satisfying at least one of the modified Amsterdam criteria.3 To avoid ascertainment bias, they used a statistic based on the proportion of carriers among unaffected individuals, which allowed an estimation of the overall cancer risk (but not separate estimations for specific types). They obtained risk estimates of 43% by age 38 and 62% by age 51. Neither study provided confidence intervals but these intervals were probably large due to the small number of families in the first study and the relatively young ages of unaffected individuals tested for the mutation in the second one. Quehenberger et al13 used a method based on the same principles as ours in that they conditioned the likelihood of the observed genotypes on the observed phenotypes and on the event that at least one cancer patient was a mutation carrier. We could expect that our estimates would be very close to theirs. Indeed, there was only a slight difference in that we found a higher risk of CRC and a smaller risk of endometrial cancer. However, because of the large confidence intervals in the two studies and of the absence of difference found by Quehenberger et al,13 the penetrance values were not estimated separately for MLH1 and MSH2. Our results, combined to those of the three studies described above, confirm that most studies have overestimated the risks of colorectal cancer in HNPCC syndrome. Regarding the risk of endometrial cancer, we found a much lower estimate than previous studies but our results were not strongly different from those of Quehenberger et al,13 who found a risk of 31.5% (confidence interval: 11.1–70.3%). In our study, the upper bound of the confidence interval was 20%, which enables us to conclude that previous studies might have overestimated this risk, probably because endometrial cancer, although not ‘officially’ included in the recommended criteria, has been known to be associated with the syndrome for a long time, and this factor might have played a role in referring patients from physicians to oncogeneticists.
A considerable advantage of the GRL, as well as other retrospective methods, is that it is valid regardless of the inclusion criteria. It can thus be applied to samples of families selected according to different criteria. This property should be used in the future to pool large amounts of data of HNPCC families from different studies, to obtain reliable and precise estimates of risks. This would also permit us to estimate the risk of other HNPCC-associated cancers, scarcely known at present, and help organising the management of families and the surveillance of carrier relatives. Such a study is presently ongoing in France. It aims at collecting data from all the families tested for MLH1, MSH2 and MSH6 mutations. It will also allow us to detect a possible genetic heterogeneity among families according to the mutation involved, and to test for the role of other familial factors, either genetic or not, that could influence cancer risk in carriers.
Currently, carriers of MMR mutations in HNPCC families frequently undergo early colonoscopic screening at the age of 20 or 25 years. This should be considered when defining uninformative censoring events for unaffected relatives. Observation time was censored at age of first colonoscopy in the study by Quehenberger et al13 and at current age in the present one. These procedures could lead, in the first case, to shorter observation times for most individuals and, in the second case, to overlooking removal of precancerous lesions such as polyps. However, the clinical events observed during colorectal surveillance should be taken into account. The age at first diagnosis of an adenomatous polyp or the age at last colonoscopy in the absence of polyp detection should be more appropriate censoring times as more complete surveillance information is used to define the observation times. This could increase the power of the studies and the accuracy of the estimations of cancer risks.
References
Lynch HT, de la Chapelle A : Genetic susceptibility to non-polyposis colorectal cancer. J Med Genet 1999; 36: 801–818.
Vasen HF, Mecklin JP, Khan PM, Lynch HT : The International Collaborative Group on Hereditary Non-Polyposis Colorectal Cancer ICG-HNPCC). Dis Colon Rectum 1991; 34: 424–425.
Vasen HF, Watson P, Mecklin JP, Lynch HT : New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative Group on HNPCC. Gastroenterology 1999; 116: 1453–1456.
Aarnio M, Mecklin JP, Aaltonen LA, Nyström-Lahti M, Järvinen HJ : Life-time risk of different cancers in hereditary non-polyposis colorectal cancer (HNPCC) syndrome. Int J Cancer (Pred Oncol) 1995; 64: 430–433.
Voskuil DW, Vasen HF, Kampman E, Van't Veer P, the National Collaborative Group on HNPCC: Colorectal cancer risk in HNPCC families: development during lifetime and in successive generations. Int J Cancer 1997; 72: 205–209.
Vasen HFA, Wijnen JT, Menko FH et al: Cancer risk in families with hereditary nonpolyposis colorectal cancer diagnosed by mutation analysis. Gastroentology 1996; 110: 1020–1027.
Lin KM, Shashidharan M, Thorson AG et al: Cumulative incidence of colorectal and extracolonic cancers in MLH1 and MSH2 mutation carriers of hereditary nonpolyposis colorectal cancer. J Gastrointest Surg 1979; 2: 67–71.
Aarnio M, Sankila R, Pukkala E et al: Cancer risk in mutation carriers of DNA-mismatch-repair genes. Int J Cancer 1999; 81: 214–218.
Vasen HF, Stormorken A, Menko FH et al: MSH2 mutation carriers are at higher risk of cancer than MLH1 mutation carriers: a study of hereditary nonpolyposis colorectal cancer families. J Clin Oncol 2001; 19: 4074–4080.
Carayol J, Khlat M, Maccario J, Bonaïti-Pellié C : Hereditary non-polyposis colorectal cancer: current risks of colorectal cancer largely overestimated. J Med Genet 2002; 39: 335–339.
Dunlop MG, Farrington SM, Carothers AD et al: Cancer risk associated with germline DNA mismatch repair gene mutations. Hum Mol Genet 1997; 6: 105–110.
Parc Y, Boisson C, Thomas G, Olschwang S : Cancer risk in 348 French MSH2 or MLH1 gene carriers. J Med Genet 2003; 40: 208–213.
Quehenberger F, Vasen HFA, van Houwelingen HC : Risk of colorectal and endometrial cancer for carriers of mutations of the hMLH1 and hMSH2 gene: correction for ascertainment. J Med Genet 2005; 42: 491–496.
Carayol J, Bonaïti-Pellié C : Estimating penetrance from family data using a retrospective likelihood when ascertainment depends on genotype and age of onset. Genet Epidemiol 2004; 27: 109–117.
Kraft P, Thomas DC : Bias and efficiency in family-based gene-characterization studies: Conditional, Prospective, Retrospective, and joint likelihoods. Am J Hum Genet 2000; 66: 1119–1131.
Sposto R : Cure model analysis in cancer: an application to data from the Children's Cancer Group. Stat Med 2002; 21: 293–312.
Planté-Bordeneuve V, Carayol J, Ferreira A et al: Genetic study of transthyretin amyloid neuropathies: carrier risks among French and Portuguese families. J Med Genet 2003; 40: e120.
Lalouel JM : GEMINI – a computer program for optimization of general non linear functions. Technical report no 14. Salt Lake City: university of Utah, Department of Medical Biophysics and Computing, 1979.
Wang Q, Lasset C, Desseigne F et al: Prevalence of germline mutations of hMLH1, hMSH2, hPMS1, hPMS2 and hMSH6 genes in 75 French kindreds with nonpolyposis colorectal cancer. Hum Genet 1999; 105: 79–85.
Remontet L, Esteve J, Bouvier AM et al: Cancer incidence and mortality in France over the period 1978–2000. Rev Epidemiol Sante Publique 2003; 51: 3–30.
Ponz de Leon M, Benatti P, Di Gregorio C et al: Genetic testing among high-risk individuals in families with hereditary nonpolyposis colorectal cancer. Br J Cancer 2004; 90: 882–887.
Acknowledgements
We acknowledge with gratitude the support of the Fondation de France and the Ligue Nationale Contre le Cancer through Jérôme Carayol's fellowship and the support of the Ligue Contre le Cancer du Rhône et de l’Ardèche for the management of the HNPCC families database. We thank Catherine Huber for helpful advice and Marie Dominique Reynaud for editing this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Alarcon, F., Lasset, C., Carayol, J. et al. Estimating cancer risk in HNPCC by the GRL method. Eur J Hum Genet 15, 831–836 (2007). https://doi.org/10.1038/sj.ejhg.5201843
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201843
Keywords
This article is cited by
-
MTHFR C677T and A1298C polymorphism’s effect on risk of colorectal cancer in Lynch syndrome
Scientific Reports (2023)
-
Colorectal cancer surveillance in Portuguese families with lynch syndrome: a cohort study
International Journal of Colorectal Disease (2018)
-
Cancer predisposition genes: molecular mechanisms and clinical impact on personalized cancer care: examples of Lynch and HBOC syndromes
Acta Pharmacologica Sinica (2016)
-
ACG Clinical Guideline: Genetic Testing and Management of Hereditary Gastrointestinal Cancer Syndromes
American Journal of Gastroenterology (2015)
-
Guidelines on Genetic Evaluation and Management of Lynch Syndrome: A Consensus Statement by the US Multi-Society Task Force on Colorectal Cancer
American Journal of Gastroenterology (2014)
