
Abstract
Purpose To report a series of 15 primary lacrimal sac tumours and present an algorithm in managing this rare condition.
Design Noncomparative interventional case series.
Methods A retrospective review of the clinical, radiological, and pathological records of 15 patients with primary lacrimal sac tumours.
Main outcome measures Histologic evaluation and clinical follow-up, including tumour clearance and recurrence, were assessed.
Results Of the patients, 11 were female and the median age at presentation for the whole group was 59 years (range 22–94 years). The commonest tumour was non-Hodgkins B-cell lymphoma (five cases), followed by two cases each of squamous cell carcinoma and transitional cell carcinoma, one case of benign transitional papilloma, haemangiopericytoma, adenoid cystic carcinoma, mucoepidermoid carcinoma, mixed (squamous/transitional) carcinoma, and a highly malignant undifferentiated tumour. Treatment modalities included surgery, radiotherapy and chemotherapy and, with a median follow-up of 30 months (range 2 months to 17 years), two patients had died from metastatic disease but nine patients remained without evidence of recurrent tumour.
Conclusions Primary lacrimal sac tumours are extremely rare, require long-term follow-up for recurrence and metastasis, and can be fatal.
Similar content being viewed by others

Introduction
Almost 300 lacrimal sac tumours have been reported, most being primary epithelial tumours (73%) of which 75% are malignant.1,2,3,4,5,6 Mesenchymal primary tumours, such as fibrous histiocytoma, fibroma, haemangioma, haemangiopericytoma, angiosarcoma, or lipoma, are less common (14%) and the rarer tumours include lymphomas (8%), malignant melanomas (4%), and neural tumours (1%).6,7
Secondary tumours may originate either from adjacent structures, such as the paranasal sinuses and orbit, or as metastases—although the latter are rarely confined to the lacrimal sac alone.3 Inflammatory lesions, such as nonspecific chronic inflammation (‘pseudotumour’) or granulomatous disease, are not neoplasms but may present as lacrimal sac masses.
Lacrimal sac tumours are rare but potentially life-threatening, so that early diagnosis and treatment is particularly important. We report the presentation, diagnosis, management, and outcome of a series of 15 patients with primary lacrimal sac tumours, and present an algorithm for managing this unusual condition.
Methods
A retrospective review of the clinical records, histology, and radiology was performed for 15 patients with primary lacrimal sac tumours from the Lacrimal Clinic, Moorfields Eye Hospital.
Results
The median follow-up for the present series was 30 months (range 2 months to 17 years), with no evidence of disease recurrence in nine patients (Table 1). Local recurrence was seen in a transitional papilloma (case A) and a transitional cell carcinoma (case H), the latter treated successfully with endoscopic excision biopsy. One patient with lymphoma (case K) developed cervical lymphatic involvement at 2 years after radiotherapy for the lacrimal sac mass, this recurrence responding well to chemotherapy, and one patient is alive with pulmonary metastases at 16 years after orbital exenteration for adenoid cystic carcinoma (case I). Two deaths from metastatic disease occurred within 2 months of diagnosis (cases B and N), and one patient died at 41 months without recurrent disease (case F).
Case reports
Case A
Examination of a 50-year-old woman who presented with 1 year of left epiphora revealed left lower canalicular block but no signs of a lacrimal mass. During external dacryocystorhinostomy 8 months later, a lacrimal sac polyp was excised and later shown to be a well-differentiated transitional cell papilloma with no evidence of invasion. Further endonasal recurrences at 1 and 6 months after primary surgery were ablated endoscopically with argon laser and, with regular endoscopic examination, the patient has remained disease-free for 5 years.
Case B
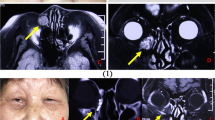
On treatment with systemic steroids and penicillamine for rheumatoid arthritis and Felty's syndrome, a 76-year-old man was referred with an 18-month history of an enlarging, ulcerating left inner canthal mass. At 5 months prior to referral, he had presented with a lacrimal sac fistula, but skin biopsy at the referring hospital had demonstrated chronic inflammatory cells with no histological evidence of malignancy. Although microbiological samples had cultured a mixed growth of Staphylococcus aureus and Bacteroides fragilis, treatment with topical and systemic antibiotics had given little improvement.
At referral, there was a large ulcerating lesion extending inferomedially into his orbit and tethering the globe (Figure 1a). Left infraorbital hypoaesthesia was present and CT scan showed an extensive orbital mass eroding the maxilla and ethmoid, biopsies confirming the clinical diagnosis of squamous cell carcinoma with a marked scirrhous component (Figure 1b). Since the tumour was too extensive for surgical resection, the patient underwent radical radiotherapy and died from cerebral metastases 2 months later.

(case B) (a) A large ulcerating inner canthal mass of 18 months duration, in which the histology (b) reveals squamous cell carcinoma—with islands of squamous cells showing nuclear atypia, set in a stroma with a predominantly chronic inflammatory cell infiltrate (haematoxylin & eosin, × 400).
Case C
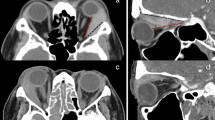
A 53-year-old man, presenting at his referring hospital with 5 years of left epiphora and a patent lacrimal drainage system, received treatment for blepharitis and repair of his lower punctual ectropion. A left medial canthal mass was noted and dacryocystography (DCG) showed passage of contrast to the nose, but a filling defect in the lacrimal sac (Figure 2a). Orbital CT confirmed a lacrimal sac mass with extension along the medial wall of the left orbit (Figure 2b) and incision biopsy demonstrated invasive transitional cell carcinoma with some squamous differentiation (Figure 2c).

(case C) (a) Dacryocystography demonstrates a distended left lacrimal sac with intrinsic filling defect (white arrows) and (b) CT scan reveals a soft tissue mass extending from the lacrimal sac into the medial part of the left orbit. (c) Histology demonstrates transitional cell carcinoma, with pleomorphic cells and prominent nucleoli (haematoxylin & eosin, × 200).
After referral to Moorfields Eye Hospital, he underwent dacryocystectomy with wide excision of adjacent orbital periosteum, ethmoids, and lateral nasal wall. Following a course of postoperative radiotherapy, the patient remains disease-free at 4 years of follow-up.
Case D
Following an episode of dacryocystitis, an 83-year-old lady was found to have a firm rubbery mass in the right lacrimal sac, and involving both upper and lower canaliculi (Figure 3a). The mass extended above the canthal tendon and there was no evidence of systemic spread. DCG showed obstruction in the lacrimal sac and CT confirmed a soft tissue mass extending along the medial wall of the orbit, without bony erosion (Figure 3b).

(case D) (a) Patient presenting with a right inner canthal mass, extending into both the upper and lower lids, and involving both canaliculi. (b) The mass was shown to extend along the medial orbital wall on CT scan and (c) incisional biopsy demonstrated a low-grade marginal zone lymphoma of the lacrimal sac (haematoxylin & eosin, × 200).
Canalicular biopsy demonstrated a low-grade B-cell marginal zone lymphoma (Figure 3c), without any evidence of systemic involvement on oncological workup. The patient was successfully treated by six 1-week courses of chlorambucil, with no sign of recurrence at 5 years' follow-up.
Case E
A 34-year-old lady attended with 15 months of right epiphora and having noticed a right inner canthal mass for 6 months. While initially thought to be a lacrimal sac mucocoele, at surgery a tumour mass was evident and excised intact. While histology showed a well-circumscribed low-grade mucoepidermoid carcinoma (Figure 4), tumour extension to the excision margins was considered to be an indication for external beam radiotherapy and the patient remains disease-free at 3 years after surgery.

(case E) Mucoepidermoid carcinoma of the lacrimal sac, with islands of epithelial tumour cells set in a mucinous background (haematoxylin & eosin, × 200), with areas of mucin production (inset; periodic acid-Schiff, × 400).
Discussion
Benign papillomas tend to present at a younger age than carcinomas (the latter occurring from 41 to 75 years in one study) and, despite a male preponderance in one Chinese series, no significant racial or sexual predilection is thought to exist.3,8 In our series of 15 patients, 11 (73%) were female and the median age at presentation was 59 years (range 22–94 years).
Most lacrimal sac tumours present insidiously with symptoms of associated chronic dacryostenosis or dacryocystitis and, in one study, 43% were found inadvertently at the time of dacryocystorhinostomy.1 Common complaints include epiphora (present in all of our cases) or a lacrimal sac mass (in 12 (80%) of our patients); tumour masses located above the medial canthal tendon are almost always a tumour (cases C and D).2 Epistaxis, punctal bleeding (either spontaneously or on sac irrigation), or pain is very rare with sac tumours and was absent in all cases reported here.
Lacrimal sac tumours are usually firm and incompressible, as compared to the fluctuance of inflammatory swellings. Proptosis or nonaxial globe displacement occurs late with sac tumours, when orbital invasion occurs, and early globe displacement favours a primary sinus tumour as the cause of epiphora.1 Skin telangiectasia may occur over lacrimal sac tumours and, at a later stage, skin ulceration and invasion of adjacent structures may occur (Figure 1a).1 Involvement of the regional (preauricular, submandibular, or cervical) lymph nodes occurs late in the condition and distant metastasis is rare.2
Assessment
In a patient with a lacrimal sac mass, a specific history of blood-stained tears, epistaxis or a prior naso-sinus disease should be sought. The differential diagnosis includes dacryocystitis, a secondary neoplasm from adjacent structures or, more rarely, a specific inflammation (such as Wegener's granuloma) or infective process (such as tuberculosis or fungal infection). Location of the tumour relative to the canthal tendon is particularly important, the consistency of the mass and the presence of punctal discharge should be noted, and evidence of regional lymphadenopathy should be sought, together with examination of the nasal space and sinuses.
With benign tumours, the lacrimal system often remains patent to irrigation, whereas carcinomas are generally associated with complete obstruction.1,3 Dacryocystography in the presence of an intrinsic tumour often shows a filling defect of the sac lumen (Figure 2a) or a distended sac with uneven or mottled contrast media. Orbital and sinus CT is imperative where tumour is suspected and will provide evidence of expansion or erosion of the lacrimal sac fossa, or invasion into neighbouring structures (Figure 2b and Figure 3b). Systemic evaluation, both clinically and with chest X-ray and haematological tests, should be undertaken and a diagnostic tissue biopsy is mandatory prior to definitive treatment.
Therapeutic options
Traditional treatment of suspected lacrimal sac tumours advocates incisional biopsy of the sac under frozen section followed by dacryocystectomy once diagnostic confirmation of tumour is achieved, with subsequent definitive therapy including radical resection, radiotherapy, or chemotherapy.1,2,5,9 This policy will, however, lead to unnecessary morbidity in the absence of tumour and inadequate initial clearance of more extensive lesions. We do not, therefore, employ this strategy and have developed an alternative algorithm for treating these rare tumours.
When the diagnosis of lacrimal malignancy is clinically obvious, as with large ulcerative tumours (case B, Figure 1), incisional biopsy is performed to determine subsequent definitive therapy. If the only suggestion of tumour is a filling defect on DCG, malignancy would be unusual and more likely causes are polyps, mucosal folds, or sequestered mucous. In such cases, open dacryocystorhinostomy with biopsy of any abnormal tissue is performed. Locally invasive tumours, such as papillomas, may be readily monitored for recurrence by nasal endoscopy (case A), while aggressive tumours may be treated with radical surgery or radiotherapy (case H).
CT imaging of the orbit and sinuses is organized if a lacrimal sac mass is hard, extending above the canthal tendon or displacing the globe—especially in the presence of ocular or nasal bleeding. Incisional biopsy is performed where the mass extends beyond the lacrimal sac fossa (case C), but otherwise an attempted intact excision of the tumour is made (case E). With incisional biopsy, a deep sample from the lacrimal sac is taken and the incision closed with a nylon suture; decisions about definitive therapy are based upon definitive histology rather than frozen section analysis. If there is no evidence of tumour, dacryocystorhinostomy is performed at a later date.
Definitive treatment for epithelial tumours of the lacrimal sac consists of surgery, radiotherapy, and occasionally chemotherapy. Where epithelial and mesenchymal tumours are localized to the lacrimal sac fossa, intact excision of the tumour (and the periosteum of the fossa and as much of the nasolacrimal duct as possible) is undertaken through a dacryocystorhinostomy incision (case E).1,2,7 Resection of the adjacent orbital and lateral nasal wall may be required with more extensive tumours (case H) or, in advanced cases, it may be necessary to perform orbital exenteration, resection of paranasal sinuses, or cervical lymph node dissection (cases F and I). Postoperative radiotherapy (used in 8/15 of this series) is recommended for malignant epithelial tumours, although irradiation of benign papillomas might induce malignant transformation. Recurrent lesions may be treated with further surgery or radiotherapy.
Although found in one-third of our cases, lacrimal sac lymphomas are rare and their primary treatment consists of radiotherapy, with or without chemotherapy.6,10,11 Malignant melanomas of the lacrimal sac are extremely rare, have a poor prognosis with an average survival of 18 months, and the various treatments (such as extensive surgical resection, radiotherapy, or chemotherapy) may delay recurrence, but probably do not improve survival.12,13
Pathological features and prognosis
Lacrimal sac tumours may conveniently be grouped into epithelial and nonepithelial types: papillomas are the commonest benign epithelial tumours, while oncocytic adenomas and benign mixed tumours are rare.5 Transitional cell papillomas consist of stratified columnar epithelium containing scattered goblet cells and cilia (case A), squamous papillomas show acanthotic, stratified squamous epithelium (with occasional foci of dyskeratosis) resting on thickened basement membrane-like material, and mixed cell papillomas show features common to both types; all can display an exophytic or endophytic growth pattern, the latter having a higher rate of recurrence or of malignant transformation.2,4,6 Benign papillomas have a tendency to recur (as shown in case A), especially where of inverted form—with a 10–40% recurrence.1
Lacrimal sac carcinomas are believed to arise de novo or, less commonly, from a preceding papilloma.2,4 Squamous and transitional cell carcinomas are the most common, but other types include adenocarcinoma, oncocytic adenocarcinoma, mucoepidermoid, poorly differentiated, and adenoid cystic carcinoma; carcinomas have a quoted recurrence rate of 50% (mortality increasing with recurrence), and 37.5% mortality has been reported after wide surgical excision and radiotherapy.1,3 Squamous cell carcinomas (cases B and F) can exhibit a wide range of differentiation—atypical squamous cells (with abundant eosinophilic cytoplasm, pleomorphic nuclei, prominent nucleoli, and mitotic figures) showing invasion of deeper tissue layers (Figure 1b). Transitional cell carcinoma (cases C and H) resembles that of the urinary bladder, with pleomorphic cells and prominent nucleoli (Figure 2c). Adenoid cystic carcinoma can display a cribriform or basaloid pattern— the former resembling ‘Swiss cheese’, with sharply demarcated aggregates of small tightly packed malignant cells and rounded cystic foci (case I).14 Mucoepidermoid carcinoma (case E; Figure 4) has both epidermoid and mucous-secreting cells with mucin-filled cystic spaces, while oncocytic adenocarcinoma shows oncocytic cells with nuclear atypia, arranged in an infiltrative pseudoglandular pattern.15,16
Nonepithelial lacrimal sac tumours include mesenchymal lesions, lymphoid lesions, malignant melanoma, and granulocytic sarcoma.7 Fibrous histiocytomas, showing a mixture of spindle-shaped fibroblastic cells and plump histiocyte-like cells, are the commonest of mesenchymal tumours and usually benign.17 Haemangiopericytoma (case G) has metastatic potential, and typically demonstrates a vascular pattern of sinusoidal spaces, between which are spindle-shaped pericytes.18 A spectrum of lymphoid lesions of the lacrimal sac may occur, from atypical lymphoid hyperplasia to non-Hodgkins B-cell lymphomas.10,11 Lymphoma (Figure 3) occurred in five cases (33%) of the present series, this being a higher proportion than the 8% reported in a previous series of 117 patients.7 Malignant melanoma, granulocytic sarcoma, lipoma, and neurofibroma are all very rare nonepithelial lacrimal sac tumours and not observed in our series. Recurrence and mortality rates for nonepithelial tumours vary: benign fibrous histiocytoma has a good prognosis if completely excised, while the malignant potential of haemangiopericytoma can be unpredictable.2,18 Lymphomas respond to radiotherapy and chemotherapy, with outcome depending on the extent and type of tumour, whereas malignant melanoma carries a bad prognosis, which is little altered despite aggressive treatment.10,12,13 Encouragingly, all of the nonepithelial tumours in the present series (five lymphomas and one haemangiopericytoma; follow-up from 16 months to 9 years) had a favourable prognosis.
Some lacrimal sac tumours show mixed features—such as squamous and transitional cell differentiation in case O—while other malignant tumours may be unclassified—such as case N, in which the cell of origin was unclear and the tumour was considered a ‘highly malignant undifferentiated tumour’. In any case, all lacrimal sac tumours require careful lifelong follow-up, as recurrence and metastasis may occur many years after initial treatment.
References
Flanagan JC, Stokes DP . Lacrimal sac tumours. Ophthalmology 1978; 85: 1282–1287.
Ryan SJ, Font RL . Primary epithelial neoplasms of the lacrimal sac. Am J Ophthalmol 1973; 76: 73–88.
Schenck NL, Ogura HJ, Pratt LL . Cancer of the lacrimal sac. Ann Otol Rhinol Laryngol 1973; 82: 153–161.
Spencer WH . Ophthalmic Pathology. A Text and Atlas. Vol 3, WB Saunders Company: Philadelphia, 1986, pp. 2317–2328.
Hornblass A, Jakobiec FA, Bosniak S, Flanagan J . The diagnosis and management of epithelial tumours of the lacrimal sac. Ophthalmology 1980; 87: 476–490.
Stefanyszyn MA, Hidayat AA, Pe'er JJ, Flanagan JC . Lacrimal sac tumours. Ophthalmic Plast Reconstr Surg 1994; 10: 169–184.
Pe'er JJ, Stefanyszyn M, Hidayat AA . Nonepithelial tumours of the lacrimal sac. Am J Ophthalmol 1994; 118: 650–658.
Ni C, D'Amico DJ, Fan CQ, Kuo PK . Tumours of the lacrimal sac. Int Ophthalmol Clin 1982; 22: 121–140.
Hurwitz JJ . Lacrimal sac tumors. In: Hurwitz JJ (ed). The Lacrimal System. Lippincott-Raven: Philadelphia, 1996, pp 195–198.
Nakamura K, Uehara S, Omagari J, Kunitake N, Kimura M, Makino Y et al. Primary non-Hodgkin's lymphoma of the lacrimal sac. Cancer 1997; 80: 2151–2155.
Kheterpal S, Chan SY, Batch A, Kirkby GR . Previously undiagnosed lymphoma presenting as recurrent dacryocystitis. Arch Ophthalmol 1994; 112: 519–520.
Lloyd WC, Leone CR . Malignant melanoma of the lacrimal sac. Arch Ophthalmol 1984; 102: 104–107.
Levine MR, Dinar Y, Davies R . Malignant melanoma of the lacrimal sac. Ophthalmic Surg Lasers 1996; 27: 318–320.
Kincaid MC, Meis JM, Lee MW . Adenoid cystic carcinoma of the lacrimal sac. Ophthalmology 1989; 96: 1655–1658.
Blake J, Mullaney J, Gillan J . Lacrimal sac mucoepidermoid carcinoma. Br J Ophthalmol 1986; 70: 681–685.
Perlman JI, Specht CS, McLean IW, Wolfe SA . Oncocytic adenocarcinoma of the lacrimal sac: report of a case with paranasal sinus and orbital extension. Ophthalmic Surg 1995; 26: 377–379.
Choi G, Lee U, Won NH . Fibrous histiocytoma of the lacrimal sac. Head Neck 1997; 19: 72–75.
Carnevali L, Trimarchi F, Rosso R, Stringa M . Haemangiopericytoma of the lacrimal sac: a case report. Br J Ophthalmol 1988; 72: 782–785.
Acknowledgements
We thank Professor P Luthert and Dr I Cree, of the Institute of Ophthalmology, London, for their preparation and interpretation of the histopathological illustrations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Parmar, D., Rose, G. Management of lacrimal sac tumours. Eye 17, 599–606 (2003). https://doi.org/10.1038/sj.eye.6700516
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6700516
Keywords
This article is cited by
-
Therapierefraktäre Epiphora nach Dakryozystorhinostomie – Stellenwert von Bildgebung und Histopathologie
Die Ophthalmologie (2023)
-
First-Onset Dacryocystitis: Characterization, Treatment, and Prognosis
Ophthalmology and Therapy (2022)
-
Iodine-125 interstitial brachytherapy for malignant lacrimal sac tumours: an innovative technique
Eye (2021)
-
Clinical analysis of 90 cases of malignant lacrimal sac tumor
Graefe's Archive for Clinical and Experimental Ophthalmology (2018)
-
‘Preoperative imaging should be performed for all cases of acquired nasolacrimal duct obstruction’—No
Eye (2017)
