
Abstract
Acute oral administration of selective serotonin re-uptake inhibitors (SSRIs) increases plasma cortisol by facilitating brain serotonin activity. Recently, salivary cortisol sampling has grown in popularity as a noninvasive means of assessing HPA axis activity. The aim of the present study was to find out whether acute oral administration of the SSRI, citalopram, increases salivary cortisol in healthy volunteers and whether the increase produced by an equivalent dose of its active isomer, escitalopram, is greater. A total of 15 healthy subjects were tested on three occasions receiving either oral citalopram (20 mg), escitalopram (10 mg), or placebo in a double-blind, randomized, crossover design. Salivary cortisol and plasma cortisol and prolactin were measured for 240 min after each treatment. Relative to placebo, both citalopram and escitalopram increased salivary and plasma cortisol levels with no evidence of consistent differences between them. Plasma prolactin concentration was not altered by either active treatment. Plasma and salivary cortisol responses after citalopram but not escitalopram correlated significantly. The present study does not support an enhanced effect of escitalopram on 5-HT-mediated neuroendocrine responses.
Similar content being viewed by others


INTRODUCTION
Drugs that potentiate serotonin (5-HT) function can produce increases in the secretion of cortisol and prolactin (Raap and Van de Kar, 1999). For example, intravenous administration of selective serotonin re-uptake inhibitors (SSRIs) produces a reliable increase in plasma prolactin as well as in plasma and salivary cortisol (Laakmann et al, 1990; Seifritz et al, 1996). The effects of acute oral administration of clinical doses of SSRIs on these neuroendocrine responses are less striking (Meltzer and Nash, 1988), but increases in plasma cortisol with citalopram (20 mg orally) and paroxetine (20–40 mg orally) have been described (Hennig and Netter, 2002; Reist et al, 1996; Kojima et al, 2003; Carpenter et al, 2003). However, effects of oral SSRIs on salivary cortisol have not yet been reported.
The active isomer of citalopram, escitalopram, has recently been marketed for the treatment of depression. Animal experimental studies suggest that the effects of escitalopram on 5-HT release and on certain 5-HT-mediated responses are greater than equivalent doses of citalopram (Sanchez et al, 2003; Mork et al, 2003). This might be associated with greater clinical efficacy and an earlier onset of action in depressed patients (Gorman et al, 2002). The aim of the present study was to assess the effect of oral citalopram (20 mg) and escitalopram (10 mg) on plasma prolactin and salivary cortisol in healthy volunteers. We hypothesized that both drugs would increase salivary cortisol, but that, from the animal experimental data outlined above, the effect of escitalopram would be greater and that, correlating with its suggested earlier onset of action, acute escitalopram but not citalopram would increase plasma prolactin (see Cowen and Sargent, 1997).
SUBJECTS AND METHODS
Subjects
In total, 15 healthy volunteers (11 female, four male) mean age 43.6 years (range 24–65 years) gave informed consent to the study which was approved by the local ethics committee. On the basis of the structured clinical interview for DSM-IV the subjects were determined to be free of current Axis 1 disorder. They had no current significant physical illness and had been free of medication for at least 3 months. Female subjects were tested in the early follicular phase of their menstrual cycle.
Neuroendocrine Testing
Subjects attended the laboratory at midday, having fasted from breakfast, and an indwelling venous cannula was inserted. This time of day was used to avoid the usual sharp decline in cortisol levels over the early morning (Carpenter et al, 2003). Subjects were tested reclining and were not allowed to sleep. After a 30-min rest period subjects received (a) citalopram (20 mg orally), (b) escitalopram (10 mg orally), or (c) placebo, in a double-blind, random order, crossover design. Blood samples for prolactin, cortisol, and drug levels were carried out at 30-min intervals for the next 240 min. Saliva samples for cortisol were taken at the same time points and were collected with a Salivette® (Sarstedt, Numbrecht). The mean interval between the three tests was 15.4±2.0 days. Citalopram and escitalopram were manufactured by Lundbeck and obtained from the hospital pharmacy.
Biochemical Measurements
Plasma was separated from heparinized blood samples by centrifugation and stored at −30°C. Plasma cortisol and salivary cortisol concentrations were determined by ‘in-house’ double antibody radioimmunoassay. Antibodies were obtained from Bioclin (Cardiff) and IDS (Tyne and Wear) and iodine-labelled cortisol from Amersham (Bucks). The intra- and interassay coefficients of variation over the range encompassed by the standard curve were 4.3 and 5.8% for plasma cortisol and 3 and 10% for salivary cortisol, respectively. The lower limit of detection of cortisol in saliva was 0.5 nmol/l. Plasma prolactin was determined by an immunoradiometric assay kit (Coat-a count, DPC, Lanberis, Wales). Intra- and interassay coefficients of variation <3 and <6%, respectively. Citalopram was assayed by high-performance liquid chromatography with a UV detector set to a wavelength of 240 nm as previously described (Clement et al, 2001). Samples were extracted into 2% ammonia solution in methanol with propanolol (150 ng) being used as an internal standard. The mobile phase consisted of 0.05 M ammonium phosphate buffer (pH 3.5) in acetonitrile (50:41, v/v). Peak height ratios of citalopram vs propranalol were measured by the Borwin system.
Statistics
Salivary cortisol data were log-transformed to approximate more satisfactorily to a normal distribution (Kolmogorov–Smirnov test). Salivary cortisol and plasma cortisol and prolactin were plotted against time and analyzed with a two-way repeated measures analysis of variance (ANOVA) with ‘drug’ (citalopram vs escitalopram vs placebo) and ‘time’ (sampling time) as the main within-subjects factors. A Huynh–Feldt correction was used where the assumption of sphericity was violated. For clarity, uncorrected degrees of freedom have been reported. Paired t-tests (two-tailed) were used as post hoc tests. Endocrine responses and drug levels were also measured as area under the curve (AUC) by the trapezoid method with subtraction of baseline secretion (extrapolated from time ‘0’). Correlations were carried out using Pearson's product moment. All values are given as mean±standard error of the mean (SEM) unless otherwise specified. All statistical analyses were performed using the Statistical Package for Social Sciences (SPSS) version 11.5.
RESULTS
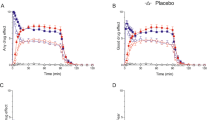
The treatments were well tolerated and no subject experienced significant nausea. Exploratory analyses of the endocrine responses showed no main or interactive effects of gender (all p-values >0.1), which was therefore omitted as a factor in subsequent analyses. The ANOVA of plasma prolactin showed a main effect of time (F=10.16; df=8,122; p<0.001), but no main effect of drug (F=0.30; df=2,28; p=0.75) or drug by time interaction (F=1.10; df=16,224; p=0.41). On all test occasions, plasma prolactin rose modestly from baseline over the time course of the sampling period (Figure 1).

Mean±SEM plasma prolactin in 15 healthy volunteers who were randomly allocated to receive oral doses of either citalopram 20 mg (▪), escitalopram 10 mg (▴), or placebo (•) in a three-way crossover design. There is no difference between the three treatments (F=1.10; p=0.41, ANOVA).
In contrast to their lack of effect on prolactin, both citalopram and escitalopram increased salivary cortisol (Figure 2). The ANOVA of salivary cortisol showed a main effect of drug (F=5.31; df=2,28; p=0.016) of time (F=7.10; df=8,112; p<0.001) and a significant drug by time interaction (F=2.17; df=16,224; p=0.038). Post hoc testing of cortisol levels showed a number of significant differences at various times between placebo and citalopram and escitalopram. However, there were no differences between citalopram and escitalopram at any time point (Figure 1). The mean AUC of salivary cortisol response to citalopram was significantly greater than that to placebo (−9.0±3.6 vs −15.2±3.9 nmol h/l, p=0.022). The mean AUC salivary cortisol response to escitalopram (−8.9±6.4 nmol h/l) was very similar to that of citalopram but did not differ significantly from either the response to citalopram (p=0.67) or placebo (p=0.51). Post hoc analyses of the AUC responses with the nonparametric Wilcoxon test gave essentially similar results.

Mean±SEM salivary cortisol in 15 healthy volunteers who were randomly allocated to receive oral doses of either citalopram 20 mg (▪), escitalopram 10 mg (▴), or placebo (•) in a three-way crossover design. Results are shown as antilog of log transformed data. Citalopram significantly greater than placebo, **p<0.01, *p<0.05; escitalopram significantly greater than placebo, ††p<0.01, †P<0.05.
Citalopram and escitalopram also increased plasma cortisol (Figure 3). The ANOVA showed a main effect of drug (F=17.03; df=2,28; p<0.001) and time (F=4.26; df=8,112; p=0.011). However, there was no significant drug by time interaction (F=1.51; df=16,224; p=0.15). Despite the lack of a significant interaction term we carried out post hoc paired t-tests to test for any possible differences in response between two active treatments. As with the salivary cortisol measures, however, post hoc t-tests revealed no difference in plasma cortisol between citalopram and escitalopram at any time point. The mean AUC of plasma cortisol response to escitalopram was significantly greater than that to placebo (−6.1±3.0 vs −17.9±4.3 μg h/100 ml, p=0.023). The mean AUC plasma cortisol response to citalopram (−10.3±5.2 μg h/100 ml) was somewhat less than that seen with escitalopram but did not differ significantly from either the response to escitalopram (p=0.36) or placebo (p=0.13). Post hoc analyses of the AUC responses with the nonparametric Wilcoxon test gave similar results.

Mean±SEM plasma cortisol in 15 healthy volunteers who were randomly allocated to receive oral doses of either citalopram 20 mg (▪), escitalopram 10 mg (▴), or placebo (•) in a three-way crossover design. Citalopram significantly greater than placebo, **p<0.01, *p<0.05; escitalopram significantly greater than placebo, ††p<0.01, †p<0.05.
Plasma levels of both citalopram and escitalopram started to rise within about 120 min of oral administration and were still rising to a small extent at the end of the sampling period (Figure 4). No citalopram was detected in any baseline sample. As expected, plasma levels of citalopram were about double those of escitalopram. There were no significant correlations between the plasma AUC of citalopram and escitalopram and the AUC of salivary and plasma cortisol responses (all p-values >0.05). A significant correlation was seen between plasma and salivary cortisol responses to citalopram (r=0.64; p=0.01). However, this was not the case with the salivary and plasma cortisol responses to escitalopram (r=0.21; p=0.45).

Mean±SEM of plasma citalopram concentration in 15 healthy volunteers who were randomly allocated to receive oral doses of either citalopram 20 mg, escitalopram 10 mg, or placebo in a three-way crossover design. Only data from citalopram 20 mg (▴) and escitalopram 10 mg (▪) are shown.
DISCUSSION
The present data confirm that acute oral administration of citalopram increases HPA axis activity (Raap and Van de Kar, 1999; Hennig and Netter, 2002). The acute facilitatory effect of SSRIs on the HPA axis is thought to be mediated by increased release of corticotrophin-releasing hormone (CRH) in the paraventricular nucleus of the hypothalamus, which leads to increased corticotropin (ACTH) release at pituitary level (Raap and Van de Kar, 1999). This, in turn, results in greater secretion of cortisol from the adrenal gland into the plasma, which can be detected by salivary monitoring. Potentially, this might provide a simple and noninvasive means of assessing 5-HT neuroendocrine responses in both volunteers and patient groups.
Of currently available SSRIs, citalopram may be the most suitable for this purpose because it takes a relatively short time (2–4 h) to reach peak plasma levels after oral administration (Gutierrez and Abramowitz, 2000). It should be noted, however, that our sampling period, which ended 4 h after treatment ingestion, may not have quite captured the peak of level of citalopram and escitalopram in plasma and the same may obviously be true of the cortisol responses. In the report by Hennig and Netter (2002), peak plasma cortisol responses to citalopram were apparent about 3 h after oral citalopram intake.
Despite the increase in cortisol secretion after citalopram, there was no effect of either citalopram or escitalopram on plasma prolactin levels. After intravenous administration of citalopram, however, increases in plasma prolactin are seen (Seifritz et al, 1996; Attenburrow et al, 2001). This suggests that it is possible to increase prolactin release after acute citalopram treatment but that this effect requires higher plasma levels of citalopram than are produced by oral administration. A similar phenomenon is seen with the serotonergic antidepressant, clomipramine, where intravenous administration produces reliable increases in plasma prolactin whereas oral administration does not (Laakmann et al, 1977, 1984).
A possible explanation of this difference is that prolactin release may be more buffered by inhibitory serotonin autoreceptor activity (Raap and Van de Kar, 1999). For example, increases in plasma prolactin levels are seen after 3 weeks of oral paroxetine administration but not after 1 week, despite the presence of similar plasma levels of paroxetine on both occasions (Cowen and Sargent, 1997). This might be consistent with gradual autoreceptor desensitization, which leads to increased 5-HT-mediated prolactin release over continued treatment. If this is the case one might speculate that the kinetic profile produced by intravenous administration is more likely to override serotonin autoreceptor inhibition than oral dosing.
The ability of the racemic mixture of citalopram to inhibit the re-uptake of 5-HT resides exclusively in the S-enantiomer, escitalopram. Escitalopram has recently been marketed as an antidepressant with some evidence that it might be more clinically efficacious than the parent compound even at equipotent doses (Gorman et al, 2002). In support of this, animal experimental studies have indicated that equivalent doses of escitalopram are more effective than citalopram at increasing extracellular 5-HT as measured by in vivo microdialysis (Mork et al, 2003). This had led to the proposal that the R-enantiomer of citalopram may contain some pharmacological activity that attenuates the ability of the S-enantiomer to increase extracelluar 5-HT (Mork et al, 2003). If this is the case it might be expected that escitalopram would cause a greater increase in the cortisol response than an equivalent dose of citalopram.
Indeed, from the AUC data, the effect of escitalopram on plasma cortisol appeared a little more robust than that of citalopram; however, with the salivary cortisol responses the reverse was the case, suggesting that the effects of escitalopram and citalopram on 5-HT-mediated cortisol release at these doses do not show consistent differences. In support of this, the post hoc t-tests showed no difference in salivary and plasma cortisol levels between citalopram and escitalopram at any time point (Figures 2 and 3). Nevertheless, it must be acknowledged that we studied only single doses of citalopram and escitalopram and also that our sampling time may not have continued for long enough to capture the peak of the cortisol responses. In the former respect it is worth noting that Mork et al (2003) found in their microdialysis study that while of 2 mg/kg escitalopram produced a greater increase in extracellular 5-HT than 4 mg/kg of citalopram, the effects of 1 mg/kg escitalopram and 2 mg/kg of citalopram were not different from each other. It is possible, therefore, that administration of higher doses of citalopram and escitalopram might reveal a greater effect of escitalopram on cortisol release.
The method we employed to measure citalopram does not distinguish the S- and R-enantiomers. As expected, therefore, plasma levels of citalopram were about twice as high after citalopram 20 mg than escitalopram 10 mg. In both experimental studies and clinical trials it is generally reckoned that the equivalent dose of escitalopram is about half that of citalopram (Mork et al, 2003; Gorman et al, 2002). However, pharmacokinetic studies in humans suggest that after citalopram administration, the ratio of S-citalopram to R-citalopram in plasma is about 0.5–0.7 (Sidhu et al, 1997; Mork et al, 2003). Thus, the dosing regime in the current study might have led to relatively more active S-enantiomer being present in plasma after escitalopram 10 mg than after citalopram 20 mg. However, this, of course, cannot account for the failure of escitalopram to increase cortisol more than citalopram because such an effect would favor escitalopram over the racemic mixture of citalopram.
As noted above, the AUC cortisol data were rather contradictory relative to the ANOVA findings and showed a significant effect of escitalopram but not citalopram to increase plasma cortisol while the reverse was the case with the salivary cortisol data. We believe that these inconsistencies are due to the greater inherent variability and lower power of AUC measures relative to ANOVA. Indeed, in numerical terms, the AUC salivary cortisol response to escitalopram was almost the same as that of citalopram but because of greater variance was not statistically significantly different from placebo. For this reason, as noted above, we think it better to put more weight on the fact that post hoc t-tests failed to show differences between cortisol responses to citalopram and escitalopram. The observed power of the ANOVA of salivary cortisol to detect a drug by time difference between active treatment and placebo was 0.82 on the SPSS analysis. However, clearly, the power to detect smaller differences between citalopram and escitalopram would have been substantially less.
Another important methodological issue is how closely the salivary cortisol responses to citalopram and escitalopram mirror the cortisol responses in plasma; that is, how far one measure can be taken as a surrogate for the other. In the case of intravenous citalopram the relationship between these two measures is very strong (r=0.91) (Bhagwagar et al, 2002). With oral citalopram a significant but lesser correlation was obtained (r=0.64), but with escitalopram the correlation (r=0.21) was not significant. We do not have an obvious explanation for these differences. Presumably, with intravenous citalopram it is more likely that the totality of the cortisol response will have occurred during the time frame of a conventional neuroendocrine challenge study while this is less likely to be the case with an oral citalopram challenge (see above). However, the current data suggest that after oral SSRI challenge caution should be used when extrapolating results from salivary cortisol measures to plasma, particularly in the case of escitalopram.
Gender is known to influence some models of 5-HT neuroendocrine function (Ghaziuddin et al, 2003), and in the present study we tested both male and female subjects. Although the ANOVA showed no main or interactive effects of gender, the sample size was too small to draw firm conclusions about whether or not endocrine responses to citalopram and escitalopram differ between men and women. A potential influence of the menstrual cycle on SSRI-mediated cortisol release also requires further study (O'Keane et al, 1991).
In conclusion, our study confirms that acute administration of SSRIs stimulates the HPA axis and that this effect can be detected through monitoring of salivary cortisol levels. Within its limitations, the present study does not support the proposal that escitalopram has a more potent effect on 5-HT neurotransmission than equivalent doses of citalopram.
References
Attenburrow MJ, Mitter PR, Whale R, Terao T, Cowen PJ (2001). Low-dose citalopram as a 5-HT neuroendocrine probe. Psychopharmacology 155: 323–326.
Bhagwagar Z, Hafizi S, Cowen PJ (2002). Acute citalopram administration produces correlated increases in plasma and salivary cortisol. Psychopharmacology 163: 118–120.
Carpenter LL, Anderson GM, Siniscalchi JM, Chappell PB, Price LH (2003). Neuropsychopharmacology 28: 339–347.
Clement EM, Odontiadis J, Franklin M (2001). Measurement of citalopram in plasma by HPLC with UV detection. J Psychopharmacol 15(Suppl): A64.
Cowen PJ, Sargent PA (1997). Changes in plasma prolactin during SSRI treatment: evidence for a delayed increase in 5-HT neurotransmission. J Psychopharmacol 11: 345–348.
Ghaziuddin N, Welch K, Greden J (2003). Central serotonergic effects of m-chlorophenylpiperazine (mCPP) among normal control adolescents. Neuropsychopharmacology 28: 133–139.
Gorman JM, Korotzer A, Su G (2002). Efficacy comparison of escitalopram and citalopram in the treatment of major depressive disorder: pooled analysis of placebo-controlled trials. CNS Spectrums 7: 40–44.
Gutierrez MM, Abramowitz W (2000). Pharmacokinetic comparison of oral solution and tablet formulations of citalopram: a single-dose, randomized, crossover study. Clin Ther 22: 1525–1532.
Hennig J, Netter P (2002). Oral application of citalopram (20 mg) and its usefulness for neuroendocrine challenge tests. Int J Neuropsychopharmacol 5: 67–71.
Kojima H, Terao T, Iwakawa M, Soya A, Inoue N, Shiraishi Y et al (2003). Paroxetine as a 5-HT neuroendocrine probe. Psychopharmacology 167: 97–102.
Laakmann G, Gugath M, Kuss HJ, Zygan K (1984). Comparison of growth hormone and prolactin stimulation induced by chlorimipramine and desipramine in man in connection with chlorimipramine metabolism. Psychopharmacology 82: 62–67.
Laakmann G, Hinz A, Voderholzer U, Daffner C, Muller OA, Neuhauser H et al (1990). The influence of psychotropic drugs and releasing hormones on anterior pituitary hormone secretion in healthy subjects and depressed patients. Pharmacopsychiatry 23: 18–26.
Laakmann G, Schumacher G, Benkert O, von Wender K (1977). Stimulation of growth hormone secretion by desipramine and clorimipramine in man. J Clin Endocrinol Metab 44: 1010–1013.
Meltzer HY, Nash JF (1988). Serotonin and mood: neuroendocrine aspects. In: Ganten D, Pfaff D (eds). Current Topics in Neuroendocrinology. Springer-Verlag: New York. pp 183–210.
Mork A, Kreilgaard M, Sanchez C (2003). The R-enantiomer of citalopram counteracts escitalopram-induced increase in extracellular 5-HT in the frontal cortex of freely moving rats. Neuropharmacology 45: 167–173.
O'Keane V, O'Hanlon M, Webb M, Dinan T (1991). D-fenfluramine/prolactin response throughout the menstrual cycle: evidence for an oestrogen induced alteration. Clin Endocrinol 34: 289–292.
Raap DK, Van de Kar LD (1999). Minireview: selective serotonin reuptake inhibitors and neuroendocrine function. Life Sci 65: 1217–1235.
Reist C, Helmeste D, Albers L, Chhay H, Tang SW (1996). Serotonin indices and impulsivity in normal volunteers. Psychiatry Res 60: 177–184.
Sanchez C, Gruca P, Bien E, Papp M (2003). R-citalopram counteracts the effect of escitalopram in a rat conditioned fear stress model of anxiety. Pharmacol Biochem Behav 75: 903–907.
Seifritz E, Baumann P, Muller MJ, Annen O, Amey M, Hemmeter U et al (1996). Neuroendocrine effects of a 20-mg citalopram infusion in healthy males. Neuropsychopharmacology 14: 253–263.
Sidhu J, Priskorn M, Poulsen M, Segonzac A, Groullier G, Larsen F (1997). Steady-state pharmacokinetics of the enantiomers of citalopram and its metabolites in humans. Chirality 9: 686–692.
Acknowledgements
We thank D Laver and A Reed for technical assistance and R Hockney for nursing care. The study was supported by the Medical Research Council. PJC is an MRC Clinical Scientist.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nadeem, H., Attenburrow, MJ. & Cowen, P. Comparison of the Effects of Citalopram and Escitalopram on 5-Ht-Mediated Neuroendocrine Responses. Neuropsychopharmacol 29, 1699–1703 (2004). https://doi.org/10.1038/sj.npp.1300475
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1300475
Keywords
This article is cited by
-
Quantification of Cerebral Blood Flow as Biomarker of Drug Effect: Arterial Spin Labeling phMRI After a Single Dose of Oral Citalopram
Clinical Pharmacology & Therapeutics (2011)
-
Specific effects of escitalopram on neuroendocrine response
Psychopharmacology (2009)
-
Time trial performance in normal and high ambient temperature: is there a role for 5-HT?
European Journal of Applied Physiology (2009)
-
The Effects of Increased Central Serotonergic Activity on Prepulse Inhibition and Habituation of the Human Startle Response
Neuropsychopharmacology (2007)
