
Abstract
The widespread use of the selective serotonin reuptake inhibitors (SSRIs) has been accompanied by numerous reports describing a potential association with hyperprolactinemia. Antipsychotics are commonly known to elevate serum prolactin (PRL) through blockade of dopamine receptors in the pituitary. However, there is little awareness of the mechanisms by which SSRIs stimulate PRL release. Hyperprolactinemia may result in overt symptoms such as galactorrhea, which may be accompanied by impaired fertility. Long-term clinical sequelae include decreased bone density and the possibility of an increased risk of breast cancer. Through literature review, we explore the possible pathways involved in serotonin-induced PRL release. While the classic mechanism of antipsychotic-induced hyperprolactinemia directly involves dopamine cells in the tuberoinfundibular pathway, SSRIs may act on this system indirectly through GABAergic neurons. Alternate pathways involve serotonin stimulation of vasoactive intestinal peptide (VIP) and oxytocin (OT) release. We conclude with a comprehensive review of clinical sequelae associated with hyperprolactinemia, and the potential role of SSRIs in this phenomenon.
Similar content being viewed by others

INTRODUCTION
Hyperprolactinemia is an undesirable effect of several classes of psychotropic medications. While it is well recognized in relation to antipsychotic use (Conner and Fried, 1998; Dickson and Glazer, 1999; Kleinberg et al, 1971; Meltzer et al, 1979), there is limited awareness of this adverse effect of selective serotonin reuptake inhibitors (SSRIs). A growing number of case reports and small studies have described PRL abnormalities and/or manifestations such as galactorrhea, amenorrhea, and breast tenderness in association with the use of SSRIs in women (Peterson, 2001; Amsterdam et al, 1997; Bronzo and Stahl, 1993; Morrison et al, 2001; Arya and Taylor, 1995; Iancu et al, 1992; Attenburrow et al, 2001; Bonin et al, 1997; Jeffries et al, 1992; Spigset and Mjorndal, 1997; Cowen and Sargent, 1997; Dulchin et al, 2001; Laine et al, 1997; Urban and Veldhuis, 1991). Possible long-term clinical consequences of hyperprolactinemia, including decreased bone density and a potential increased risk of breast cancer, are only beginning to be investigated (Klibanski et al, 1980; Wang et al, 2002). Subtle impacts on fertility mediated by PRL due to its effects on circulating gonadotropins have also not been studied in patients exposed to SSRIs.
In the first part of this review, we examine the related neuroanatomy and neurobiological mechanisms by which SSRIs may cause hyperprolactinemia. Next, we review the clinical manifestations of hyperprolactinemia. Finally, we outline questions for further research to determine the incidence of, risk factors for, and significance of SSRI-induced hyperprolactinemia.
THE SSRIS AND PROLACTIN
The stimulus for the development of the SSRIs originated in the serotonin hypothesis of depression. This hypothesis was proposed by Carlsson, Van Praag, Asberg, and others (Agurell, 1983; Asberg et al, 1976), based on studies showing a low CSF 5-HIAA response to probenecid in depressed individuals (Van Praag, 1977), decreased central 5-HT in the brains of suicide victims (Pare et al, 1969), and reports of antidepressant effects of tryptophan, a serotonin precursor (Berger, 1975). Additional support for the serotonin hypothesis of depression came from the demonstration of reversal of monoamine antidepressant action in patients pretreated with a 5-HT synthesis inhibitor (Shopsin et al, 1976).
Neuroendocrine challenge tests later helped to substantiate the serotonin hypothesis of depression (Coccaro et al, 1989). These tests are employed to provide an index of central serotonergic function, based on the premise that 5-HT stimulation leads to the release of pituitary hormones such as ACTH and PRL(Yatham and Steiner, 1993). Neuroendocrine probes of serotonergic function include 5-HT precursors (L-tryptophan, 5-hydroxytryptophan), releasing agents (fenfluramine), reuptake inhibitors (clomipramine), and receptor agonists (m-CPP) among others. In humans, the observation that oral administration of fenfluramine to healthy volunteers induced a dose-related increase in PRL secretion provided an index of central serotonergic function (Quattrone et al, 1983), a phenomenon that has been reliably reproduced many times over. Fenfluramine, a serotonin-releasing agent, which also inhibits the reuptake of synaptic 5-HT and stimulates postsynaptic 5-HT2A and 2C receptors (Newman et al, 1998) induces a rapid surge of PRL in laboratory animals and humans (Lu and Meites, 1973; Pinder et al, 1975; Slater et al, 1976). In contrast to healthy controls, a blunted PRL response occurs in some depressed patients when challenged with fenfluramine (Siever et al, 1984; Cleare et al, 1995; Coccaro et al, 1989; Mann et al, 1995; O'Keane and Dinan, 1991; Lopez-Ibor et al, 1988; Mitchell and Smythe, 1990), suggesting abnormal central serotonergic transmission in this condition. However, many studies have failed to show similar results (Asnis et al, 1988; Abel et al, 1997; Maes et al, 1991; Weizman et al, 1988; Kavoussi et al, 1998). The main criticism of studies documenting a positive correlation between a blunted PRL response and depression is the use of heterogeneous patient populations with other psychiatric comorbidities (Newman et al, 1998). More recent studies indicate that blunted PRL response to fenfluramine is associated with impulsive aggression and suicidal behavior, traits associated with depression (Coccaro et al, 1989, 1997a, 1997b; Fava et al, 2000; New and Siever, 2002; Placidi et al, 2001). These findings suggest that serotonin dysfunction may be a marker of traits associated with some forms of depression and other psychiatric conditions, rather than a causal factor of major depression. In patients chronically treated with SSRIs, a normalization of the blunted PRL response would be expected. However, results have been conflicting with some studies showing enhanced PRL response (Kasper et al, 1990; O'Keane et al, 1992), and others finding no change or even decrements in the response (Dulchin et al, 2001; Kavoussi et al, 1999). These studies underscore how individual differences in serotonin responsivity are reflected in variable PRL responsivity across patient samples.
The exact mechanisms through which SSRIs achieve their therapeutic and neuroendocrine effects are not known. SSRIs elevate serotonin levels at the synapse through reuptake blockade of the serotonin transporter (ser-T) (Hyttel, 1984; Tatsumi et al, 1997) which interrupts the normal negative feedback control of presynaptic serotonin release and increases serotonin at the synapse (Dubovsky, 1994). With the exception of fluoxetine, SSRIs generally have little interaction with postsynaptic 5-HT receptors and are mainly thought to work through this presynaptic mechanism. While several SSRIs differentially interact with other neurotransmitter systems (Goodnick and Goldstein, 1998; Hyttel, 1984; Tatsumi et al, 1997), including dopamine (sertraline) and norepinephrine (paroxetine) stimulation of PRL likely involves serotonergic mechanisms, since all SSRIs have been implicated in hyperprolactinemia, regardless of their effects on these other transmitter systems (Attenburrow et al, 2001; Bronzo and Stahl, 1993; Spigset and Mjorndal, 1997; Cowen and Sargent, 1997; Peterson, 2001; Morrison et al, 2001).
Prolactin's Physiologic Role
PRL is a polypeptide hormone, which is secreted in a pulsatile fashion and is structurally related to GH and human placental lactogen (hPL) (Niall et al, 1971). PRL release follows a circadian rhythm, with increased secretion in the early evening hours (Sassin et al, 1972; Waldstreicher et al, 1996). Peak PRL levels occur in the early morning secondary to its increased secretion with prolonged sleep. PRL levels also fluctuate with the menstrual cycle, peaking with ovulation (Seppala, 1978). Physiological increases in plasma PRL levels are seen with stress, pregnancy, sleep, exercise, meals, sexual intercourse, and breastfeeding (see Review by Yazigi (Yazigi et al, 1997)). PRL's actions are mediated at the PRL receptor, which is most densely concentrated in the choroid plexus where active uptake of serum PRL occurs. To a lesser degree, the PRL receptor is also distributed in the paraventricular nucleus (PVN) and other hypothalamic sites (Bakowska and Morrell, 1997; Chiu and Wise, 1994; Crumeyrolle-Arias et al, 1993; Walsh et al, 1987) with generally higher densities found in females (Chiu and Wise, 1994; Muccioli et al, 1991; Pi and Grattan, 1998).
In addition to mammotropic and lactogenic functions (Frantz, 1978), PRL plays a role in developing neuroendocrine and behavioral adaptations in the maternal brain (Grattan, 2001), and influences reproductive function in both males and females (Doherty et al, 1981; Harlan et al, 1983; Witcher and Freeman, 1985). Elevated levels of PRL interfere with the normal pulsatile secretion of LH and FSH, leading to the inhibition of gonadal function (Seppala, 1978; Bohnet et al, 1976; Greenspan, 2001). In humans, elevated PRL levels are associated with hypogonadism and infertility (see below) (Gomez et al, 1977; Katz and Adashi, 1990), while subtle perturbations in PRL dynamics have been linked to infertility of unknown etiology (see below) (Subramanian et al, 1997).
PRL also has a broad role in complex behaviors outside reproduction and lactation, including grooming behaviors (Drago et al, 1983), food intake (Noel and Woodside, 1993; Li et al, 1995) and the response to acute physiological stress (Drago et al, 1990; Holsboer and Barden, 1996; Neill, 1970; Torner et al, 2001). Various forms of stress are associated with increased PRL release, including ether stress (Johnston and Negro-Vilar, 1986), restraint stress (Torner et al, 2001; Gala, 1990; Neill, 1970; Rossier et al, 1980; Seggie and Brown, 1975) thermal stress (Vaha-Eskeli et al, 1991), social conflict in mice (Huhman et al, 1995), and academic stress in humans (Malarkey et al, 1991). PRL circadian rhythm disturbances occur in patients with premenstrual dysphoric disorder, which may be linked to chronobiologic abnormalities in this condition (Parry et al, 1996). Evidence from animal models indicates that PRL interfaces with the HPA axis, blocking stress-induced increases in corticosterone. Consistent with these neuroendocrine effects, PRL exerts anxiolytic effects in these animals (Torner et al, 2001). In humans, low scores on the Hamilton anxiety scale correlated with high PRL levels in healthy lactating women (Asher et al, 1995).
Related to its role in the stress response, PRL participates in the regulation of the immune system (Gala, 1990). PRL enhances thymic function in PRL-deficient mice (Chen et al, 1972) and stimulates T lymphocyte mitogenesis (Viselli et al, 1991; Shiu et al, 1983). PRL involvement in the immune response is further supported by increased lymphocytic PRL gene expression in the setting of graft rejection (Shen et al, 1992). Taken together, these data indicate that PRL is an important part of the physiological response to stress.
ANATOMY OF PRL RESPONSE: HYPOTHALAMIC–PITUITARY CONNECTIONS
The following overview aims to orient the reader to specific hypothalamic–pituitary pathways implicated in PRL release, which can be influenced by serotonin. It is not intended as a comprehensive review of the vast network of hypothalamic–pituitary connections.
Hypothalamus
PRL is secreted by the anterior pituitary, which is regulated by the hypothalamus. The hypothalamus orchestrates the control of the neuroendocrine system, including PRL release, through a complex series of connections. Different hypothalamic nuclei are composed of cells with unique characteristics and functions. Differential outputs of the various hypothalamic nuclei control autonomic, endocrine, and behavioral responses. Furthermore, many hypothalamic cells also project outside the pituitary, influencing diverse regions in the brain and brainstem (Buijs and Van Heerikhuize, 1982; Sofroniew et al, 1981).
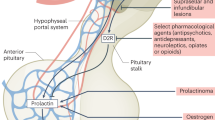
The hypothalamus has three main subdivisions: the periventricular zone, the medial hypothalamus, and the lateral hypothalamus, which are arranged medial-to-lateral from the midline. The periventricular and medial hypothalamic subdivisions are the main regions associated with PRL homeostasis (Figure 1). The periventricular zone is apposed to the ventricular wall, and includes the periventricular nucleus and the arcuate nucleus. Both of these nuclei contain dopaminergic cells, which tonically inhibit PRL release via projections to the anterior pituitary (AP) (Macleod, 1976). This system is known as the ‘tuberoinfundibular dopamine pathway’ (TIDA) (Schofield and Everitt, 1981). PRL release associated with antipsychotic drugs is mediated by this pathway, through blockade of D2 receptors located on the lactotrophs (Bunzow et al, 1988).

Schematics representing the pathways through which serotonin may stimulate PRL secretion. See text for details. PVN, paraventricular nucleus; MFB, medial forebrain bundle; F, fornix; PRL, prolactin; ARC, arcuate nucleus; DA, dopamine; PRF, prolactin-releasing factor; VIP, vasoactive intestinal peptide; OT, oxytocin; TIDA, tuberoinfundibular dopamine pathway; OC, optic chiasm; 5-HT, serotonin; GABA, gamma-aminobutyric acid.
The medial hypothalamus contains several prominent nuclei which are implicated in PRL regulation. The paraventricular nucleus (PVN) and supraoptic nucleus (SON) are associated with a variety of functions, which include regulation of metabolism and body temperature, control of water and food intake, and cardiovascular and gastrointestinal functioning (Lechan and Jackson, 1982; Crawley and Kiss, 1985; Leibowitz, 1978; Sawchenko et al, 1981; Porter and Brody, 1985). The PVN contains neurosecretory magnocellular cells, which produce oxytocin (OT) and vasopressin (AVP), and parvocellular cells, which synthesize corticotropin releasing factor (CRF), vasoactive intestinal peptide (VIP), thyroid releasing hormone (TRH), and other neuropeptides (Bloom et al, 1982; Kiss, 1988; Lechan and Jackson, 1982; Merchenthaler et al, 1982; Mezey and Kiss, 1985). The magnocellular OT and AVP cells project to the posterior lobe of the pituitary, while the parvocellular division projects to the AP via the median eminence. OT, which is found only in mammals, is associated with reproductive and maternal behaviors, and is also implicated in a variety of social behaviors (Hughes et al, 1987; Insel, 1992; Winslow and Insel, 1991). It is specifically involved in the initiation of parturition and milk ejection, and is itself a stimulator of PRL secretion (see below) (Samson et al, 1986). VIP, also a major mediator of PRL secretion, is found in cells of the parvocellular PVN (see below) (Ceccatelli et al, 1989). The SON, which has developmental and cellular similarities to the PVN, contains OT-positive cells (Saper, 1990; Van de Kar et al, 1995).
Pituitary
The pituitary is composed of anterior, posterior, and intermediate lobes. The AP arises from the ectoderm of the primitive pharynx known as Rathke's pouch, and is considered a glandular structure, composed of secretory cells (Sheng and Westphal, 1999). PRL is secreted by lactotrophs, and gonadotrophs, somatotrophs, thyrotrophs, and corticotrophs synthesize LH and FSH, GH, TSH, and ACTH, respectively (Laycock, 1983; Perez et al, 1995; Horvath and Kovacs, 1994). PRL and GH are also secreted by mammosomatotrophs which are found in very small numbers in the normal pituitary (Horvath and Kovacs, 1994). Releasing factors produced in the hypothalamus are secreted into the blood supply of the AP, diffusing locally to stimulate release of their respective hormones. Paracrine and autocrine mechanisms are also thought to play an important role in pituitary regulation (Denef, 1986).
The neurohypophysis, also known as posterior pituitary (PP), derives from neural tissue of the primitive hypothalamus, and is devoid of secretory cells (Stopa et al, 1993). OT and AVP produced in the PVN and SON are transported anterogradely and stored in the axon terminals found in the PP. These neurohormones are subsequently released in the systemic circulation in response to a variety of peripheral stimuli (Strand, 1999). Milk ejection in response to suckling is one example in which peripheral stimulation, in this case represented by afferents in the breast tissue, leads to the release of OT (Crowley and Armstrong, 1992).
Although the AP and PP have different embryonic origins, by week 16 of development they are fused together. These two pituitary lobes communicate via short portal vessels, which guarantee that hormones and releasing factors will diffuse between them via their shared blood supply (Liu and Ben-Jonathan, 1994). Thus, hormones released into the PP can influence the AP directly via communicating vessels as well as through the systemic circulation.
NEUROHORMONES AND PEPTIDES INVOLVED IN PRL SECRETION
A number of different agents are involved in the fine balance of stimulation vs inhibition of PRL release (McCann et al, 1984). These are organized according to documented ‘direct’ and ‘indirect’ effects on PRL release, based on in vitro and in vivo studies. Prolactin-inhibiting factors (PIFs) directly decrease PRL secretion, while prolactin releasing factors (PRFs) directly stimulate PRL secretion from lactotrophs. PRFs may also participate in indirect pathways. Neurotransmitters and neuropeptides that rely on intermediaries to stimulate PRL release are considered ‘modulators’ or ‘indirect regulators’ of PRL secretion (Ben-Jonathan, 1994). Indirect regulators can also have important direct effects on PRL gene expression, despite their inability to directly stimulate PRL secretion.
It is important to note that there are a myriad of substances that affect PRL release, although their physiological significance in PRL homeostasis is unclear. These substances include angiotensin II, somatostatin, substance P, neurotensin, galanin, endothelin, calcitonin, and atrial natriuretic peptide, which are not included in this review (for discussion, see Freeman (Freeman et al, 2000)).
PIFs and Other Inhibiting Agents
Dopamine
Dopamine is considered the primary physiologic PIF, and exerts tonic inhibition via two main pathways (Everitt et al, 1984; Martinez de la Escalera and Weiner, 1992). The main pathway is the TIDA, described above. Dopamine cell bodies in the arcuate nucleus send projections to the median eminence to release DA into the portal vessels, through which it reaches the lactotrophs in the anterior pituitary. PRL release is inhibited by the binding of dopamine to D2 receptors on the lactotrophs (Caron et al, 1978; Meller et al, 1991). The other inhibitory pathway, known as the tuberohypophyseal tract (Bjorklund et al, 1973), also originates in the arcuate nucleus but involves DA release into the blood supply of the posterior pituitary. Through that pathway, dopamine can reach the lactotrophs via short portal vessels. When tonic inhibition exerted by dopamine is overcome, PRL release ensues.
Others
In addition to its role in thyroid function, triiodothyronine (T3) is an inhibitor of human PRL gene transcription (Pernasetti et al, 1997), and is likely to participate in the physiological regulation of PRL secretion. Interestingly, TRH, the releasing factor for T3, exerts the opposite effect, acting as a PRF (see below). PRL exerts a short-loop negative feedback on its own release by stimulating TIDA cells through binding to PRL receptors on these neurons (Grattan, 2001).
PRL Stimulation
PRFs
VIP: VIP stimulates PRL release in vivo and in vitro (Ben-Jonathan, 1994). At the cellular level, VIP directly binds to its receptors on the lactotroph cell membrane, stimulating adenyl cyclase activity, and PRL gene transcription and release (Wanke and Rorstad, 1990). Systemic administration of VIP to rhesus monkeys and man causes a significant elevation of plasma PRL (Frawley and Neill, 1981; Lightman and Young, 1987). Conversely, passive immunoneutralization with VIP antisera reduces or abolishes the normal PRL surge associated with ether stress, suckling, and direct serotonergic stimulation (Shimatsu et al, 1984; Abe et al, 1985) and (Kaji et al, 1985). Afferent sources of VIP include the suprachiasmatic nucleus (SCN) and the PVN (Lam, 1991). However, VIP immunoreactive granules have also been localized in the lactotrophs (Segerson et al, 1989; Morel et al, 1982). VIP can thus regulate PRL release via hypothalamic afferents, and also through direct paracrine and autocrine mechanisms in the AP.
Oxytocin: OT, which is associated with parturition and maternal behaviors, also directly causes PRL release in vitro and in vivo (Mogg and Samson, 1990). Consistent with this, OT receptor mRNA expression is highly specific and restricted to lactotrophs in the AP (Breton et al, 1995). In lactating rats, elevation of OT plasma levels occurs just before the accompanying PRL surge associated with suckling. Administration of antioxytocin serum significantly reduces this PRL surge (Samson et al, 1986). A link between OT and VIP's action on PRL release is suggested by experiments in which animals injected with anti-OT serum prior to intraventricular administration of VIP fail to show elevation of plasma PRL. These results indicate that that OT participates in VIP-induced PRL release (Samson et al, 1989).
TRH: Thyrotropin-releasing hormone stimulates the release of PRL from the AP (Aizawa and Hinkle, 1985). Besides stimulating the secretion of PRL in vivo and in vitro, TRH has also been shown to stimulate PRL gene expression (Benker et al, 1990). TRH binds to membrane receptors on the lactotrophs, activating the phospholipase C signaling pathway (Bjoro et al, 1990). This pathway culminates in the activation of protein kinase C, with consequent increased PRL gene expression (Benker et al, 1990).
Modulators
Estrogen: Estrogen is a well-known modulator of PRL (Ben-Jonathan, 1994). Daily estrogen use in females increases basal and stimulated PRL secretion within 2–3 days (Greenspan, 2001). Estrogen's influence on PRL occurs through regulation of PRL gene expression (Schaufele, 1999) and indirect stimulation of PRL release (Chen and Meites, 1970; Grosvenor, 1960). The lactotroph expresses both nuclear estrogen receptor α and β subtypes (ERα and ERβ), as well as a truncated form of ERα (Mitchner et al, 1999). Based on information to date, ERβ is most significantly involved in estrogen-induced PRL gene expression (Mitchner et al, 1999). Estrogen binding to its β subtype receptor stimulates the PRL gene promoter, resulting in transcription of the PRL gene (Schaufele, 1999).
In addition to its direct actions on PRL gene expression, estrogen also interacts with PRFs at the hypothalamic level (Benker et al, 1990). Estrogen treatment stimulates PRL release by influencing hypothalamic VIP (Lam et al, 1990), and also OT gene expression (Shughrue et al, 2002).
Opioids: Endogenous opioid peptides such as enkephalins and dynorphins are involved in the regulation of PRL secretion during pregnancy, lactation, and stress (Ferland et al, 1978; Rossier et al, 1980; Sagrillo and Voogt, 1991). The suckling-induced PRL surge is blocked by treatment with naloxone, a nonselective opioid antagonist (Selmanoff and Gregerson, 1986). Naloxone also decreases stress- and estrogen-induced PRL secretion (Dupont et al, 1980; Petraglia et al, 1987).
Opioids act on a hypothalamic level, although a direct effect on the pituitary has not been completely excluded (Enjalbert et al, 1979). Endogenous opioid peptides and synthetic opiates suppress TH activity and m-RNA levels in the TIDA neurons (Arbogast and Voogt, 1998; Deyo et al, 1979; Reymond et al, 1983; Andrews and Grattan, 2003). Therefore, one established mechanism of opioid-induced PRL secretion is through inhibition of TIDA neuronal activity, probably modulated by the opioid receptor subtypes μ and κ (see review, Ben-Jonathan, 1994; Soaje and Deis, 1994). Contacts between opioid-containing terminals and TIDA neurons include pro-enkephalin, pro-dynorphin and pro-opiomelanocortin terminals (Fitzsimmons et al, 1992).
Serotonin: It has long been recognized, based on classic experiments mentioned above, that acute injections of 5-HT or its precursor, 5-HTP, stimulate PRL release (Kamberi et al, 1971). Conversely, blocking either serotonin synthesis or transmission results in a blunted PRL release (Kordon et al, 1973; Gil-ad et al, 1976; Kamberi et al, 1971). These effects depend on one or more PRFs, making serotonin an indirect, albeit potent, modulator of PRL release (Ben-Jonathan, 1994; Freeman et al, 2000) (Figure 1).
VIP and OT are among the best-studied candidates for serotonin's downstream actions on PRL release (McCann et al, 1984; Shimatsu et al, 1982, 1984; Van de Kar et al, 1995, 2001). VIP and OT are both found in the PVN, implicating this structure as an important anatomic substrate of PRL release (Mezey and Kiss, 1985; Sofroniew et al, 1981; Swanson and Sawchenko, 1983). Consequently, the PVN is among the best-studied sites of serotonin-induced PRL release, and is known to contain the postsynaptic 5-HT1A, 5-HT2A, and 5-HT2C receptor subtypes (Van de Kar et al, 2001; Jorgensen et al, 1992, 2003; Appel et al, 1990; Rittenhouse et al, 1993; Van de Kar et al, 1989). 5-HT2A/2C agonists increase Fos immunoreactivity (an index of synaptic activity) in the PVN (Van de Kar et al, 2001) ,while ablation of the PVN results in a markedly decreased PRL response to serotonin agonists (Bagdy, Rittenhouse #7066, Van de Kar #7052).
Recruitment of VIP neurons in the serotonin-induced PRL response is implied by the fact that VIP antiserum significantly blunts the usual PRL surge associated with serotonergic stimulation in vivo (Kaji et al, 1985; Shimatsu et al, 1982,). While it is hypothesized that VIP neurons of the PVN are directly stimulated by serotonergic fibers (Kiss et al, 1984; Lam, 1991; Reichlin, 1988), to date, serotonergic terminals have not been demonstrated on these VIP-expressing cells (Kiss et al, 1984).
Oxytocin, another PRF found in the PVN (magnocellular subdivision), is also stimulated by serotonin agonists in male and female animals (Moos, F l983; Saydoff, J l991; Van de Kar et al, 1995). Recent studies show that OT is also stimulated by fenfluramine in healthy men (Lee et al, 2003). These data, combined with the observation that bilateral PVN lesions inhibit the effect of the 5-HT-receptor agonists on PRL release, suggest that serotonin may stimulate PRL secretion through the OT system (Arey and Freeman, 1989; Van de Kar et al, 1995). In support of this idea, D-fenfluramine administration induces Fos- immunoreactivity in oxytocin-containing cells in the PVN (Javed et al, 1999).
Taken together, pharmacologic and anatomic data indicate that the PVN represents a major regulatory site of serotonin-induced PRL release, although the relative role of VIP and OT pathways in this response is not established. Furthermore, while ablation of the PVN blunts the PRL response to 5-HT agonists, it does not entirely abolish it (Bagdy, 1996; Rittenhouse et al, 1993), demonstrating that other pathways contribute to serotonin-induced PRL release.
An alternate path for serotonin-induced PRL release is inhibition of the tuberoinfundibular dopamine cells (TIDA). However, there is little synaptic contact between serotonergic fibers and the dopamine cells, indicating that if direct inhibition of dopamine cells occurs, it is through volume transmission of serotonin in the region (Kiss and Halasz, 1986). There is more direct evidence for serotonergic stimulation of GABAergic neurons in the vicinity of the TIDA dopamine cells, based on the presence of 5-HT1A receptors on these cells (Mirkes and Bethea, 2001). Assuming that these GABAergic cells are interneurons, their stimulation by 5HT would result in inhibition of TIDA cells, releasing the tonic inhibition of PRL (see Figure 1). Consistent with this idea, several pharmacologic studies show that GABA agonists injected in the hypothalamus promote PRL secretion (Ondo and Dom, 1986; Fuchs et al, 1984; Wagner et al, 1994), as does alprazolam administration (Bondolfi et al, 1997; Shioiri et al, 1996; Zemishlany et al, 1990). However, intraventricular injection of GABA has yielded differential effects depending on the dose utilized (with a reduction in plasma PRL seen with low (physiologic) doses, and increases with higher doses) (McCann et al, 1984; Vijayan and McCann, 1978). Several benzodiazepines have also been reported to decrease PRL secretion (Jarvinen et al, 1992). These collective results suggest that PRL levels are differentially influenced by GABAergic drugs depending on variables such as drug potency (affinity for the GABA receptor) and dose.
Hyperprolactinemia: Established pharmacologic causes of hyperprolactinemia include D2 receptor antagonists, tricyclic antidepressants, methyldopa, reserpine, and verapamil. Diseases related to hyperprolactinemia include prolactinomas and tumors compressing the pituitary stalk, hypothyroidism, and decreased clearance from renal insufficiency (Conner and Fried, 1998; Katz and Adashi, 1990; Klein et al, 1964; Quigley et al, 1979; Wieck and Haddad, 2003; Yazigi et al, 1997). The upper normal value for serum PRL in most laboratories is 20 ng/ml (nanograms per milliliter) for men and 25 ng/ml for women. Serum PRL levels between 20 and 200 ng/ml are generally associated with any cause of hyperprolactinemia, and values above 200 ng/ml are usually seen with prolactinomas (Gomez et al, 1977; Melmed, 2001; Schlechte et al, 1989).
The classic manifestations of hyperprolactinemia are galactorrhea, amenorrhea, infertility, and decreased libido in women, and erectile dysfunction, hypogonadism, and infertility in males (Gomez et al, 1977; Carter et al, 1978; Segal et al, 1979; Seppala, 1978). Long-term clinical consequences of hyperprolactinemia are less obvious, and include osteopenia in men and women (Greenspan et al, 1986; Jackson et al, 1986; Klibanski et al, 1988) and the possibility of an increased risk of breast cancer in women (Welsch and Nagasawa, 1977; Kwa et al, 1981). PRL levels associated with impaired fertility, decreased bone density and breast cancer have not been established. However, these conditions have a major public health impact and are therefore of special interest.
Altered Fertility
Menstrual disturbances associated with hyperprolactinemia range from luteal phase dysfunction with normal menses to frank amenorrhea (Bohnet et al, 1976; Katz and Adashi, 1990; Yazigi et al, 1997). Subtle abnormalities in PRL secretion may impact fertility. In a study of women with infertility of unknown etiology, the normal midcycle increase of PRL was absent in the patients, but maintained in healthy controls. All subjects had normal basal PRL levels, implicating loss of normal pulsatility in fertility problems (Subramanian et al, 1997). Consistent with this, altered fertility associated with only modestly elevated PRL levels is well recognized with antipsychotic drug use (Dickson and Glazer, 1999; Kinon et al, 2003; Smith, 2003), but has not been studied with respect to SSRIs. Aberrant PRL levels can interfere with female fertility through both CNS and peripheral mechanisms. At the hypothalamic level, high PRL interferes with LH pulsatility due to abnormalities in GnRH secretion (Sauder et al, 1984). Another mechanism is a direct inhibitory effect of PRL on ovarian follicular development and steroidogenesis (McNatty, 1979). Corpus luteum deficiency is also a mechanism of infertility due to PRL's luteolytic effect (Kredentser et al, 1981).
Decreased Bone Density
Bone loss and the long-term development of osteoporosis are deleterious effects of hyperprolactinemia in both men and women. There is little evidence supporting a direct role for PRL in bone homeostasis (Schlechte et al, 1983; Ciccarelli et al, 1988). Rather, elevated PRL levels disrupt the normal pulsatile secretion of LH and FSH, leading to hypogonadism (Greenspan, 2001; Bohnet et al, 1976; Seppala, 1978; Greenspan et al, 1986; Quigley et al, 1979; Sauder et al, 1984), and subsequent osteopenia (Klibanski et al, 1988, 1980; Schlechte et al, 1987, 1992; Koppelman et al, 1984; Di Somma et al, 1998; Carter et al, 1978). Hypoestrogenemia as a requisite for osteopenia in women is supported by studies showing that hyperprolactinemic women with normal menstrual cycles do not show decreased bone density (Ciccarelli et al, 1988; Klibanski et al, 1988; Schlechte et al, 1992; Wardlaw and Bilezikian, 1992). One study reports that hyperprolactinemic women who were amenorrheic for less than a year did not have significant bone loss (Cann et al, 1984), and several studies indicate an absence of continued bone loss even when exposure to elevated PRL levels occurs over several years (Biller et al, 1992; Koppelman et al, 1984; Schlechte et al, 1992). However, there are no definitive data describing what duration or degree of elevated PRL levels are necessary to result in decreased bone density.
Bone loss, once established, may persist after resolution of hyperprolactinemia, indicating that even transient elevations in PRL may be an important risk factor for osteoporosis (Biller et al, 1992; Di Somma et al, 1998; Schlechte et al, 1987, 1983). Hyperprolactinemia in adolescents is associated with persistent bone loss (Coelho et al, 1999). Since the achievement of peak bone mass represents an important protection against osteoporosis (Bonjour, 1996), adolescents are an important population in which to assess drug-induced PRL abnormalities. In a small uncontrolled study of subjects on antipsychotics for at least 6 months, bone mineral density and PRL levels were inversely correlated (Abraham et al, 2003).
Breast Cancer
A subject of controversy is the role of PRL in the genesis of breast cancer. Although it has been demonstrated that PRL increases the growth of malignant breast cells in vitro (Welsch and Nagasawa, 1977), the clinical significance of such findings is an area of much debate.
The best epidemiologic study evaluating the association between elevated PRL and breast cancer was a nested case–control study conducted within the prospective Nurses' Health Study cohort of 121 700 women (Hankinson et al, 1999). In this study, 306 women diagnosed with breast cancer were matched with 448 controls. All participants had blood samples collected between the period of 1989–1990, at which point they were 43–69 years of age and all postmenopausal. Case patients were women diagnosed with breast cancer after blood collection. A statistically significant higher risk of breast cancer was found in women who had PRL levels above 9.7 ng/ml (relative risk of 2.03 with a 95% CI (1.24–3.31)), even when several established breast cancer risk factors were controlled for. In another prospective study with 40 postmenopausal breast cancer patients, Wang et al found a positive but nonsignificant association between elevated PRL levels and the risk of developing breast cancer (Wang et al, 1992).
There is also an association between elevated PRL levels and poor survival in breast cancer patients. PRL levels above 12.6 ng/ml were linked to a poor survival prognosis in postmenopausal subjects who had undergone mastectomy for breast cancer (Wang et al, 1995). Plasma PRL levels above 32 ng/ml were seen in 8% of 149 women with metastatic breast cancer, in contrast to none of the 221 control subjects (p<0.001) (Holtkamp et al, 1984). In the same study, mean survival time after mastectomy in the group with normal PRL was 154 months, compared with 89 months in the hyperprolactinemic group. Patients on PRL elevating or lowering drugs were excluded from the study.
Drug-induced risks from antidepressant medications are only beginning to be investigated. A recently published case–control study investigated the association between prior antidepressant use and risk of breast cancer (Moorman et al, 2003). Overall, there was no increased risk of breast cancer with ever-use when all antidepressant categories were taken into account (tricyclics, monoamine oxidase inhibitors, SSRIs). However, there was a trend towards increased risk with use of SSRIs for 36 months or longer. In another case–control study, Kelly et al also found a borderline statistically significant increased risk for breast cancer associated with SSRI use, noting that these results were ‘not reassuring’(Kelly et al, 1999). It is not entirely clear whether a possible association between antidepressants and increased risk of breast cancer is related to elevation in PRL levels or direct tumor growth-promoting properties of certain antidepressants (Brandes et al, 1992). However, the results from these studies warrant further investigation of the potential association between serotonin-enhancing drugs, PRL, and breast cancer.
CLINICAL RELEVANCE OF SSRI-INDUCED HYPERPROLACTINEMIA: EMERGING DATA
While the link between acute serotonin stimulation and PRL release has been long established, the clinical effects of chronic serotonin stimulation on PRL has only been investigated recently. A review of the literature retrieved 13 single case reports describing nonpuerperal lactation associated with the use of SSRIs in women (Bronzo and Stahl, 1993; Morrison et al, 2001; Peterson, 2001; Arya and Taylor, 1995; Iancu et al, 1992; Bonin et al, 1997; Jeffries et al, 1992; Lesaca, 1996; Bondolfi et al, 1997; Davenport and Velamoor, 2002; Gonzalez et al, 2000; Otero et al, 2002; Pablos et al, 2001). In these case reports, the patients were all females, most of them premenopausal. Plasma PRL levels ranged between 28 and 60 ng/ml. A common theme of the single case reports is the onset of galactorrhea, usually associated with amenorrhea, shortly after initiation of treatment with an SSRI. In all reports, symptoms promptly subsided with discontinuation of the drug. All SSRIs are implicated in these reports.
There have also been several uncontrolled studies assessing changes in PRL level with therapy with SSRIs (Amsterdam et al, 1997; Attenburrow et al, 2001; Bronzo and Stahl, 1993; Morrison et al, 2001; Peterson, 2001; Spigset and Mjorndal, 1997; Cowen and Sargent, 1997; Dulchin et al, 2001; Laine et al, 1997; Urban and Veldhuis, 1991). All reports showed varied degrees of basal PRL elevation with SSRI treatment. Amsterdam et al (1997) examined the incidence of mammoplasia (breast enlargement) in 59 women taking SSRIs or venlafaxine, finding the highest rate with paroxetine compared to the other antidepressants. Paroxetine-treated patients exhibited statistically significant elevations in PRL levels, although all subjects on fluoxetine, sertraline or venlafaxine showed nonsignificant elevation of their basal PRL. Although significant effects were only seen for paroxetine, this may be related to the relatively greater number of patients on paroxetine (n=28), compared to the other drugs (fluoxetine (n=8), sertraline (n=4), and venlafaxine (n=19)), resulting in inadequate power to detect changes in the latter groups.
The incidence and prevalence of hyperprolactinemia in patients taking SSRIs will be important to pursue in future controlled studies. Based on cumulative case reports, all SSRIs have the potential to cause elevation of basal PRL. This observation was recently confirmed by the French Pharmacovigilance Database Study, an epidemiological study that investigated the rates of hyperprolactinemia induced by multiple prescription medications from 1985 to 2000 (Petit et al, 2003). Of the total of 159 cases of drug-induced hyperprolactinemia, 17% had been induced by SSRIs, which included sertraline (OR=odds ratio 15.74), fluoxetine (OR 49), paroxetine (OR 8.10), fluvoxamine (OR 5.96), and citalopram (OR 3.62). Citalopram was the only SSRI not to reach statistical significance. The available data indicate that SSRI-induced hyperprolactinemia is a class-related effect.
RETHINKING HYPERPROLACTINEMIA
The small number of case reports may be an indication that SSRI-induced hyperprolactinemia is not a common phenomenon. An alternative explanation is that symptoms may go unrecognized by treating clinicians either because they are subtle, or patients tend not to report them. Based on case reports, women seem to be at higher risk for SSRI-induced hyperprolactinemia. Because recognition of galactorrhea and amenorrhea induced by SSRIs leads to prompt discontinuation of the drug without further deleterious consequences, other than the interruption of treatment, the issue of SSRI-induced hyperprolactinemia has been generally seen as a benign one. However, classic definitions of clinical hyperprolactinemia, based on primary pituitary pathology, may not be entirely applicable for drug-induced PRL hypersecretion. It remains to be answered if minimal elevations of PRL, which do not qualify as hyperprolactinemia by current laboratory criteria, impact gonadal function, with consequent effects on fertility and bone density. As mentioned above, subtle alterations in PRL have been demonstrated in women with unexplained infertility (Subramanian et al, 1997). It is thus important that future studies measuring changes in PRL take into consideration the individual baseline levels of the study subjects. It is possible that a specific PRL level that would not cause any problems in one individual may induce clinically important pathology in another. Given the large number of patients taking SSRIs (Frank et al, 2001), the incidence of PRL-associated effects may be significant, even if a small percentage of patients are affected. Moreover, SSRIs are commonly prescribed to postmenopausal women, a population in which the diagnosis of hyperprolactinemia is difficult to recognize, given their hypoestrogenic state, and lack of obvious clinical manifestations. It is unknown whether even mild PRL elevations would add to the risk of osteoporosis and breast cancer in that population.
This review highlights possible mechanisms and manifestations of serotonergic stimulation of PRL, considering them in the context of SSRI use. There are a number of different pathways through which SSRIs may cause PRL abnormalities, including release of a PRF such as VIP or OT, and the inhibition of the TIDA system. The wide-ranging effects of PRL on reproduction, lactation, stress, and immune responses, suggest a fundamental role in maintaining the homeostasis of the organism. This view is supported by the anatomy of this system, which involves complex, and perhaps redundant, pathways mediating PRL regulation.
Future studies should characterize changes in PRL associated with SSRI use, and determine the magnitude and duration of PRL elevations and their association with chronic sequelae such as blunted fertility, decreased bone density, and elevated risk of breast cancer. Measuring pre- and post-treatment PRL levels in well-characterized patient groups will begin to address these issues, advancing our understanding of serotonin–prolactin interactions.
References
Abe H, Engler D, Molitch ME, Bollinger-Gruber J, Reichlin S (1985). Vasoactive intestinal peptide is a physiological mediator of prolactin release in the rat. Endocrinology 116: 1383–1390.
Abel KM, O'Keane V, Murray RM, Cleare AJ (1997). Serotonergic function and negative and depressive symptomatology in schizophrenia and major depression. Psychoneuroendocrinology 22: 539–548.
Abraham G, Halbreich U, Friedman RH, Josiassen RC (2003). Bone mineral density and prolactin associations in patients with chronic schizophrenia. Schizophrenia Res 59: 17–18.
Agurell S (1983). The research and development of a 5-HT selective reuptake blocker. Preclinical aspects. Acta Psychiatr Scand, Suppl 308: 19–24.
Aizawa T, Hinkle PM (1985). Thyrotropin-releasing hormone rapidly stimulates a biphasic secretion of prolactin and growth hormone in GH4C1 rat pituitary tumor cells. Endocrinology 116: 73–82.
Amsterdam JD, Garcia-Espana F, Goodman D, Hooper M, Hornig-Rohan M (1997). Breast enlargement during chronic antidepressant therapy. J Affect Disord 46: 151–156.
Andrews ZB, Grattan DR (2003). Opioid receptor subtypes involved in the regulation of prolactin secretion during pregnancy and lactation. J Neuroendocrinol 15: 227–236.
Appel NM, Mitchell WM, Garlick RK, Glennon RA, Teitler M, De Souza EB (1990). Autoradiographic characterization of (+-)-1-(2,5-dimethoxy-4-and 2 over black square]; [1 and 2 over black square]25I] iodophenyl)-2-aminopropane (and 2 over black square]; [1 and 2 over black square]25I]DOI) binding to 5-HT2 and 5-HT1c receptors in rat brain. J Pharmacol Exp Ther 255: 843–857.
Arbogast LA, Voogt JL (1998). Endogenous opioid peptides contribute to suckling-induced prolactin release by suppressing tyrosine hydroxylase activity and messenger ribonucleic acid levels in tuberoinfundibular dopaminergic neurons. Endocrinology 139: 2857–2862.
Arey BJ, Freeman ME (1989). Hypothalamic factors involved in the endogenous stimulatory rhythm regulating prolactin secretion. Endocrinology 124: 878–883.
Arey BJ, Freeman ME (1992). Activity of vasoactive intestinal peptide and serotonin in the paraventricular nucleus reflects the periodicity of the endogenous stimulatory rhythm regulating prolactin secretion. Endocrinology 131: 736–742.
Arya DK, Taylor WS (1995). Lactation associated with fluoxetine treatment. Austr NZ J Psychiatry 29: 697.
Asberg M, Thoren P, Traskman L, Bertilsson L, Ringberger V (1976). ‘Serotonin depression’—a biochemical subgroup within the affective disorders? Science 191: 478–480.
Asher I, Kaplan B, Modai I, Neri A, Valevski A, Weizman A (1995). Mood and hormonal changes during late pregnancy and puerperium. Clin Exp Obstet Gynecol 22: 321–325.
Asnis GM, Eisenberg J, van Praag HM, Lemus CZ, Friedman JM, Miller AH (1988). The neuroendocrine response to fenfluramine in depressives and normal controls. Biol Psychiatry 24: 117–120.
Attenburrow MJ, Mitter PR, Whale R, Terao T, Cowen PJ (2001). Low-dose citalopram as a 5-HT neuroendocrine probe. Psychopharmacology 155: 323–326.
Bagdy G (1996). Role of the hypothalamic paraventricular nucleus in 5-HT1A, 5-HT2A and 5-HT2C receptor-mediated oxytocin, prolactin and ACTH/corticosterone responses. Behav Brain Res 73: 277–280.
Bakowska JC, Morrell JI (1997). Atlas of the neurons that express mRNA for the long form of the prolactin receptor in the forebrain of the female rat. J Compar Neurol 386: 161–177.
Ben-Jonathan N (1994). Regulation of prolactin secretion. In Imura H (ed), The Pituitary Gland, Second ed. Raven Press, Ltd.: New York. pp 261–283.
Benker G, Jaspers C, Hausler G, Reinwein D (1990). Control of prolactin secretion. Klin Wochen 68: 1157–1167.
Berger FM (1975). Commentary. Depression and antidepressant drugs. Clin Pharmacol Ther 18: 241–248.
Biller BM, Baum HB, Rosenthal DI, Saxe VC, Charpie PM, Klibanski A (1992). Progressive trabecular osteopenia in women with hyperprolactinemic amenorrhea. [comment]. J Clin Endocrinol Metab 75: 692–697.
Bjorklund A, Moore RY, Nobin A, Stenevi U (1973). The organization of tubero-hypophyseal and reticulo-infundibular catecholamine neuron systems in the rat brain. Brain Res 51: 171–191.
Bjoro T, Sand O, Ostberg BC, Gordeladze JO, Torjesen P, Gautvik KM et al. (1990). The mechanisms by which vasoactive intestinal peptide (VIP) and thyrotropin releasing hormone (TRH) stimulate prolactin release from pituitary cells. Biosci Rep 10: 189–199.
Bloom FE, Battenberg EL, Rivier J, Vale W (1982). Corticotropin releasing factor (CRF): immunoreactive neurones and fibers in rat hypothalamus. Regul Peptides 4: 43–48.
Bohnet HG, Dahlen HG, Wuttke W, Schneider HP (1976). Hyperprolactinemic anovulatory syndrome. J Clin Endocrinol Metab 42: 132–143.
Bondolfi G, Rubin C, Bryois C, Eap CB (1997). Galactorrhoea induced by a pharmacodynamic interaction between citalopram, alprazolam and tramadol: a case report. Therapie 52: 76–77.
Bonin B, Vandel P, Sechter D, Bizouard P (1997). Paroxetine and galactorrhea. Pharmacopsychiatry 30: 133–134.
Bonjour JPaR R (1996). Bone acquisition in adolescence. In Marcus R (ed), Osteoporosis. Academic Press: San Diego. pp 465–476.
Brandes LJ, Arron RJ, Bogdanovic RP, Tong J, Zaborniak CL, Hogg GR et al. (1992). Stimulation of malignant growth in rodents by antidepressant drugs at clinically relevant doses. Cancer Res 52: 3796–3800.
Breton C, Pechoux C, Morel G, Zingg HH (1995). Oxytocin receptor messenger ribonucleic acid: characterization, regulation, and cellular localization in the rat pituitary gland. Endocrinology 136: 2928–2936.
Bronzo MR, Stahl SM (1993). Galactorrhea induced by sertraline [see comments.]. Am J Psychiatry 150: 1269–1270.
Buijs RM, Van Heerikhuize JJ (1982). Vasopressin and oxytocin release in the brain—a synaptic event. Brain Res 252: 71–76.
Bunzow JR, Van Tol HHM, Grandy DK, Albert P, Salon J, Christle M et al. (1988). Cloning and expression of a rat D2 dopamine receptor cDNA. Nature 336: 783–787.
Cann CE, Martin MC, Genant HK, Jaffe RB (1984). Decreased spinal mineral content in amenorrheic women. JAMA 251: 626–629.
Caron MG, Beaulieu M, Raymond V, Gagne B, Drouin J, Lefkowitz RJ et al. (1978). Dopaminergic receptors in the anterior pituitary gland. Correlation of [3H]dihydroergocryptine binding with the dopaminergic control of prolactin release. J Biol Chem 253: 2244–2253.
Carter JN, Tyson JE, Tolis G, Van Vliet S, Faiman C, Friesen HG (1978). Prolactin-screening tumors and hypogonadism in 22 men. N Engl J Med 299: 847–852.
Ceccatelli S, Eriksson M, Hokfelt T (1989). Distribution and coexistence of corticotropin-releasing factor-, neurotensin-, enkephalin-, cholecystokinin-, galanin- and vasoactive intestinal polypeptide/peptide histidine isoleucine-like peptides in the parvocellular part of the paraventricular nucleus. Neuroendocrinology 49: 309–323.
Chen CL, Meites J (1970). Effects of estrogen and progesterone on serum and pituitary prolactin levels in ovariectomized rats. Endocrinology 86: 503–505.
Chen HW, Meier H, Heiniger HJ, Huebner RJ (1972). Tumorigenesis in strain DW-J mice and induction by prolactin of the group-specific antigen of endogenous C-type RNA tumor virus. J Natl Cancer Inst 49: 1145–1154.
Chiu S, Wise PM (1994). Prolactin receptor mRNA localization in the hypothalamus by in situ hybridization. J Neuroendocrinol 6: 191–199.
Ciccarelli E, Savino L, Carlevatto V, Bertagna A, Isaia GC, Camanni F (1988). Vertebral bone density in non-amenorrhoeic hyperprolactinaemic women. Clin Endocrinol 28: 1–6.
Cleare AJ, Bearn J, Allain T, McGregor A, Wessely S, Murray RM et al. (1995). Contrasting neuroendocrine responses in depression and chronic fatigue syndrome. J Affect Disord 34: 283–289.
Coccaro EF, Kavoussi RJ, Cooper TB, Hauger RL (1997a). Central serotonin activity and aggression: inverse relationship with prolactin response to d-fenfluramine, but not CSF 5-HIAA concentration, in human subjects. Am J Psychiatry 154: 1430–1435.
Coccaro EF, Kavoussi RJ, Trestman RL, Gabriel SM, Cooper TB, Siever LJ (1997b). Serotonin function in human subjects: intercorrelations among central 5-HT indices and aggressiveness. Psychiatry Res 73: 1–14.
Coccaro EF, Siever LJ, Klar HM, Maurer G, Cochrane K, Cooper TB et al. (1989). Serotonergic studies in patients with affective and personality disorders. Correlates with suicidal and impulsive aggressive behavior. [erratum appears in Arch Gen Psychiatry 1990 Feb;47(2):124.]. Arch Gen Psychiatry 46: 587–599.
Coelho R, Silva C, Maia A, Prata J, Barros H (1999). Bone mineral density and depression: a community study in women. J Psychosom Res 46: 29–35.
Conner P, Fried G (1998). Hyperprolactinemia; etiology, diagnosis and treatment alternatives. Acta Obstet Gynecol Scand 77: 249–262.
Cowen PJ, Sargent PA (1997). Changes in plasma prolactin during SSRI treatment: evidence for a delayed increase in 5-HT neurotransmission. J Psychopharmacol 11: 345–348.
Crawley JN, Kiss JZ (1985). Paraventricular nucleus lesions abolish the inhibition of feeding induced by systemic cholecystokinin. Peptides 6: 927–935.
Crowley WR, Armstrong WE (1992). Neurochemical regulation of oxytocin secretion in lactation. Endocrine Rev 13: 33–65.
Crumeyrolle-Arias M, Latouche J, Jammes H, Djiane J, Kelly PA, Reymond MJ et al (1993). Prolactin receptors in the rat hypothalamus: autoradiographic localization and characterization. Neuroendocrinology 57: 457–466.
Davenport E, Velamoor R (2002). A case of paroxetine-induced galactorrhea. Can J Psychiatry—Rev Can Psychiatrie 47: 890–891.
Denef C (1986). Paracrine interactions in the anterior pituitary. Clin Endocrinol Metab 15: 1–32.
Deyo SN, Swift RM, Miller RJ (1979). Morphine and endorphins modulate dopamine turnover in rat median eminence. Proc Natl Acad Sci USA 76: 3006–3009.
Di Somma C, Colao A, Di Sarno A, Klain M, Landi ML, Facciolli G et al. (1998). Bone marker and bone density responses to dopamine agonist therapy in hyperprolactinemic males. J Clin Endocrinol Metab 83: 807–813.
Dickson RA, Glazer WM (1999). Neuroleptic-induced hyperprolactinemia. Schizophrenia Res 35: S75–S86.
Doherty PC, Bartke A, Smith MS (1981). Differential effects of bromocriptine treatment on LH release and copulatory behavior in hyperprolactinemic male rats. Horm Behav 15: 436–450.
Drago F, Bohus B, Gispen WH, Scapagnini U, De Wied D (1983). Prolactin-enhanced grooming behavior: interaction with ACTH. Brain Res 263: 277–282.
Drago F, Pulvirenti L, Spadaro F, Pennisi G (1990). Effects of TRH and prolactin in the behavioral despair (swim) model of depression in rats. Psychoneuroendocrinology 15: 349–356.
Dubovsky SL (1994). Beyond the serotonin reuptake inhibitors: rationales for the development of new serotonergic agents. J Clin Psychiatry 55: 34–44.
Dulchin MC, Oquendo MA, Malone KM, Ellis SP, Li S, Mann JJ (2001). Prolactin response to dl-fenfluramine challenge before and after treatment with paroxetine. Neuropsychopharmacology 25: 395–401.
Dupont A, Barden N, Cusan L, Merand Y, Labrie F, Vaudry H (1980). beta-Endorphin and met-enkephalins: their distribution, modulation by estrogens and haloperidol, and role in neuroendocrine control. Fed Proc 39: 2544–2550.
Enjalbert A, Ruberg M, Fiore L, Arancibia S, Priam M, Kordon C (1979). Effect of morphine on the dopamine inhibition of pituitary prolactin release in vitro. Eur J Pharmacol 53: 211–212.
Everitt BJ, Hokfelt T, Wu JY, Goldstein M (1984). Coexistence of tyrosine hydroxylase-like and gamma-aminobutyric acid-like immunoreactivities in neurons of the arcuate nucleus. Neuroendocrinology 39: 189–191.
Fava M, Vuolo RD, Wright EC, Nierenberg AA, Alpert JE, Rosenbaum JF (2000). Fenfluramine challenge in unipolar depression with and without anger attacks. Psychiatry Res 94: 9–18.
Ferland L, Kledzik GS, Cusan L, Labrie F (1978). Evidence for a role of endorphins in stress- and suckling-induced prolactin release in the rat. Mol Cell Endocrinol 12: 267–272.
Fitzsimmons MD, Olschowka JA, Wiegand SJ, Hoffman GE (1992). Interaction of opioid peptide-containing terminals with dopaminergic perikarya in the rat hypothalamus. Brain Res 581: 10–18.
Frank L, Revicki DA, Sorensen SV, Shih YC (2001). The economics of selective serotonin reuptake inhibitors in depression: a critical review. CNS Drugs 15: 59–83.
Frantz AG (1978). Prolactin. N Engl J Med 298: 201–207.
Frawley LS, Neill JD (1981). Stimulation of prolactin secretion in rhesus monkeys by vasoactive intestinal polypeptide. Neuroendocrinology 33: 79–83.
Freeman ME, Kanyicska B, Lerant A, Nagy G (2000). Prolactin: structure, function, and regulation of secretion. Physiol Rev 80: 1523–1631.
Fuchs E, Mansky T, Stock KW, Vijayan E, Wuttke W (1984). Involvement of catecholamines and glutamate in GABAergic mechanism regulatory to luteinizing hormone and prolactin secretion. Neuroendocrinology 38: 484–489.
Gala RR (1990). The physiology and mechanisms of the stress-induced changes in prolactin secretion in the rat. Life Sci 46: 1407–1420.
Gil-ad I, Zambotti F, Carruba MO, Vicentini L, Muller EE (1976). Stimulatory role for brain serotoninergic system on prolactin secretion in the male rat. Proc Soc Exp Biol Med 151: 512–518.
Gomez F, Reyes FI, Faiman C (1977). Nonpuerperal galactorrhea and hyperprolactinemia. Clinical findings, endocrine features and therapeutic responses in 56 cases. Am J Med 62: 648–660.
Gonzalez E, Minguez L, Sanguino RM (2000). Galactorrhea after paroxetine treatment. Pharmacopsychiatry 33: 118.
Goodnick PJ, Goldstein BJ (1998). Selective serotonin reuptake inhibitors in affective disorders—I. Basic pharmacology. J Psychopharmacol 12: S5–S20.
Grattan DR (2001). The actions of prolactin in the brain during pregnancy and lactation. Progr Brain Res 133: 153–171.
Greenspan FSaG DG (2001). Hypothalamus and pituitary. Basic and Clinical Endocrinology, sixth edition ed Lange Medical Books/McGraw-Hill: New York. pp 100–162.
Greenspan SL, Neer RM, Ridgway EC, Klibanski A (1986). Osteoporosis in men with hyperprolactinemic hypogonadism. Ann Intern Med 104: 777–782.
Grosvenor CE (1960). Pituitary lactogenic hormone concentration during pregnancy in the rat. Endocrinology 66: 96–99.
Hankinson SE, Willett WC, Michaud DS, Manson JE, Colditz GA, Longcope C et al. (1999). Plasma prolactin levels and subsequent risk of breast cancer in postmenopausal women. J Natl Cancer Inst 91: 629–634.
Harlan RE, Shivers BD, Pfaff DW (1983). Midbrain microinfusions of prolactin increase the estrogen-dependent behavior, lordosis. Science 219: 1451–1453.
Holsboer F, Barden N (1996). Antidepressants and hypothalamic–pituitary–adrenocortical regulation. Endocrine Rev 17: 187–205.
Holtkamp W, Nagel GA, Wander HE, Rauschecker HF, von Heyden D (1984). Hyperprolactinemia is an indicator of progressive disease and poor prognosis in advanced breast cancer. Int J Cancer 34: 323–328.
Horvath E, Kovacs K (1994). Morphology of adenohypophysial cells and pituitary adenomas. In Imura H (ed), The Pituitary Gland, 2nd edn. Raven Press, Ltd.: New York. pp 29–62.
Hughes AM, Everitt BJ, Lightman SL, Todd K (1987). Oxytocin in the central nervous system and sexual behaviour in male rats. Brain Res 414: 133–137.
Huhman KL, Mougey EH, Moore TO, Meyerhoff JL (1995). Stressors, including social conflict, decrease plasma prolactin in male golden hamsters. Horm Behav 29: 581–592.
Hyttel J (1984). Experimental pharmacology of selective 5-HT reuptake inhibitors: differences and similarities. Clin Neuropharmacol 7: 866–867.
Iancu I, Ratzoni G, Weitzman A, Apter A (1992). More fluoxetine experience. J Am Acad Child Adoles Psychiatry 31: 755–756.
Insel TR (1992). Toward a neuroanatomy of obsessive–compulsive disorder. Arch Gen Psychiatry 49: 739–744.
Jackson JA, Kleerekoper M, Parfitt AM (1986). Symptomatic osteoporosis in a man with hyperprolactinemic hypogonadism. Ann Intern Med 105: 543–545.
Jarvinen A, Rago L, Mannisto PT (1992). Effects of central and peripheral type benzodiazepine ligands on thyrotropin and prolactin secretion. Neuropeptides 21: 183–191.
Javed A, Kamradt MC, Van de Kar LD, Gray TS (1999). D-Fenfluramine induces serotonin-mediated Fos expression in corticotropin-releasing factor and oxytocin neurons of the hypothalamus, and serotonin-independent Fos expression in enkephalin and neurotensin neurons of the amygdala. Neuroscience 90: 851–858.
Jeffries J, Bezchlibnyk-Butler K, Remington G (1992). Amenorrhea and galactorrhea associated with fluvoxamine in a loxapine-treated patient. J Clin Psychopharmacol 12: 296–297.
Johnston CA, Negro-Vilar A (1986). Maturation of the prolactin and proopiomelanocortin-derived peptide responses to ether stress and morphine: neurochemical analysis. Endocrinology 118: 797–804.
Jorgensen H, Knigge U, Warberg J (1992). Involvement of 5-HT1, 5-HT2, and 5-HT3 receptors in the mediation of the prolactin response to serotonin and 5-hydroxytryptophan. Neuroendocrinology 55: 336–343.
Jorgensen H, Riis M, Knigge U, Kjaer A, Warberg J (2003). Serotonin receptors involved in vasopressin and oxytocin secretion. J Neuroendocrinol 15: 242–249.
Kaji H, Chihara K, Kita T, Kashio Y, Okimura Y, Fujita T (1985). Administration of antisera to vasoactive intestinal polypeptide and peptide histidine isoleucine attenuates ether-induced prolactin secretion in rats. Neuroendocrinology 41: 529–531.
Kamberi IA, Mical RS, Porter JC (1971). Effects of melatonin and serotonin on the release of FSH and prolactin. Endocrinology 88: 1288–1293.
Kasper S, Vieira A, Schmidt R, Richter P (1990). Multiple hormone responses to stimulation with dl-fenfluramine in patients with major depression before and after antidepressive treatment. Pharmacopsychiatry 23: 76–84.
Katz E, Adashi EY (1990). Hyperprolactinemic disorders. Clin Obstet Gynecol 33: 622–639.
Kavoussi RJ, Hauger RL, Coccaro EF (1999). Prolactin response to d-fenfluramine in major depression before and after treatment with serotonin reuptake inhibitors. Biol Psychiatry 45: 295–299.
Kavoussi RJ, Kramer J, Hauger RL, Coccaro EF (1998). Prolactin response to D-fenfluramine in outpatients with major depression. Psychiatry Res 79: 199–205.
Kelly JP, Rosenberg L, Palmer JR, Rao RS, Strom BL, Stolley PD et al. (1999). Risk of breast cancer according to use of antidepressants, phenothiazines, and antihistamines. Am J Epidemiol 150: 861–868.
Kinon BJ, Gilmore JA, Liu H, Halbreich UM (2003). Hyperprolactinemia in response to antipsychotic drugs: characterization across comparative clinical trials. Psychoneuroendocrinology 28: 69–82.
Kiss J, Halasz B (1986). Synaptic connections between serotoninergic axon terminals and tyrosine hydroxylase-immunoreactive neurons in the arcuate nucleus of the rat hypothalamus. A combination of electron microscopic autoradiography and immunocytochemistry. Brain Res 364: 284–294.
Kiss J, Leranth C, Halasz B (1984). Serotoninergic endings on VIP-neurons in the suprachiasmatic nucleus and on ACTH-neurons in the arcuate nucleus of the rat hypothalamus. A combination of high resolution autoradiography and electron microscopic immunocytochemistry. Neurosci Lett 44: 119–124.
Kiss JZ (1988). Dynamism of chemoarchitecture in the hypothalamic paraventricular nucleus. Brain Res Bull 20: 699–708.
Klein J, Segal R, Pichel Warner R (1964). Galactorrhea due to imipramine. N Engl J Med 271: 510–512.
Kleinberg D, Noel G, Frantz A (1971). Chlorpromazine stimulation and L-DOPA suppression of plasma prolactin in man. J Clin Endocrinol Metab 33: 873–876.
Klibanski A, Biller BM, Rosenthal DI, Schoenfeld DA, Saxe V (1988). Effects of prolactin and estrogen deficiency in amenorrheic bone loss. J Clin Endocrinol Metab 67: 124–130.
Klibanski A, Neer RM, Beitins IZ, Ridgway EC, Zervas NT, McArthur JW (1980). Decreased bone density in hyperprolactinemic women. N Engl J Med 303: 1511–1514.
Koppelman MC, Kurtz DW, Morrish KA, Bou E, Susser JK, Shapiro JR et al. (1984). Vertebral body bone mineral content in hyperprolactinemic women. J Clin Endocrinol Metab 59: 1050–1053.
Kordon C, Blake CA, Terkel J, Sawyer CH (1973). Participation of serotonin-containing neurons in the suckling-induced rise in plasma prolactin levels in lactating rats. Neuroendocrinology 13: 213–223.
Kredentser JV, Hoskins CF, Scott JZ (1981). Hyperprolactinemia—a significant factor in female infertility. Am J Obstet Gynecol 139: 264–267.
Kwa HG, Cleton F, Wang DY, Bulbrook RD, Bulstrode JC, Hayward JL et al. (1981). A prospective study of plasma prolactin levels and subsequent risk of breast cancer. Int J Cancer 28: 673–676.
Laine K, Anttila M, Heinonen E, Helminen A, Huupponen R, Maki-Ikola O et al. (1997). Lack of adverse interactions between concomitantly administered selegiline and citalopram. Clin Neuropharmacol 20: 419–433.
Lam KS (1991). Vasoactive intestinal peptide in the hypothalamus and pituitary. Neuroendocrinology 53: 45–51.
Lam KS, Srivastava G, Lechan RM, Lee T, Reichlin S (1990). Estrogen regulates the gene expression of vasoactive intestinal peptide in the anterior pituitary. Neuroendocrinology 52: 417–421.
Laycock J (1983). Essential Endocrinology, 2nd edn Oxford University Press.
Lechan RM, Jackson IM (1982). Immunohistochemical localization of thyrotropin-releasing hormone in the rat hypothalamus and pituitary. Endocrinology 111: 55–65.
Lee R, Garcia F, van de Kar LD, Hauger RD, Coccaro EF (2003). Plasma oxytocin in response to pharmaco-challenge to D-fenfluramine and placebo in healthy men. Psychiatry Res 118: 129–136.
Leibowitz SF (1978). Paraventricular nucleus: a primary site mediating adrenergic stimulation of feeding and drinking. Pharmacol Biochem Behav 8: 163–175.
Lesaca TG (1996). Sertraline and galactorrhea. J Clin Psychopharmacol 16: 333–334.
Li C, Kelly PA, Buntin JD (1995). Inhibitory effects of anti-prolactin receptor antibodies on prolactin binding in brain and prolactin-induced feeding behavior in ring doves. Neuroendocrinology 61: 125–135.
Lightman SL, Young III WS (1987). Vasopressin, oxytocin, dynorphin, enkephalin and corticotrophin-releasing factor mRNA stimulation in the rat. J Physiol 394: 23–39.
Liu JW, Ben-Jonathan N (1994). Prolactin-releasing activity of neurohypophysial hormones: structure–function relationship. Endocrinology 134: 114–118.
Lopez-Ibor Jr JJ, Saiz-Ruiz J, Iglesias LM (1988). The fenfluramine challenge test in the affective spectrum: a possible marker of endogeneity and severity. Pharmacopsychiatry 21: 9–14.
Lu KH, Meites J (1973). Effects of serotonin precursors and melatonin on serum prolactin release in rats. Endocrinology 93: 152–155.
Macleod RM (1976). Regulation of prolactin secretion. Front Neuroendocrinol 4: 169–194.
Maes M, D'Hondt P, Suy E, Minner B, Vandervorst C, Raus J (1991). HPA-axis hormones and prolactin responses to dextro-fenfluramine in depressed patients and healthy controls. Progr Neuro-Psychopharmacol Biol Psychiatry 15: 781–790.
Malarkey WB, Hall JC, Pearl DK, Kiecolt-Glaser JK, Glaser R (1991). The influence of academic stress and season on 24-h concentrations of growth hormone and prolactin. J Clin Endocrinol Metab 73: 1089–1092.
Mann JJ, McBride PA, Malone KM, DeMeo M, Keilp J (1995). Blunted serotonergic responsivity in depressed inpatients. Neuropsychopharmacology 13: 53–64.
Martinez de la Escalera G, Weiner RI (1992). Dissociation of dopamine from its receptor as a signal in the pleiotropic hypothalamic regulation of prolactin secretion. Endocrine Rev 13: 241–255.
McCann SM, Lumpkin MD, Mizunuma H, Khorram O, Samson WK (1984). Recent studies on the role of brain peptides in control of anterior pituitary hormone secretion. Peptides 5: 3–7.
McNatty KP (1979). Relationship between plasma prolactin and the endocrine microenvironment of the developing human antral follicle. Fertility Sterility 32: 433–438.
Meller E, Puza T, Miller JC, Friedhoff AJ, Schweitzer JW (1991). Receptor reserve for D2 dopaminergic inhibition of prolactin release in vivo and in vitro. J Pharmacol Exp Ther 257: 668–675.
Melmed S (2001). Disorders of the anterior pituitary and hypothalamus. In Braunwald E (ed), Harrison's Principles of Internal Medicine, 15th edn, Vol. 2. McGraw-Hill: New York. pp 2029–2052.
Meltzer HY, Goode DJ, Schyve PM, Young M, Fang VS (1979). Effect of clozapine on human serum prolactin levels. Am J Psychiatry 136: 1550–1555.
Merchenthaler I, Vigh S, Petrusz P, Schally AV (1982). Immunocytochemical localization of corticotropin-releasing factor (CRF) in the rat brain. Am J Anat 165: 385–396.
Mezey E, Kiss JZ (1985). Vasoactive intestinal peptide-containing neurons in the paraventricular nucleus may participate in regulating prolactin secretion. Proc Natl Acad Sci USA 82: 245–247.
Mirkes SJ, Bethea CL (2001). Oestrogen, progesterone and serotonin converge on GABAergic neurones in the monkey hypothalamus. J Neuroendocrinol 13: 182–192.
Mitchell P, Smythe G (1990). Hormonal responses to fenfluramine in depressed and control subjects. J Affect Disord 19: 43–51.
Mitchner NA, Garlick C, Steinmetz RW, Ben-Jonathan N (1999). Differential regulation and action of estrogen receptors alpha and beta in GH3 cells. Endocrinology 140: 2651–2658.
Mogg RJ, Samson WK (1990). Interactions of dopaminergic and peptidergic factors in the control of prolactin release. Endocrinology 126: 728–735.
Moorman PG, Grubber JM, Millikan RC, Newman B (2003). Antidepressant medications and their association with invasive breast cancer and carcinoma in situ of the breast. Epidemiology 14: 307–314.
Morel G, Besson J, Rosselin G, Dubois PM (1982). Ultrastructural evidence for endogenous vasoactive intestinal peptide-like immunoreactivity in the pituitary gland. Neuroendocrinology 34: 85–89.
Morrison J, Remick RA, Leung M, Wrixon KJ, Bebb RA (2001). Galactorrhea induced by paroxetine. Can J Psychiatry—Rev Can Psychiatrie 46: 88–89.
Muccioli G, Ghe C, Di Carlo R (1991). Distribution and characterization of prolactin binding sites in the male and female rat brain: effects of hypophysectomy and ovariectomy. Neuroendocrinology 53: 47–53.
Neill JD (1970). Effect of ‘stress’ on serum prolactin and luteinizing hormone levels during the estrous cycle of the rat. Endocrinology 87: 1192–1197.
New AS, Siever LJ (2002). Neurobiology and genetics of borderline personality disorder. Psychiatric Ann 32: 329–336.
Newman ME, Shapira B, Lerer B (1998). Evaluation of central serotonergic function in affective and related disorders by the fenfluramine challenge test: a critical review. Int J Neuropsychopharmacol 1: 49–69.
Niall HD, Hogan ML, Sauer R, Rosenblum IY, Greenwood FC (1971). Sequences of pituitary and placental lactogenic and growth hormones: evolution from a primordial peptide by gene reduplication. Proc Natl Acad Sci USA 68: 866–870.
Noel MB, Woodside B (1993). Effects of systemic and central prolactin injections on food intake, weight gain, and estrous cyclicity in female rats. Physiol Behav 54: 151–154.
O'Keane V, Dinan TG (1991). Prolactin and cortisol responses to d-fenfluramine in major depression: evidence for diminished responsivity of central serotonergic function. [comment]. Am J Psychiatry 148: 1009–1015.
O'Keane V, McLoughlin D, Dinan TG (1992). D-fenfluramine-induced prolactin and cortisol release in major depression: response to treatment. J Affect Disord 26: 143–150.
Ondo JG, Dom R (1986). The arcuate nucleus: a site for gamma-aminobutyric acid regulation of prolactin secretion. Brain Res 381: 43–48.
Otero MG, Ripoll AR, Pares GG, Basagana G (2002). Galactorrea asociada a paroxetina. Med Clin 118: 758–759.
Pablos EG, Martin LM, Fernandez MH, Andres RMS (2001). Un caso de galactorrea tras tratamiento con citalopram. Actas Esp Psiquiatr 29: 414.
Pare CM, Yeung DP, Price K, Stacey RS (1969). 5-hydroxytryptamine, noradrenaline, and dopamine in brainstem, hypothalamus, and caudate nucleus of controls and of patients committing suicide by coal-gas poisoning. Lancet 2: 133–135.
Parry BL, Hauger R, LeVeau B, Mostofi N, Cover H, Clopton P et al. (1996). Circadian rhythms of prolactin and thyroid-stimulating hormone during the menstrual cycle and early versus late sleep deprivation in premenstrual dysphoric disorder. Psychiatry Res 62: 147–160.
Perez FM, Rose JC, Schwartz J (1995). Anterior pituitary cells: getting to know their neighbors. Mol Cell Endocrinol 111: C1–C6.
Pernasetti F, Caccavelli L, Van de Weerdt C, Martial JA, Muller M (1997). Thyroid hormone inhibits the human prolactin gene promoter by interfering with activating protein-1 and estrogen stimulations. Mol Endocrinol 11: 986–996.
Peterson MC (2001). Reversible galactorrhea and prolactin elevation related to fluoxetine use. Mayo Clin Proc 76: 215–216.
Petit A, Piednoir D, Germain ML, Trenque T (2003). Hyperprolactinemies d'origine medicamenteuse: etude cas/non-cas dans la banque nationale de pharmacovigilance. Therapie 58: 159–163.
Petraglia F, Vale W, Rivier C (1987). Beta-endorphin and dynorphin participate in the stress-induced release of prolactin in the rat. Neuroendocrinology 45: 338–342.
Pi XJ, Grattan DR (1998). Distribution of prolactin receptor immunoreactivity in the brain of estrogen-treated, ovariectomized rats. J Compar Neurol 394: 462–474.
Pinder RM, Brogden RN, Sawyer PR, Speight TM, Avery GS (1975). Fenfluramine: a review of its pharmacological properties and therapeutic efficacy in obesity. Drugs 10: 241–323.
Placidi GP, Oquendo MA, Malone KM, Huang YY, Ellis SP, Mann JJ (2001). Aggressivity, suicide attempts, and depression: relationship to cerebrospinal fluid monoamine metabolite levels. [comment]. Biol Psychiatry 50: 783–791.
Porter JP, Brody MJ (1985). Neural projections from paraventricular nucleus that subserve vasomotor functions. Am J Physiol 248: R271–R281.
Quattrone A, Tedeschi G, Aguglia U, Scopacasa F, Direnzo GF, Annunziato L (1983). Prolactin secretion in man: a useful tool to evaluate the activity of drugs on central 5-hydroxytryptaminergic neurons. Studies with fenfluramine. Br J Clin Pharmacol 16: 471–475.
Quigley ME, Judd SJ, Gilliland GB, Yen SS (1979). Effects of a dopamine antagonist on the release of gonadotropin and prolactin in normal women and women with hyperprolactinemic anovulation. J Clin Endocrinol Metab 48: 718–720.
Reichlin S (1988). Neuroendocrine significance of vasoactive intestinal polypeptide. Ann NY Acad Sci 527: 431–449.
Reymond MJ, Kaur C, Porter JC (1983). An inhibitory role for morphine on the release of dopamine into hypophysial portal blood and on the synthesis of dopamine in tuberoinfundibular neurons. Brain Res 262: 253–258.
Rittenhouse PA, Levy AD, Li Q, Bethea CL, Van de Kar LD (1993). Neurons in the hypothalamic paraventricular nucleus mediate the serotonergic stimulation of prolactin secretion via 5-HT1c/2 receptors. Endocrinology 133: 661–667.
Rossier J, French E, Rivier C, Shibasaki T, Guillemin R, Bloom FE (1980). Stress-induced release of prolactin: blockade by dexamethasone and naloxone may indicate beta-endorphin mediation. Proc Natl Acad Sci USA 77: 666–669.
Sagrillo CA, Voogt JL (1991). Endogenous opioids mediate the nocturnal prolactin surge in the pregnant rat. Endocrinology 129: 925–930.
Samson WK, Bianchi R, Mogg RJ, Rivier J, Vale W, Melin P (1989). Oxytocin mediates the hypothalamic action of vasoactive intestinal peptide to stimulate prolactin secretion. Endocrinology 124: 812–819.
Samson WK, Lumpkin MD, McCann SM (1986). Evidence for a physiological role for oxytocin in the control of prolactin secretion. Endocrinology 119: 554–560.
Saper CB (1990). Hypothalamus. In Paxinos G (ed), The Human Nervous System. Academic Press, Inc: San Diego, CA. pp 389–413.
Sassin JF, Frantz AG, Weitzman ED, Kapen S (1972). Human prolactin: 24-h pattern with increased release during sleep. Science 177: 1205–1207.
Sauder SE, Frager M, Case GD, Kelch RP, Marshall JC (1984). Abnormal patterns of pulsatile luteinizing hormone secretion in women with hyperprolactinemia and amenorrhea: responses to bromocriptine. J Clin Endocrinol Metab 59: 941–948.
Sawchenko PE, Gold RM, Leibowitz SF (1981). Evidence for vagal involvement in the eating elicited by adrenergic stimulation of the paraventricular nucleus. Brain Res 225: 249–269.
Schaufele F (1999). Regulation of estrogen receptor activation of the prolactin enhancer/promoter by antagonistic activation function-2-interacting proteins. Mol Endocrinol 13: 935–945.
Schlechte J, Dolan K, Sherman B, Chapler F, Luciano A (1989). The natural history of untreated hyperprolactinemia: a prospective analysis. J Clin Endocrinol Metab 68: 412–418.
Schlechte J, el-Khoury G, Kathol M, Walkner L (1987). Forearm and vertebral bone mineral in treated and untreated hyperprolactinemic amenorrhea. J Clin Endocrinol Metab 64: 1021–1026.
Schlechte J, Walkner L, Kathol M (1992). A longitudinal analysis of premenopausal bone loss in healthy women and women with hyperprolactinemia. [comment]. J Clin Endocrinol Metab 75: 698–703.
Schlechte JA, Sherman B, Martin R (1983). Bone density in amenorrheic women with and without hyperprolactinemia. J Clin Endocrinol Metab 56: 1120–1123.
Schofield SPM, Everitt BJ (1981). The organization of catecholamine-containing neurons in the brain of the rhesus monkey (Macaca mulatta). J Anat 132: 391–418.
Segal S, Yaffe H, Laufer N, Ben-David M (1979). Male hyperprolactinemia:effects on fertility. Fertility Sterility 32: 556–561.
Segerson TP, Lam KS, Cacicedo L, Minamitani N, Fink JS, Lechan RM et al. (1989). Thyroid hormone regulates vasoactive intestinal peptide (VIP) mRNA levels in the rat anterior pituitary gland. Endocrinology 125: 2221–2223.
Seggie JA, Brown GM (1975). Stress response patterns of plasma corticosterone, prolactin, and growth hormone in the rat, following handling or exposure to novel environment. Can J Physiol Pharmacol 53: 629–637.
Selmanoff M, Gregerson KA (1986). Suckling-induced prolactin release is suppressed by naloxone and simulated by beta-endorphin. Neuroendocrinology 42: 255–259.
Seppala M (1978). Prolactin and female reproduction. Ann Clin Res 10: 164–170.
Shen GK, Montgomery DW, Ulrich ED, Mahoney KR, Zukoski CF (1992). Up-regulation of prolactin gene expression and feedback modulation of lymphocyte proliferation during acute allograft rejection. Surgery 112: 387–393; discussion 393–4.
Sheng HZ, Westphal H (1999). Early steps in pituitary organogenesis. Trends Genet 15: 236–240.
Shimatsu A, Kato Y, Matsushita N, Katakami H, Yanaihara N, Imura H (1982). Stimulation by serotonin of vasoactive intestinal polypeptide release into rat hypophysial portal blood. Endocrinology 111: 338–340.
Shimatsu A, Kato Y, Ohta H, Tojo K, Kabayamma Y, Inoue T et al. (1984). Involvement of hypothalamic vasoactive intestinal polypeptide (VIP) in prolactin secretion induced by serotonin in rats. Proc Soc Exp Biol Med 175: 414–416.
Shioiri T, Kita N, Takahashi S (1996). Two cases of alprazolam-induced hyperprolactinemia in patients with panic disorder. Int Clin Psychopharmacol 11: 149–152.
Shiu RP, Elsholtz HP, Tanaka T, Friesen HG, Gout PW, Beer CT et al. (1983). Receptor-mediated mitogenic action of prolactin in a rat lymphoma cell line. Endocrinology 113: 159–165.
Shopsin B, Friedman E, Gershon S (1976). Parachlorophenylalanine reversal of tranylcypromine effects in depressed patients. Arch Gen Psychiatry 33: 811–819.
Shughrue PJ, Dellovade TL, Merchenthaler I (2002). Estrogen modulates oxytocin gene expression in regions of the rat supraoptic and paraventricular nuclei that contain estrogen receptor-beta. Progr Brain Res 139: 15–29.
Siever LJ, Murphy DL, Slater S, de la Vega E, Lipper S (1984). Plasma prolactin changes following fenfluramine in depressed patients compared to controls: an evaluation of central serotonergic responsivity in depression. Life Sci 34: 1029–1039.
Slater S, de La Vega CE, Skyler J, Murphy DL (1976). Plasma prolactin stimulation by fenfluramine and amphetamine. Psychopharmacol Bull 12: 26–27.
Smith S (2003). Effects of antipsychotics on sexual and endocrine function in women: implications for clinical practice. J Clin Psychopharmacol 23: S27–S32.
Soaje M, Deis RP (1994). A modulatory role of endogenous opioids on prolactin secretion at the end of pregnancy in the rat. J Endocrinol 140: 97–102.
Sofroniew MV, Weindl A, Schrell U, Wetzstein R (1981). Immunohistochemistry of vasopressin, oxytocin and neurophysin in the hypothalamus and extrahypothalamic regions of the human and primate brain. Acta Histochemica–Suppl 24: 79–95.
Spigset O, Mjorndal T (1997). The effect of fluvoxamine on serum prolactin and serum sodium concentrations: relation to platelet 5-HT2A receptor status. J Clin Psychopharmacol 17: 292–297.
Stopa EG, LeBlanc VK, Hill DH, Anthony EL (1993). A general overview of the anatomy of the neurohypophysis. Ann NY Acad Sci 689: 6–15.
Strand FL (1999). Neuropeptides: Regulators of Physiological Processes, Vol, 1. MIT Press: Cambridge, MA.
Subramanian MG, Kowalczyk CL, Leach RE, Lawson DM, Blacker CM, Ginsburg KA et al. (1997). Midcycle increase of prolactin seen in normal women is absent in subjects with unexplained infertility. Fertility Sterility 67: 644–647.
Swanson LW, Sawchenko PE (1983). Hypothalamic integration: organization of the paraventricular and supraoptic nuclei. Annu Rev Neurosci 6: 269–324.
Tatsumi M, Groshan K, Blakely RD, Richelson E (1997). Pharmacological profile of antidepressants and related compounds at human monoamine transporters. Eur J Pharmacol 340: 249–258.
Torner L, Toschi N, Pohlinger A, Landgraf R, Neumann ID (2001). Anxiolytic and anti-stress effects of brain prolactin: improved efficacy of antisense targeting of the prolactin receptor by molecular modeling. J Neurosci 21: 3207–3214.
Urban RJ, Veldhuis JD (1991). A selective serotonin reuptake inhibitor, fluoxetine hydrochloride, modulates the pulsatile release of prolactin in postmenopausal women. Am J Obstet Gynecol 164: 147–152.
Vaha-Eskeli K, Erkkola R, Irjala K, Viinamaki O (1991). Effect of thermal stress on serum prolactin, cortisol and plasma arginine vasopressin concentration in the pregnant and non-pregnant state. Eur J Obstet Gynecol Reprod Biol 42: 1–8.
Van de Kar LD, Javed A, Zhang Y, Serres F, Raap DK, Gray TS (2001). 5-HT2A receptors stimulate ACTH, corticosterone, oxytocin, renin, and prolactin release and activate hypothalamic CRF and oxytocin-expressing cells. J Neurosci 21: 3572–3579.
Van de Kar LD, Lorens SA, Urban JH, Bethea CL (1989). Effect of selective serotonin (5-HT) agonists and 5-HT2 antagonist on prolactin secretion. Neuropharmacology 28: 299–305.
Van de Kar LD, Rittenhouse PA, Li Q, Levy AD, Brownfield MS (1995). Hypothalamic paraventricular, but not supraoptic neurons, mediate the serotonergic stimulation of oxytocin secretion. Brain Res Bull 36: 45–50.
Van Praag HM (1977). New evidence of serotonin-deficient depressions. Neuropsychobiology 3: 56–63.
Vijayan E, McCann SM (1978). The effects of intraventricular injection of gamma-aminobutyric acid (GABA) on prolactin and gonadotropin release in conscious female rats. Brain Res 155: 35–43.
Viselli SM, Stanek EM, Mukherjee P, Hymer WC, Mastro AM (1991). Prolactin-induced mitogenesis of lymphocytes from ovariectomized rats. Endocrinology 129: 983–990.
Wagner EJ, Goudreau JL, Moore KE, Lookingland KJ (1994). GABAergic regulation of tuberoinfundibular dopaminergic neurons in the male rat. Brain Res 659: 194–200.
Waldstreicher J, Duffy JF, Brown EN, Rogacz S, Allan JS, Czeisler CA (1996). Gender differences in the temporal organization of proclactin (PRL) secretion: evidence for a sleep-independent circadian rhythm of circulating PRL levels—a clinical research center study. J Clin Endocrinol Metab 81: 1483–1487.
Walsh RJ, Slaby FJ, Posner BI (1987). A receptor-mediated mechanism for the transport of prolactin from blood to cerebrospinal fluid. Endocrinology 120: 1846–1850.
Wang DY, De Stavola BL, Bulbrook RD, Allen DS, Kwa HG, Fentiman IS et al. (1992). Relationship of blood prolactin levels and the risk of subsequent breast cancer. Int J Epidemiol 21: 214–221.
Wang DY, Stepniewska KA, Allen DS, Fentiman IS, Bulbrook RD, Kwa HG et al. (1995). Serum prolactin levels and their relationship to survival in women with operable breast cancer. J Clin Epidemiol 48: 959–968.
Wang PS, Walker AM, Tsuang MT, Orav EJ, Glynn RJ, Levin R et al. (2002). Dopamine antagonists and the development of breast cancer. Arch Gen Psychiatry 59: 1147–1154.
Wanke IE, Rorstad OP (1990). Receptors for vasoactive intestinal peptide in rat anterior pituitary glands: localization of binding to lactotropes. Endocrinology 126: 1981–1988.
Wardlaw SL, Bilezikian JP (1992). Hyperprolactinemia and osteopenia. [comment]. J Clin Endocrinol Metab 75: 690–691.
Weizman A, Mark M, Gil-Ad I, Tyano S, Laron Z (1988). Plasma cortisol, prolactin, growth hormone, and immunoreactive beta-endorphin response to fenfluramine challenge in depressed patients. Clin Neuropharmacol 11: 250–256.
Welsch CW, Nagasawa H (1977). Prolactin and murine mammary tumorigenesis: a review. Cancer Res 37: 951–963.
Wieck A, Haddad PM (2003). Antipsychotic-induced hyperprolactinaemia in women: pathophysiology, severity and consequences. Selective literature review. Br J Psychiatry 182: 199–204.
Winslow JT, Insel TR (1991). Social status in pairs of male squirrel monkeys determines the behavioral response to central oxytocin administration. J Neurosci 11: 2032–2038.
Witcher JA, Freeman ME (1985). The proestrous surge of prolactin enhances sexual receptivity in the rat. Biol Reprod 32: 834–839.
Yatham LN, Steiner M (1993). Neuroendocrine probes of serotonergic function: a critical review. Life Sci 53: 447–463.
Yazigi RA, Quintero CH, Salameh WA (1997). Prolactin disorders. Fertility Sterility 67: 215–225.
Zemishlany Z, McQueeney R, Gabriel SM, Davidson M (1990). Neuroendocrine and monoaminergic responses to acute administration of alprazolam in normal subjects. Neuropsychobiology 23: 124–128.
Acknowledgements
We wish to thank Dr Barbara Gracious and Dr Lawrence Guttmacher for their critical reading and comments.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Emiliano, A., Fudge, J. From Galactorrhea to Osteopenia: Rethinking Serotonin–Prolactin Interactions. Neuropsychopharmacol 29, 833–846 (2004). https://doi.org/10.1038/sj.npp.1300412
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300412
Keywords
This article is cited by
-
Omeprazole-induced galactorrhea in kidney transplant patients—a case report
Journal of Medical Case Reports (2022)
-
(±)-MDMA and its enantiomers: potential therapeutic advantages of R(−)-MDMA
Psychopharmacology (2018)
-
The Impact of Psychotropic Medications on Bone Health in Youth
Current Psychiatry Reports (2018)
-
Antidepressant use and circulating prolactin levels
Cancer Causes & Control (2016)
-
The Effects of Novel and Newly Approved Antipsychotics on Serum Prolactin Levels: A Comprehensive Review
CNS Drugs (2014)
