
Abstract
Members of the family Marseilleviridae are giant viruses that have the ability to infect amoebas. Such viruses were initially described in 2009. Since then, this family has grown, and diverse members have been found in different environments and geographic locations. Previous phylogenetic analyses suggested the existence of four marseillevirus lineages. A fourth lineage was described with the discovery of the Brazilian marseillevirus (BrMr), isolated from Pampulha Lake, Brazil. Here we describe the isolation and characterization of the Golden marseillevirus (GMar), a new marseillevirus isolated from golden mussels (Limnoperna fortunei) in South of Brazil. This new representative of Marseilleviridae has circular, double-stranded (dsDNA) that contains 360, 610 base pairs and encodes 483 open read frames (ORFs). The complete virus genome was sequenced and phylogenic analyses indicated clear differences between this virus and other marseilleviruses. In addition, this is the only marseillevirus so far that has been isolated from mussels, and this report expands the diversity of environments from which giant viruses could be recovered.
Similar content being viewed by others


Introduction
The discovery of protist-infecting giant viruses started with the isolation of Acanthamoeba polyphaga mimivirus (APMV) in 20031. Following the discovery of mimivirus, a virus named Marseillevirus was isolated in 2007 on biofilm from a cooling tower near Paris2. In 2010, Megavirus chilensis was recovered from a seawater sample from the shores of Chile3. Soon after, in 2013, the largest of the giant viruses so far discovered, Pandoravirus, was recovered from Chilean shores and from a garden pond in Australia4. More recently, Pithovirus sibericum5 and Mollivirus sibericum6 were identified in thirty-thousand-year-old Siberian permafrost. These viruses exhibit astonishing features, with large capsids and genomes comparable in size to small bacteria7. The “cell-like” features in giant viruses have led numerous scientists to suggest fundamental concepts outside the report of these particular viruses. These features include the size of the virions and genomes and the observation that many genes have no evident homologs in other microorganisms and, hence, might come from still unknown sources. The principal theoretical developments proposed that the giant viruses are characterized by a “fourth domain of life” that is different from but comparable to the three cellular domains8. The discovery of these giant viruses has crossed some boundaries between viruses and cellular life9, although ribosomes remain a distinctive attribute of living microorganisms10. According to the Baltimore system, giant viruses are placed among other dsDNA viruses: the nucleocytoplasmic large-DNA viruses (NCLDVs). Although dsDNA viruses rarely appear to have a single evolutionary origin, the NCLDVs all contain five core genes and share 50 likely ancestral genes10. Initially, NCLDVs included four known families of large-DNA viruses infecting eukaryotes: Poxyviridae, Asfaviridae, Iridoviridae, and Phycodnaviridae. The list of NCLDVs increased with the discovery of mimiviruses1 and, more recently, marseilleviruses2,11.
The first member of Marseilleviridae was isolated from the water of a cooling tower in Paris in 20072. Subsequently, another family member, Lausannevirus, was discovered in water samples collected from the river Seine in 201112. In 2013, the Cannes8 virus was discovered in southeastern France in water from a cooling tower13. Tunisvirus and Fontaine Saint-Charles virus were discovered in freshwater from decorative fountains in Tunisia and France, respectively14,15. The Insectomime virus was isolated from the internal organs and digestive tract of a dipteran larva known as the drone fly16. Recently, two other marseilleviruses were recovered: Port-Miou virus, isolated from a sample from a brackish submarine spring17, and the Brazilian marseillevirus, isolated from Pampulha Lake in Minas Gerais18.
As demonstrated in earlier studies, giant viruses may be isolated from diverse environments. Recently, using molecular and virological methods, one study has demonstrated that oysters are excellent sources for isolating mimiviruses, possibly due to their structure, which allows for the bioaccumulation of microorganisms like viruses and, probably, amoebas19. This has brought our attention to golden mussels (Limnoperna fortunei). These mollusks, which are native to the freshwater systems of China, Southeast Asia, are a mytilid invasive species of the Plata Basin, accidentally introduced in 1991 by ballast water20. After its introduction to the region, the geographical distribution of L. fortunei included Guaíba Lake at Rio Grande do Sul (southern Brazil), where it was first reported in 199821.
In this manuscript, we demonstrate that mussels are possible sources for the isolation of marseilleviruses. The isolation of marseillevirus from mussels is important in order to increase the number of strategies for isolation using filter-feeding organisms. Here, we report the isolation, identification, and genomic analysis of the Golden marseillevirus (GMar), a new marseillevirus isolated from golden mussels collected in southern Brazil.
Methods
Study Area and Samples
Forty specimens of golden mussels (L. fortunei) were collected from Guaíba Lake, Rio Grande do Sul, Brazil, in July 2014 (30°01′59″S, 51°13′48″W; Fig. 1). The mussels were collected from a two meters deep location, and they were attached to a metal grid that had been submerged for six months before the date of collection. Due to the high human density and the concentration of industries around the lake, this region receives a large amount of domestic and industrial waste from three main rivers that form the lake’s origins: Gravataí, Sinos, and Caí rivers18.

(A) Map of South America with emphasis on Brazil. (B) Region of Porto Alegre, capital of Rio Grande do Sul. The point of collection of the golden mussel specimens is marked with an [X]. (Data obtained from the IBGE database. Edited in QGis 2.10.1 http://www.qgis.org/).
Preparation of samples for virus isolation
Forty golden mussels were submerged for 15 min in 70% ethanol for superficial shell decontamination. Subsequently, the valves were opened and the inner water was collected and diluted in 1 mL of saline buffer (PBS). The samples were pooled, totaling eight pools. These pools were homogenized with 1 mL of PBS, centrifuged at 10,000 × g, filtered through a 0.45-μm membrane and 10 U/μL of Penicillin-GIBCO® by Life Technologies were added to the filtrate to avoid bacterial contamination.
Isolation of GMar in free-living amoebas
Cells of Acanthamoeba polyphaga (genotype T4) were cultivated in 10 mL of Peptona-Yeast Extract-Glucose (PYG) medium at 30 °C in 25-cm3 culture flasks, supplemented with 50 μg of gentamicin. After 48 h, the cells were harvested and centrifuged. The pellet was re-suspended in sterile PAS (Page’s amoeba saline), and 104 amoebas per well were cultured in 24-well microplates. After 24 h, 100 μL of the inoculum, prepared as mentioned above, were used to infect the A. polyphaga monolayers, which were then incubated for three days at 30 °C. This step was repeated by sub-culturing the primary culture onto fresh amoeba monolayers. Up to five blind passages were performed. The amoeba cells were assessed daily for the presence of viruses and for cytopathic effects on the cell such as lysis and cellular rounding. To evaluate the replication of GMar in different amoeba, monolayers of Acanthamoeba castellani was infected too.
One-step growth curves
After the virus isolation, infectivity assays were performed by inoculating the A. polyphaga monolayers in the 24-well microplates with the virus isolate at an multiplicity of infection (MOI) of 10. The viruses were allowed to adsorb for 1 h, and then the cells were washed with PAS and incubated at 30 °C. At 1–14, 24, 25 and 26 h post-infection (pi), the cultures were frozen and thawed three times and then titrated. The virus titration was performed in 96-well microplates containing approximately 4 × 104 A. polyphaga cells per well in 100 μL of the PYG medium. Virus titers were calculated according to the method of Spearmann and Kärber and expressed as the log10 culture infectious doses per 50 mL (TCID50/50 μL).
Transmission electron microscopy
After the titrations, the A. polyphaga monolayers were infected at an MOI of 10. Uninfected cell cultures were used as controls. After 7 h of infection, the monolayers were washed twice with 0.1 M PBS and fixed with 2.5% glutaraldehyde (grade I) in 0.1 M PBS for 1 h at room temperature. Following the visualization of the full cytopathic effect (CPE), the cell monolayers were scraped off of the plates and pelleted by centrifugation at 900 × g for 5 min. The amoebae were then fixed with 2% osmium tetroxide and embedded in EPON resin. Ultrathin sections were stained with 2% uranyl acetate and examined using a Tecnai G2-Spirit FEI 2006 transmission electron microscope operating at 80 kV at the Microscopy Center, UFMG, Brazil.
DNA extraction
The supernatants of the A. polyphaga-infected cells were collected, vigorously vortexed, and centrifuged at 5,000 × g for 5 min. The cell-free virus particles were pelleted on a 25% sucrose gradient by ultracentrifugation (Sorvall Combi) at 33,000 × g for 2 h at 4 °C. The pellet was re-suspended in Tris-EDTA-NaCl buffer (TEN). In order to remove the nucleic acids not protected by the capsid, the preparation was treated with 100 U of DNAse I (Roche) and 100 U of RNAse (Invitrogen) at 37 °C for 1 h. Next, the virus DNA was extracted using phenol-chloroform according to Sambrook and Russel22. The DNA was re-suspended in ultrapure water and the quality and amount of virus DNA was analyzed using a NanoSpec® and Qubit apparatus (Life Technologies).
Genome sequencing, assembly and annotation
Next-generation sequencing was performed in a MiSeq (Illumina) apparatus with pair-end applications (2 × 150 pb). The pair-end samples were prepared with a Nextera XT DNA sample prep kit. After sequencing, a total of 510,511 reads were de novo assembled using Geneious® software. The transfer RNA (tRNA) sequences were analyzed by applying the tRNAscan-SE tool. Gene predictions were performed using FgenesV23 tool. Functional annotation was performed by BLAST searches against the UniProtkB using an e-threshold of 0.01 and by the set of clusters of orthologous groups of proteins (COGs) of the NCLDV (NCVCOGs). Finally, the genome annotation was manually revised. After assembly, the topology of the genome was determined in silico and by the amplification of specific genomic fragments (data not shown).
Phylogenetic analyses and genomes comparison
Phylogenetic reconstruction based on individual and concatenated alignment of the five Megavirales core genes-namely the family B DNA polymerase, the D6/D11 helicase, the VV A18 helicase, the D5 primase-helicase, and major capsid protein. The amino acid sequences were aligned with Muscle software, and the phylogenetic tree was constructed in MEGA 7. We built a hierarchical-clustering based on the gene presence/absence patter of 5443 NCVOGs, using a MeV tool with Pearson correlation as distance metric. The average nucleotide identity using both best hits (one-way ANI) and reciprocal best hits (two-way ANI) between two genomic datasets as calculated.
Results
The isolation of GMar and the morphology of the infected cells
The inoculation of A. polyphaga monolayers (Fig. 2a) resulted in three pools with clear cytopathic effects (ECP) showed in Fig. 2b. The CPE was characterized by the formation of cell chains, cell rounding, and lysis. In addition, the viruses were also inoculated in monolayers of A. castellani in order to identify the virus specificity. Following the infection, it was noticed that the viruses were able to replicate and cause cell lysis in both species of amoebas.

Cytopathic effect observed 10 h after the inoculation of A. polyphaga monolayers with the isolated virus (200×).
Cells rounding, chain groups and lysis. (a) Negative control, uninfected amoeba culture; (b) Infected amoeba culture; FC: formation of chain groups.
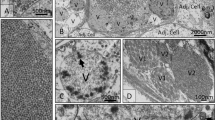
For Transmission electron microscopy (TEM), a concentrated preparation of virus-infected A. polyphaga cells revealed markedly icosahedral particles of about 200 nm in diameter. The structure of the virus particles strongly resembled the structure of marseilleviruses (Fig. 3).

Transmission electron microscopy (TEM) image of A. polyphaga infected with the GMar.
The image shows icosahedral particles inside an amoeba. A scale bar measured particles with 500 nm. We can observe different phases of viral morphogenesis.
One-step growth curve of GMar
The replicative cycle of GMar was measured at different periods after infection. After 1 h pi, we observed cell alterations in the amoeba cytoplasm, described previously like as cell rounding. At this time, high virus titers (104/50 μL) were detected. After 5 h pi, high virus titers were detected, as shown in Fig. 4 (1011/50 μL). At this time, we observed cell rounding and cell chains formation. After 10 h pi, it was possible to visualize extensive cell lysis, showed in Fig. 2 and the high titers were maintained. Figure 4 shows an exponential increase of infectious viral particles within 3–5 h pi. The Fig. 4 shows the replicative cycle of GMar. It is interesting to note that the cycle was completed in less than 24 h. After 1 h, we could detect virus and levels increased exponentially from the 3 h of infection. The highest titles were observed after 5 h.

The infectivity assays were performed by inoculating A. polyphaga with the GMar at an MOI of 10.
The viruses were allowed to adsorb for 1 h, and then the cells were washed with PBS and incubated at 30 °C. At 1–14 h and 24–26 h, the cultures were frozen and thawed three times and then titrated. A logarithmic curve expresses the titers measured at each time point pi. After 3 days of incubation, the titer (TCID50) was calculated as described by Reed and Muench24.
GMar genome features
The complete genome of three isolates that induced CPE in A. polyphaga was fully sequenced with a 217 coverage. The nucleotide sequences of the other two viruses were identical to the first one, and consequently, they were considered clones (data not show). The virus genome of one isolate (GenBank submission KT835053) is a circular, double-stranded DNA molecule composed of 360, 610 bp, which is comparable in size to the genomes, described previously, which range from 346, 754 bp to 386, 631 bp for Lausannevirus and Insectomime virus respectively. The mean guanine-cytosine is 43.1% for GMar, which is also similar to other members of this family.
A total of 483 open reading frames (ORFs) were identified after merging all coding sequence predictions. These ORFs were evenly distributed on both negative (263 ORFs) and positive (220 ORFs) strands, similar to the gene distribution on the Brazilian marseillevirus genome (261 and 230 ORFs on negative and positive strands, respectively). The GMar virus gene content consists of 48.03% uncharacterized proteins (232 of the 483 predicted proteins). The predicted proteins contained between 35 and 1293 amino acids. Moreover, we found numerous paralogous families, consisting of genes that code endonucleases (seven sequences), serine/threonine protein kinases (sixteen sequences), F-box containing protein (three sequences), putative MutH/archeal (four sequences) and ankyrin repeat (ten sequences). In addition, we found two histone-like proteins: a histone H2B/H2A fusion protein and a histone H3 containing a N-terminal Histone-like transcription factor, an archeal histone domain and C-terminal H3-like domain (252, 844- > 252, 089 and 252, 549- > 253, 574). The histone-like proteins in GMar resemble those predicted in other marseilleviruses, such as Lausannevirus12 and Brazilian marseillevirus18.
A total of 440/483 ORFs (91%) from the GMar had a significant BLASTp matches to marseilleviruses protein sequences available in the NCBI non redundant database. The 43 ORFs that displayed no identity with previously described sequences were tentatively classified as ORFans. As for the other marseilleviruses, no tRNA-related genes were detected in the GMar genome.
Comparative genome
Furthermore, we noticed a high proportion of orthologous genes (Fig. 5) in members of Marseilleviridae. Figure 5 shows that 212 orthologous genes are shared by Marseillevirus (Lineage A), Lausannevirus (Lineage B), Tunisvirus (Lineage C), Brazilian marseillevirus (Lineage D) and Golden marseillevirus. It is interesting to note that there are fourteen (14) non-shared genes, among these there are seven (7) GMar genes.

Distribution of orthologous genes clusters among marseilleviruses from one representative of each lineage (A: Marseillevirus; B: Lausannevirus; C: Tunisvirus and D: Brazilian marseillevirus) and GMar.
The graph shows the number of orthologous genes that were used in this analysis.
Phylogeny
A hierarchical clustering tree was constructed using presence/absence matrix of 5443 clusters of orthologous genes shared by NCLDVs. It shows in Fig. 6 that GMar is distant from other known lineages proposed, including the lineage D, recently discovered.

Hierarchical clustering tree based on phyletic patterns.
Phylogeny based on presence/absence matrix of 5443 NCVOG (clusters of orthologous genes shared by NCLDVs). The Pearson correlation was used as metric distance, and the scale bar means the branch time.
Phylogenetic analysis based on the core genes (DNA polymerase B, the VV A18-helicase, the D5 helicase, the D6/D11 helicase and the major capsid protein), for concatenated alignment (Fig. 7) show four clades for the previously described lineages, with a first group consisting of lineage A (Marseillevirus, Senegalvirus, Melbournevirus and Cannes8 virus). Three other clades appear to delimit the phylogeny of the Marseilleviridae family, composing the lineages previously known as B, C and D and a fifth comprising of GMar. These trees show the GMar within the family Marseilleviridae, but far from the other members already described. The analysis of nucleotide identity between the BrMr, recently founder of a new lineage and geographically closer to GMar, displays 74.96% of similarity. In addition to the concatenated tree, five trees with separate genes were also generated to support this result, based on the sequences available in the database. In spite of some variations in the positions, the clusters formed were the same, reinforcing the proposition of a new lineage to the virus here described (Supplementary data).

Phylogenetic reconstruction based on a concatenated alignment of the five core genes, DNA polymerase B, major capsid protein, VV-A18 helicase, D6/D11 helicase and D5 helicase.
The amino acid sequences were aligned using Muscle. Evolutionary history was inferred by using the Maximum Likelihood method based on the JTT matrix-based model. The analysis involved 16 amino acid sequences. All positions containing gaps and missing data were eliminated. There were a total of 2552 positions in the final dataset. Evolutionary analyses were conducted in MEGA725.
Discussion
The discovery of a new strain of marseillevirus was attained by analyzing golden mussels, invasive organisms that are found worldwide. The source of the GMar is the inner water collected from golden mussels found in a local lake in Porto Alegre, Brazil, and this is the first report on the use of this organism for the isolation of marseilleviruses. Although this is a small-scale study, our results suggest that the golden mussels seem appropriate for the recovery of GMar and, perhaps, other marseilleviruses. It is likely that the filtration process necessary for the ingestion of nutrients of these organisms may be responsible for the accumulation of such viruses in their tissues. Oysters have been previously shown to be excellent sources for the isolation of mimiviruses due to their highly efficient filtration capacity, filtering more than 400 liters of water per day19. From the findings reported here, it seems that golden mussels behave similarly. By concentrating the water contaminants, the species was a rich source of giant viruses. In the near future, evaluations of golden mussels as accumulator of other giant viruses should be pursued.
Two members of the Marseilleviridae already described (Marseillevirus and Insectomime) can replicate in A. castellani, even if they have been isolated in A. polyphaga2,16. Our isolate share the same characteristic, as cytopathic effects is observed detected in both species. When comparing the replication profile of GMar with other marseilleviruses we observed additional similarities. After 3 h pi a MET of GMar infected amoebas revealed large viral factories which occupied half of the amoeba cytoplasm26. All virus factories contained different stages of the virion morphogenesis. At four hours pi we detected high titers of virus and one hour after that, an exponential increase in virus titers was observed.
In this study, we propose the foundation of a new lineage (E) in the Marseilleviridae family, of which GMar would be the first member. This suggestion is validated by genome analyses emphasizing several divergences between GMar and other marseilleviruses. When we look at the shared orthologues genes, we observe that seven genes are unique to GMar. It is noteworthy that the core genome analysis clearly distinguishes GMar from other lineages, drawing attention to the similar conserved gene content of the presently known marseilleviruses. More remarkably, phylogeny based on gene presence/absence patterns NCVOG reflects the gene loss and gain history of the giant viruses, clustering GMar into a distinctive clade in Marseilleviridae family. Conclusively, phylogenetic analysis show the four lineages of Marseilleviridae, which are presently composed by A, B, C and D. Recently a marseillevirus isolated in a river in Tokyo27 had its genome sequenced, however phylogenetic analyses are still missing to determine to which lineage this virus belongs. In our analyses, we included this new marseillevirus, and one can see that it’s in a new branch on the tree next to the lineage A. Based on concatenated alignment of five core genes, GMar probably founds a fifth clade in the marseilleviruses phylogeny. Even though the virus here described displays notable differences when compared to the other members of the Marseilleviridae, phylogenetic analysis shows that it is distant to Mimiviridae and the other giant viruses already identified. The analysis of separate genes also reinforces and consistently maintains the GMar in a separate branch in tree, indicating the possibility of a new strain. Finally, phylogenetic analysis of both the concatenated genes how the individual genes (according to availability in the database), shows clearly the four strains already described and characterized the GMar in one branch distant.
As the knowledge about the giant viruses is something recent, and the database grows with each new study, it is obvious that very soon there will be a review of annotation of genomes available, and probably a reorganization of the family Marseilleviridae with the inclusion of new members. In brief, these data increase the knowledge about the biological and genomic characteristic of marseilleviruses, currently incomplete.
Additional Information
How to cite this article: dos Santos, R. N. et al. A new marseillevirus isolated in Southern Brazil from Limnoperna fortunei. Sci. Rep. 6, 35237; doi: 10.1038/srep35237 (2016).
References
La Scola, B. et al. A giant virus in amoebae. Science 299 (2003).
Boyer, M. et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 106(51), 21848–21853 (2009).
Arslan, D., Legendre, M., Seltzer, V., Abergel, C. & Claverie, J. M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 108, 17486–17491 (2011).
Philippe, N. et al. Pandoraviruses: Amoeba Viruses with Genomes Up to 2.5 Mb Reaching That of Parasitic Eukaryotes. Science 341, 281–286 (2013).
Legendre, M. et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 111, 4274–4279 (2014).
Legendre, M. et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 112, 5327–5335 (2015).
Yoosuf, N. et al. Complete genome sequence of Courdo11 virus, a member of the family Mimiviridae. Virus Genes 48(2), 218–223 (2014).
Boyer, M. et al. Phylogenetic and phyletic studies of informational genes in genome highlight existence of a 4 domain of life including giant viroses. PLoS One 5(12), 15530 (2010).
Yutin, N. et al. Origin of giant viruses from smaller DNA viruses not from a fourth domain of cellular life. Virology 466–467, 38–52 (2014).
Katzourakis, A. & Aswad, A. The origins of giant viruses, virophages and their relatives in host genomes. BMC Biol. 12, 51 (2014).
Aherfi, S. et al. The expanding family Marseilleviridae. Virology 466–467, 27–37 (2014).
Thomas, V. et al. Lausannevirus, a giant amoebal virus encoding histone doublets. Environ. Microbiol. 13(6), 1454–1466 (2011).
Aherfi, S. et al. Complete genome sequence of Cannes 8 virus, a new member of the proposed family “Marseilleviridae”. Virus Genes. 47, 550–555 (2013).
Aherfi, S. et al. Complete genome sequence of Tunisvirus, a new member of the proposed family Marseilleviridae. Arch Virol 159(9), 2349–2358 (2014).
Pagnier, I. et al. A Decade of Improvements in Mimiviridae and Marseilleviridae Isolation from Amoeba. Intervirology 56, 354–363 (2013).
Boughalmi, M. et al. First isolation of a Marseillevirus in the Diptera Syrphidae Eristalis tenax. Intervirology 56(6), 386–394 (2013).
Doutre, G. et al. Complete Genome Sequence of a New Member of the Marseilleviridae Recovered from the Brackish Submarine Spring in the Cassis Port-Miou Calanque, France. Genome Announc. 3(6) (2015).
Dornas, F. P. et al. A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae. Viruses 8(3) (2016).
Andrade, K. R. et al. Oysters as hot spots for mimivirus isolation. Arch Virol. 160(2), 477–82 (2015).
Oliveira, M. D. et al. Colonization and Spread of Limnoperna fortunei in South America. Springer Series in Invas. Ecol. 10 (2015).
Mansur, M. C. D., Quevedo, C. B., Santos, C. P. & Callil, C. T. Prováveis vias de introdução de Limnoperna fortunei (Dunker, 1857) (Mollusca, Bivalvia, Mytilidae) na Bacia da Laguna dos Patos, Rio Grande do Sul e novos registros de invasão no Brasil pelas Bacias do Paraná e Paraguai (ed. Silva, J. S. V. & Souza, R. C. C. L ). Água de lastro e bioinvasão. Interciência 33–38 (2004).
Sambrook, J. & Russell, D. W. Molecular Cloning: A Laboratory Manual. 3rd (ed. New York: Cold Spring Harbor Laboratory Press) 631–632 (2001).
Fgenes, V. Available online: http://linux1.softberry.com/berry.phtml?topic=virus&group=programs&subgroup=gfindv (Date of acess 15/05/2015).
Reed, L. J. & Muench, H. A simple method of estimating fifty percent endpoints. The American Journal of Hygiene. 27, 493–497 (1938).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33, 1870–1874 (2016).
Arantes, T. A. et al. The Large Marseillevirus Explores Different Entry Pathways by Forming Giant Infectious Vesicles. J. Virol. 90(11), 5246–5255 (2016).
Takemura, M. Draft Genome Sequence of Tokyovirus, a Member of the Family Marseilleviridae Isolated from the Arakawa River of Tokyo, Japan. Genome Announc. 4(3) (2016).
Acknowledgements
We acknowledge professor Jônatas Abrahão (UFMG) and his group, especially for Ana Paula Moreira Franco for their technical assistance. We thank Keli Barroso from the Laboratory of Parasitology from UFRGS, for amoebas culture. Funding from CAPES, CNPq, FAPERGS and FINEP. We also acknowledge Cintia Pinheiro from the Department of Ecology of UFRGS for the support in the collection of the golden mussels.
Author information
Authors and Affiliations
Contributions
R.N.d.S. performed the virus isolation and the data analysis; F.S.C. performed the assembly of genome and annotation; N.R.M.d.A. performed biological experiments; F.F. performed the phylogenetic analysis; R.A.C. participated in the collection of the mussels; L.C.-O. performed the DNA extraction and data analysis; F.L.A. helpful support to analyze the data and conduce the orthologous analysis; T.S.A. performed the electron microscopy; P.M.R. and A.C.F. designed the experiments and analyzed the data.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
dos Santos, R., Campos, F., Medeiros de Albuquerque, N. et al. A new marseillevirus isolated in Southern Brazil from Limnoperna fortunei. Sci Rep 6, 35237 (2016). https://doi.org/10.1038/srep35237
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35237
This article is cited by
-
Ultrastructure of the gill ciliary epithelium of Limnoperna fortunei (Dunker 1857), the invasive golden mussel
BMC Zoology (2022)
-
Viral histones: pickpocket’s prize or primordial progenitor?
Epigenetics & Chromatin (2022)
-
Giant virus biology and diversity in the era of genome-resolved metagenomics
Nature Reviews Microbiology (2022)
-
What we know and don’t know about the invasive golden mussel Limnoperna fortunei
Hydrobiologia (2022)
-
Translating the language of giants: translation-related genes as a major contribution of giant viruses to the virosphere
Archives of Virology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
