


Abstract
The minimal structural unit of a solid tumor is a single cell or a cellular compartment such as the nucleus. A closer look inside the cells reveals that there are functional compartments or even structural domains determining the overall properties of a cell such as the mechanical phenotype. The mechanical interaction of these living cells leads to the complex organization such as compartments, tissues and organs of organisms including mammals. In contrast to passive non-living materials, living cells actively respond to the mechanical perturbations occurring in their microenvironment during diseases such as fibrosis and cancer. The transformation of single cancer cells in highly aggressive and hence malignant cancer cells during malignant cancer progression encompasses the basement membrane crossing, the invasion of connective tissue, the stroma microenvironments and transbarrier migration, which all require the immediate interaction of the aggressive and invasive cancer cells with the surrounding extracellular matrix environment including normal embedded neighboring cells. All these steps of the metastatic pathway seem to involve mechanical interactions between cancer cells and their microenvironment.
The pathology of cancer due to a broad heterogeneity of cancer types is still not fully understood. Hence it is necessary to reveal the signaling pathways such as mechanotransduction pathways that seem to be commonly involved in the development and establishment of the metastatic and mechanical phenotype in several carcinoma cells. We still do not know whether there exist distinct metastatic genes regulating the progression of tumors. These metastatic genes may then be activated either during the progression of cancer by themselves on their migration path or in earlier stages of oncogenesis through activated oncogenes or inactivated tumor suppressor genes, both of which promote the metastatic phenotype. In more detail, the adhesion of cancer cells to their surrounding stroma induces the generation of intracellular contraction forces that deform their microenvironments by alignment of fibers. The amplitude of these forces can adapt to the mechanical properties of the microenvironment. Moreover, the adhesion strength of cancer cells seems to determine whether a cancer cell is able to migrate through connective tissue or across barriers such as the basement membrane or endothelial cell linings of blood or lymph vessels in order to metastasize. In turn, exposure of adherent cancer cells to physical forces, such as shear flow in vessels or compression forces around tumors, reinforces cell adhesion, regulates cell contractility and restructures the ordering of the local stroma matrix that leads subsequently to secretion of crosslinking proteins or matrix degrading enzymes. Hence invasive cancer cells alter the mechanical properties of their microenvironment.
From a mechanobiological point-of-view, the recognized physical signals are transduced into biochemical signaling events that guide cellular responses such as cancer progression after the malignant transition of cancer cells from an epithelial and non-motile phenotype to a mesenchymal and motile (invasive) phenotype providing cellular motility. This transition can also be described as the physical attempt to relate this cancer cell transitional behavior to a T1 phase transition such as the jamming to unjamming transition. During the invasion of cancer cells, cell adaptation occurs to mechanical alterations of the local stroma, such as enhanced stroma upon fibrosis, and therefore we need to uncover underlying mechano-coupling and mechano-regulating functional processes that reinforce the invasion of cancer cells. Moreover, these mechanisms may also be responsible for the awakening of dormant residual cancer cells within the microenvironment.
Physicists were initially tempted to consider the steps of the cancer metastasis cascade as single events caused by a single mechanical alteration of the overall properties of the cancer cell. However, this general and simple view has been challenged by the finding that several mechanical properties of cancer cells and their microenvironment influence each other and continuously contribute to tumor growth and cancer progression. In addition, basement membrane crossing, cell invasion and transbarrier migration during cancer progression is explained in physical terms by applying physical principles on living cells regardless of their complexity and individual differences of cancer types. As a novel approach, the impact of the individual microenvironment surrounding cancer cells is also included. Moreover, new theories and models are still needed to understand why certain cancers are malignant and aggressive, while others stay still benign. However, due to the broad variety of cancer types, there may be various pathways solely suitable for specific cancer types and distinct steps in the process of cancer progression.
In this review, physical concepts and hypotheses of cancer initiation and progression including cancer cell basement membrane crossing, invasion and transbarrier migration are presented and discussed from a biophysical point-of-view. In addition, the crosstalk between cancer cells and a chronically altered microenvironment, such as fibrosis, is discussed including the basic physical concepts of fibrosis and the cellular responses to mechanical stress caused by the mechanically altered microenvironment. Here, is highlighted how biophysical approaches, both experimentally and theoretically, have an impact on classical hallmarks of cancer and fibrosis and how they contribute to the understanding of the regulation of cancer and its progression by sensing and responding to the physical environmental properties through mechanotransduction processes. Finally, this review discusses various physical models of cell migration such as blebbing, nuclear piston, protrusive force and unjamming transition migration modes and how they contribute to cancer progression. Moreover, these cellular migration modes are influenced by microenvironmental perturbances such as fibrosis that can induce mechanical alterations in cancer cells, which in turn may impact the environment. Hence, the classical hallmarks of cancer need to be refined by including biomechanical properties of cells, cell clusters and tissues and their microenvironment to understand mechano-regulatory processes within cancer cells and the entire organism.
Export citation and abstract BibTeX RIS
Recommended Professor Robert H Austin2019
Abbreviations
| ADAM | A disintegrin and metalloproteinase |
| ADMA | Asymmetric dimethylarginine |
| APC | Polyposis coli protein |
| Arp2/3 | Actin-related protein 2/3 |
| CSCs | Cancer stem cells |
| DH | DBl homology |
| DNA | Deoxyribonucleic acid |
| DRFs | Diaphanous-related formins |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-mesenchymal transition |
| ERK1/2 | Extracellular regulated kinase 1/2 |
| FAK | Focal adhesion kinase |
| FDG | 18F-fluorodeoxyglucose |
| FGF | Fibroblast growth factor |
| FGFR | Fibroblast growth factor receptor |
| FLM | Filamin |
| GBM | Glioblastoma |
| GBP | GTPase binding domain |
| GEFs | Guanine nucleotide exchange factors |
| GPCRs | G-protein-coupled receptors |
| Hg | Hedgehog |
| IDH | Isocitrate dehydrogenase 1/2 |
| KLF-2 | Krüppel-like factor 2 |
| LAP | Latency associated peptide |
| L-NMMA | L-n(g)-mono-methyl-arginine |
| LINC | Linker of nucleoskeleton and cytoskeleton complex |
| LTBP-1 | Latent TGF-β1 binding protein |
| MAP | Mitogen-activated protein kinase |
| MAT | Mesenchymal to amoeboid transition |
| mDia | Mammalian homolog of Drosophila diaphanous |
| MMPs | Matrix metalloproteinases |
| MTOCs | Microtubule organizing centers |
| MT1-MMP | Membrane-type 1 matrix metalloproteinase |
| NE | Nuclear envelope |
| NO | Nitric oxide |
| NOS | Nitric oxide synthase |
| PAA | Polyacrylamide gels |
| PDGF-AB | Platelet-derived growth factor-AB |
| PDGFR | Platelet-derived growth factor receptor |
| PET | Positron emission tomography |
| PI3K | Phosphoinositide 3-kinase |
| PKA | Protein kinase A |
| PKG | Protein kinase G |
| PP2A | Protein phosphatase 2A |
| PTP1B | Protein-tyrosine phosphatase 1B |
| PTEN | Phosphatase and tensin homolog deleted on chromosome 10 |
| RTKs | Receptor tyrosine kinases |
| srABD | Spectrin-related actin-binding domain |
| TCF/LEF1 | T-cell factor/lymphoid enhancer-binding factor1 |
| TSA | Trichostatin A |
| VASP | Vasodilator-stimulated phosphoprotein |
| WASP | Wiskott-Aldrich syndrome protein |
1. Introduction
Cell motility is important for many physiological processes such as wound healing, tissue repair, immune response and development of tissues and organs as well as pathological processes such as the malignant progression of cancer. In general, a certain cancer type can turn into a malignant systemic disease, when specific cancer cells manage to migrate out of the primary solid tumor mass into the surrounding tumor stroma. The migratory capability is of critical importance for predicting the outcome of a certain cancer and the prognosis for cancer survival. The migration of cells has been intensively investigated for several decades, however, most of the knowledge is gained from artificial 2D motility assays using single cells or a collection of cells such as clusters or spheroids, which are seeded on a flat substrate. More sophisticated cell motility assays account for the environmental cues by using 3D matrix scaffolds such as collagen type I matrices, Matrigel and hydrogels based on gelatin, which is denatured collagen. Tissue and organ environmental cues rely on the structural architecture defining pore or mesh sizes, the chemical and biomolecule composition such as ions or cell matrix receptor ligands, respectively. All of which determine the mechanical properties of the extracellular matrix in vivo and in vitro migration assays and hence influence cellular behavior such as adhesion, survival, cell-division rate, migration, force transmission and generation and restructuring of the matrix by secretion of molecules such as enzymes, cross-linkers or ligands for other interacting cells.
Classical tumor biological approaches have still not managed to cover the entire complexity of solid tumors of epithelial origin (carcinomas), non-epithelial driven tumors, and the malignant progression of the disease. The field of physical driven cancer research is growing, but still needs a closer connection to tumor biology and medical driven cancer research. Since the development and ongoing establishment of the physics of cancer field many of these new directions of physically based cancer research have pronouncedly altered the field of current cancer research such as diagnosis and treatment and broadened the sometimes limited classical biological and biochemical view of cancer disease.
Besides the proposed hallmarks of cancer describing the malignant transformation of the cells (Hanahan and Weinberg 2000, Hanahan and Weinberg 2011), there is still a ninth hallmark needed that includes the mechanical properties of cancer cells as proposed (Mierke 2014). Similarly, the eight other hallmarks require the inclusion of the physical aspects. This review presents the influence of the matrix and cellular mechanical properties on the initiation and progression of cancer. In particular, it discusses how cancer cells or normal neighboring cells communicate at tumor borders and respond to external physical stimuli provided by the extracellular matrix or the adjacent cells such as fibroblasts or endothelial cells embedded into the connective tissue or lining blood or lymph vessels and how this impact cancer cell motility. Various cellular responses on the molecular, cellular, cell cluster (such as a cellular spheroid) and compartment scale are introduced. Additionally, candidates or physical parameters are identified that modify tumor initiation, tumor boundary crossing, cancer cell basement membrane crossing, cancer cell invasion and transbarrier migration involving mechano-coupling and mechano-regulatory functions of proteins. Moreover, theoretical aspects of cancer cell sorting or malignant phenotype segregation including basement membrane crossing, invasion and transbarrier migration are discussed on different length and time scales. The role of specific proteins such as focal adhesions in response to mechanical stimuli are presented as short-scale mechanisms, whereas the actomyosin contractility, intermediate filaments and microtubules cytoskeletal elements and their alteration by stroma mechanical properties such as mechanotransduction processes are presented as large-scale mechanisms.
Finally, the different approaches and the integration of different physical and biological processes that affect cell mechanical properties regulating cancer initiation, cancer cell boundary crossings and cancer progression, including interaction with chronic inflammation such as fibrosis, are highlighted and discussed. The first part introduces the major signaling pathways involved in initiation and malignant progression of cancer that are required to understand the impact of the microenvironment on the mechanical phenotype of cancer cells.
1.1. Physical Oncology background
Genetic and epigenetic alterations can lead to the initiation and subsequently to the development of cancer. These changes affect the homeostasis of the cells and lead to uncontrolled proliferation. Hence, the proliferation is then no longer suppressed by tumor suppressor genes and there exists also no repression of aberrantly dividing cells in niches that are even not their original habitat. Cancers are most commonly found to be composed of epithelial cells, which then subsequently become solid carcinomas in organs such as colon, liver, lung, kidney, skin, pancreas, bladder and breast. In contrast, sarcomas originate from mesenchymal tissues and are found in fibroblasts, endothelial cells, osteoblasts, adipocytes and myocytes. Another source of non-epithelial tumors are cells of the nervous systems that form gliomas, neuroblastomas and medulloblastomas, and hematopoietic cells that cause leukemia and lymphoma. In this review article, the focus is on solid tumors of epithelial origin (carcinomas).
Within solid tumors, altered homeostasis facilitates in general the progression of a benign proliferating cell cluster (termed hyperplasia) to a larger cell mass of proliferating cells with altered morphology, cytological detectable recognition and altered cellular organization such as cytoskeletal aberrancies (figure 1). Upon expansion of the primary tumor, the cancer cells of the solid tumor mass have no longer sufficient oxygen and nutrients, and hence the primary tumor induces the growth of novel blood vessels into the tumor mass (termed neoangiogenesis) that helps cancer cells in the tumor core to regain access to oxygen and nutrients.
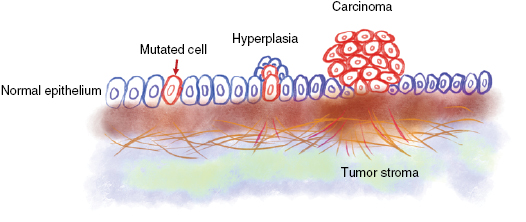
Figure 1. Origin and progression of a carcinoma. In the normal epithelium may occur a single mutation in an individual epithelial cell. The mutated cell grows to a hyperplasia and subsequently to a in situ solid primary carcinoma.
Download figure:
Standard image High-resolution imageIn physical oncology, principles of engineering and physics have been employed for a better understanding of oncology such as basic cancer biology that starts from quantitative assessment of tumor growth and progression and ends with elevated detection as well as enhanced treatment of cancer or the mathematical modeling of drug distribution in the patient, cell cycle kinetics of cancer cells and subsequently the dynamics of tumor growth. In general, physical oncology research focusses on how experimental approaches and theoretical models lead to a better knowledge of cancer complexity. Thereby it combines the detection of cancer–related changes in the patient such as the invention of more effective diagnosis with advanced cure by using more effective treatment strategies. In cancer, physical changes of tissues and cells can facilitate the growth of the primary tumor and can cause the initiation of cancer (Bhowmick et al 2004, Levental et al 2009, Ng and Brugge 2009, Cirri and Chiarugi 2012). Cancer cells invade the surrounding tissue and displace normal cells therein (figure 2), which alters the tissue physically, as it gets frequently stiffer (Gehler et al 2009, Levental et al 2009, Egeblad et al 2010, Lopez et al 2011, Plodinec et al 2012) and sometimes even softer such as in lipomas, which belong to benign tumors (Totty et al 1986). In both cases the cancer-associated 'normal' tissue becomes more heterogeneously structured, which in turn accelerates in the case of tissue stiffening the progression of cancer such as mammary tumors (Rubashkin et al 2014) and may cause resistance to drug treatments in breast cancer (Majidinia and Yousefi 2017, Senthebane et al 2017). Moreover, also tumor acidity and hypoxia can also lead to drug resistance (Tredan et al 2007). However, when the crosslinking of the extracellular matrix is impaired and hence tissue tension decreased, the microenvironment of primary mammary tumor may not stiffen (Pickup et al 2013), which in turn supports the drug treatment efficiency.
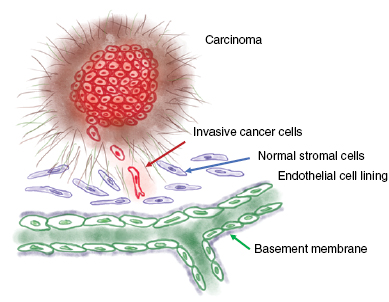
Figure 2. Dissemination of cancer cells from the primary solid carcinoma. Invasive cancer cells spread from the solid tumor mass and migrate into the surrounding tissue. Thereby these cancer cells displace normal stromal cells. Cancer cells migrate towards blood or lymphoid vessels nearby the tumor microenvironment.
Download figure:
Standard image High-resolution imagePhysical aberrancies are not limited to the tumor microenvironment's structural changes, as they cover also the disordered and hence chaotic vasculature of the primary tumor, enhanced interstitial pressure as well as tumor-based stress, tissue hypoxia, altered extracellular matrix composition and progressive stiffening of primary tumors. In cancer, physical barriers are of importance, as increased tissue stiffness can cause altered drug delivery efficiency, decrease the infiltration of tissues by immune cells in breast cancer and melanoma (Muenst et al 2016, Cohen and Blasberg 2017) and finally all these changes facilitate malignant and aggressive progression of cancer (Rubashkin et al 2014, Majidinia and Yousefi 2017, Senthebane et al 2017). Although the physical hallmarks of cancer are not yet fully clearly known, novel anticancer strategies aim the physical aberrancies that are caused by solid primary tumors to impair tumor mass growth. This is currently a quite novel and growing field in the nanomedicine discipline. Cells can manage to withstand biophysical stimulation through mechanosignaling processes employing physically coupled proteins from extracellular matrix adhesion molecules such as integrins that are connected via focal adhesion proteins to actin filaments and subsequently also components of the nucleus. However, also other components are involved such as intermediate filaments or microtubules. The process of mechanotransduction involves the activation of distinct transcriptions factors and their downstream target genes and causes a remodeling of the structure and the organization of cells as a response to physical environmental changes (Kanchanawong et al 2010, Case and Waterman 2015). In summary, there seems to exist a tight connection between the mechanical properties of cancer cells and their surrounding microenvironment.
1.2. Malignant progression of cancer
A solid primary tumor can be excised precisely by surgery, whereas the prediction of the recurrence of the solid tumor at the same site or the malignant progression of cancer including the formation of secondary tumors at distant sites is still not reliable due to the broad variability of cancer types and their microenvironment. Both, the reoccurrence of a tumor at the original place or at a targeted site involve a scenario, which is termed the metastatic cascade. In particular, the malignant progression of cancer is determined by the process of metastasis that subsequently may turn cancer into a systemic disease leading finally to patients' death. The process of metastasis is characterized by a complex composition of many steps following a linear propagation (figure 3) (Chambers et al 2002, Eger and Mikulits 2005). The metastatic cascade can be stopped at distinct points leaving the malignant cancer cells in a dormant state (Aguirre-Ghiso 2007, Quail and Joyce 2013). This state of tumor dormancy may involve various processes that can be partly facilitated by the microenvironment and encompasses at least three different types such as tumor mass dormancy, in which the proliferation is balanced by apoptosis, cellular dormancy (cells are arrested in the G0 phase of the cell cycle), or immune dormancy, in which the immunoediting causes finally a state of equilibrium (Aguirre-Ghiso 2007, Schreiber et al 2011, Hensel et al 2013). After a lag phase, these dormant cancer cells return to their motile (invasive) phenotype (Quail and Joyce 2013). It is not yet known, how this phenomenon of cancer cell arrest is facilitated in detail or how cancer cells can overcome the arrested state after some time and thus the mechanisms need still more investigation. In more detail, the microenvironment needs to be included that may drive the awakening of these dormant cancer cells. In most general cases, the initiation of the metastatic cascade begins with a change of the motility within a subpopulation of the cancer cells within a solid primary tumor (figure 3). Single cancer cells or a collection of cancer cells migrate through the barrier of the primary solid tumor mass including the tumor surrounding basement membrane (figure 3). The spreading of individual cancer cell or a collection of clustered cancer cells from the primary tumor is termed dissemination of cancer cells. These cancer cells possess the capacity to invade into the tumor surrounding microenvironment such as the stroma consisting of extracellular matrix proteins, enzymes and cell types that are on the one hand locally 'transformed' by the primary solid tumor (Bizzarri and Cucina 2014) and on the other hand by invading malignant cancer cells such as breast cancer cells (Morel et al 2008, Mierke et al 2011a).
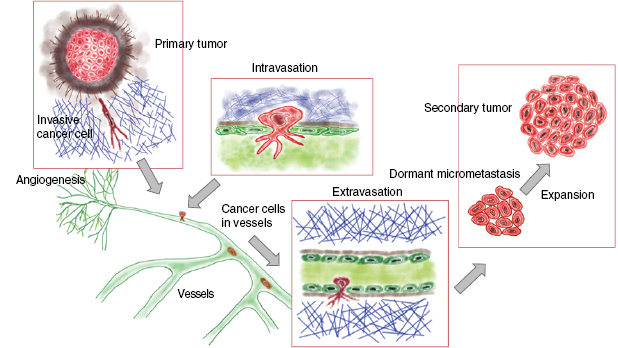
Figure 3. Heterogeneous tumors produce circulating tumor cells (CTCs). CTCs are generated from the primary solid tumor or metastatic tumors after intravasation in the peripheral blood vessels. The single cell heterogeneity originates either from genetic mutations (red, green and blue) or from epigenetic plasticity (light colored circles). Both, mutations and epigenetic alterations can occur simultaneously within the same cancer cell, Thus, metastases may consist of numerous mutations and/or epigenetic alterations.
Download figure:
Standard image High-resolution imageIn the tumor microenvironment such as the tumor stroma, endothelial cells build a neovascular system (termed tumor vasculature) by undergoing a neoangiogenesis process that has been evoked by growth factors secreted by the tumor mass to the support subsequently the primary tumor with nutrients and oxygen (figure 3). In the next step, these malignant cancer cells manage to break through the basal membrane and the vessel lining consisting of endothelial cells to finally transmigrate into blood or lymph vessels (termed intravasation). After the transbarrier break-through of cancer cells through the basal membrane and the endothelial cell lining of the vessels, they are passively transported by the vessel flow through the whole body to targeted sites suited for metastasis (figure 3). Upon reaching these targeted regions, cancer cells adhere to the luminal site of the endothelial cell lining of the vessels, start to proliferate and grow either to a secondary tumor within the vessel or transmigrate through the endothelial cell lining of the vessel (Al-Mehdi et al 2000). Thereby a new secondary tumor is built either in the vessels of the blood or lymph vasculature, or cancer cells, which have managed to transmigrate through the endothelial vessel lining (termed extravasation) into the extracellular matrix of the connective tissue, form a secondary tumor within the new tissue. In the latter case, the malignant cancer cells such as breast cancer cells, melanoma cells, prostate or bladder cancer cells which may have become even more motile after transmigration through confluent endothelial monolayers (figure 3) (Mierke 2011, Mierke et al 2008b, 2011a, 2011b). Thereafter, they migrate deeper into the targeted tissue site, proliferate, grow and assemble to secondary tumors, which finally means that the primary tumor has metastasized into another organ or tissue. How can malignant cancer cells decide in which tissue they metastasize and how do cancer cells know when they have reached the targeted tissue? What role play the mechanical properties of these tissues? However, the main question is still unanswered: Why can certain cancer cells that belong to a subpopulation of the primary tumor become malignant and migrate through the tissue? All these subsequent steps of the metastatic cascade leading finally to a malignant progression cancer require the motility of specific cancer cells and alterations of the microenvironment, which then also contribute to the promotion of the malignant phenotype of these cancer cells (Levental et al 2009, Wolf and Friedl 2009, Mierke 2011, Fischer et al 2017, Mierke et al 2017). Moreover, it has been shown that the mechanical properties of motile and invasive cells such as human breast cancer cells and murine embryonic fibroblasts are altered (Fischer et al 2017, Kunschmann et al 2017, Mierke et al 2017).
1.3. Cancer signaling pathways regulating motility are involved in mechanotransduction
The signaling pathways in cancer are usually explored in determining their role in the regulation of the motility of cancer cells. At the first glance there seems to be no functional link of these signaling pathways to the mechanical aspects of cancer cell migration and invasion. However, the cancer signaling pathways are supposed to be involved in mechanotransduction processes that transfer the extracellular signals from the environment towards the cells in order to alter their mechanical and subsequently their migratory phenotype. Some of these prominent signaling pathways of cancer progression such as PI3K/Akt have been studied in response to shear stress in endothelial cells, and indeed, it has been found they react to mechanical stress by altered gene expression (Frueh et al 2013, Maimari et al 2016). Since the process of mechanotransduction involves sensing of the mechanical properties of the microenvironment by cells and their reaction on alterations in microenvironmental mechanics, the reaction of cells to mechanical force is highly complex as more than thousand genes and even a factor ten more genes are associated by interactions and may be involved (Frueh et al 2013, Maimari et al 2016). For example, endothelial cells are subject to shear stress and involve the PI3K/Akt signal transduction pathway (Frueh et al 2013, Maimari et al 2016). Although the focus on these studies is mostly on a specific signaling pathway or only of part of it, it has evolved that these pathways are interconnected and a large crosstalk between them exists. Indeed, the signaling pathways that are involved in the stress sensing and the reaction towards shear stress in endothelial cells are Wnt, TGF-beta and Notch. These pathways play a role in cancer and especially in the malignant progression of cancer. Hence the current findings of these signaling pathways on mechanical perturbance and microenvironmental changes are discussed below.
The knowledge of the pathology of cancer is historically based on biochemical and molecular research that focusses on the deregulation of signaling pathways essential for cellular functions and cellular or tissue homeostasis. The deregulation of signaling pathways represents a hallmark of cancer (Hanahan and Weinberg 2000, Hanahan and Weinberg 2011). All factors regulating the migration modes or their switch seem to be associated with specific signaling pathways. Cancer can be simply seen as a disease that is driven by uncontrolled or inappropriate cellular growth, which is evoked by distinct defects in major signal transduction pathways and their critical components. All of which contribute to the mechanical phenotype of cancer cells. However, we have to keep in mind that there exists a broad heterogeneity of cancer types and a general conclusion from one cancer type to another can only be drawn with great caution. Moreover, the molecules that regulate the malignant progression of cancer such as increased migration and invasion of cancer cells can be end-products or components of several signaling pathways that provide the pathology of cancer. Hence, more systemic analysis of the signaling pathways may help to decipher the mechanical phenotype caused or supported by them. Another critical regulatory signaling event in the initiation and further development of cancer in various organ systems is apoptosis (Thompson 1995). At the first glance oncologists seem to be focused on proliferation and cell cycle kinetic enhancements, but the fight against cancer requires a deeper look inside the regulatory signaling pathways including the investigation of deregulated decreased apoptosis (Evan and Vousden 2001), which has also been proposed as a basic hallmark of cancer (Hanahan and Weinberg 2011).
Why is it so important to understood signal transduction pathways in cancer diseases? When a pharmacological drug is applied to the patient, it develops its function through interaction with signal transduction pathways that follow a non-linear propagation and affects multiple networks, which belong to interconnected multiple signaling transduction pathways. Each cancer type and even each patient may require a different treatment, as other signaling pathways play a role and also the microenvironment may have a different impact. The knowledge of the structure and assembly of such signaling pathways allow us to detect and select distinct novel drug targets within the pathway and helps us to understand the individual effect of a certain drug on the signal transduction pathways. Moreover, it is even necessary to extract critical components within intertwined pathways and integrate them into a single network (Brambilla and Gazdar 2009). Indeed, there have been performed extensive molecular analysis of human lung cancers in order to reveal specific genes and pathways (Ding et al 2008) that can be targeted for drug design and additionally through genome-wide screenings multiple genetic (Weir et al 2007, Thomas et al 2007) and epigenetic alterations have been identified such as over 20 alterations for each lung tumor type (Sekido et al 2003) (figure 4).
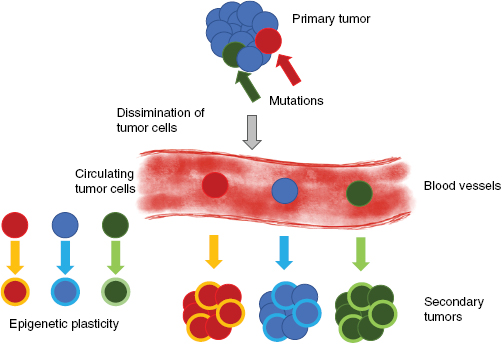
Figure 4. Heterogeneous primary tumors and CTCs. Specific cancer cells disseminate from the primary tumor into the extracellular matrix tissue environment and intravasate into peripheral blood or lymph vessels. With the vessels, the cells are termed CTCs. During cancer progression the cells may undergo epigenetic alterations (termed epigenetic plasticity) and thus secondary tumors such as metastases contain mutated and epigenetically altered, plastic cancer cells.
Download figure:
Standard image High-resolution imageBesides classical target genes for tumors, a class of small non-protein-encoding RNA molecules (termed miRNAs) have been identified as additional tumor therapeutic targets (figure 5). They consist of approximately 22 nucleotides and are capable of regulating the gene expression through up- or down-regulation of specific mRNAs, which is facilitated by direct base-pairing interactions and subsequent degradation of double stranded RNA representing an intrinsic defense mechanism of cells (Cowland et al 2007, Barbarotto et al 2008, Boyd 2008). Multiple miRNAs possess expression patterns that are tissue-specific (termed miRNA fingerprints). These miRNA expression patterns are dysregulated in many cancer types such as lung cancer (Takamizawa et al 2004, Nana-Sinkam and Geraci 2006, Yanaihara et al 2006). miRNA have been identified as tissue-based biomarkers and may be altered during cancer initiation and progression. When miRNAs are released by cancer cells present in the blood system, they can serve as potential tumor markers for diagnosis. Approximately 500 human miRNAs have been found and many are still not yet revealed, and they have been suggested either to target the function of oncogenes and tumor suppressors or act themselves as oncogenes and tumor suppressors that are deregulated in distinct cancer types such as leukemia or lung cancer (Hammond 2015, Svoronos et al 2016). However, only a small group of miRNAs have been identified as possible targets for cancer therapy such as the miR-34 miRNA, which has been found to be reduced in expression or contain minimal deletions in distinct lung cancer cell lines (Calin et al 2004, Bommer et al 2007) (figure 5).
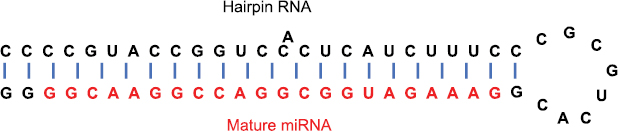
Figure 5. Structure of a mature miRNA. The mature miRNA consists of 22 nt and forms a hairpin-loop RNA with targeted mRNAs. Mature miRNAs can act either as oncogenes or tumor suppressors.
Download figure:
Standard image High-resolution imageThe following signal transduction pathways introduce briefly into the field of cancer research with a focus on their connection to mechanical properties of cells and their microenvironment. They explain through which signal transduction events certain downstream targets in these pathways are deregulated and subsequently affect the regulation of cellular motility.
1.3.1. Introduction into major signaling pathways affected by cancer regulating mechanotransduction.
The insights into cancer biology of humans have established cancer as a disease, which is facilitated by genetic mutations. Several recent molecular biological technical advances have revealed many new insights into the field of human cancer biology, which has been manifested cancer as a disease based on distinct mutations. Many tumor models are focusing on individual or family germline mutations such as BRCA1 and BRCA2 mutations in breast/ovarian cancer and have been related to hereditary cancer syndromes (Tyrer et al 2004), which are inherited by autosomal dominant mechanism.
Beside the well-known inherited family tumor syndromes that have been characterized by specific by germline mutations, the somatic genetic alterations have become the focus of cancer research, as they have been identified to be present at the onset of tumorigenesis and even shown to be increased in numbers during malignant tumor progression or during cancer treatment. As tumors adapt further somatic mutations during cancer treatment and cancer progression, it has been started to characterize the molecular and genetic profile of individual tumors. These somatic mutations are observed upon DNA damage evoked by environmental carcinogens or mutations based on replication errors during cell division. The somatic mutations can on the one hand cause gain-of-function mutations by expressing oncogenes that promote tumor formation or progression, whereas on the other hand lead to inactive tumor suppressor genes that then no longer repress excessive proliferation and survival of cells outside of their normal physiological space in a distinct tumor niche.
In general, primary tumors consist of cancer cells with hundreds or thousands of mutations, which are mainly not essential for tumor growth or progression and hence termed 'passenger' mutations (Vogelstein et al 2013). Among these enormous numbers of mutations are only two to eight mutations such as point mutations in Ras, deletions in the phosphatase and tensin homolog deleted on chromosome 10 (PTEN) or amplifications in Myc that are mutations supporting the tumor growth and progression and thus they are named 'driver' mutations (Vogelstein et al 2013). In addition, these driver mutations include large-scale rearrangements in chromosomes such as chromosome 9 and 22 function that emerge an oncogenic tyrosine kinase Abl (in certain leukemias) and gene conversions or mitotic recombinations, which subsequently lead to loss of heterozygosity mainly in certain tumor suppressor genes such as retinoblastoma protein or TP53. Epigenetic mutations represent also changes in the methylation state of promotors affecting the expression of distinct genes that play a role in oncogenesis (Sandoval and Esteller 2012, Suva et al 2013). Thus, the epigenetic silencing of genes is more often detected compared to mutational silencing of genes involved in cancer. Hence the tumor microenvironment plays an important role in regulating cancer progression (Brabek et al 2010, Mierke et al 2018).
The knowledge of the molecular tumor profile before, during and after drug treatment should enable us to fight cancer directly and adapt the concept of treatment based on the individual communication between the tumor and the drug treatment (Birner et al 2016). However, the mechanical characterization of tumors has not yet been started systematically, but may also have benefits in individual tumor treatment, as the mechanical properties affect cellular functions (Fletcher and Mullins 2010, Fischer et al 2017, Xie et al 2018). Signal transduction pathways acting in homeostatic processes are likely to be dysregulated in cancer such as altered tyrosine kinases and intracellular signaling molecules. However, single oncogenic protein alterations cannot be the target of treatments as they are involved in complex signal transduction pathways, which are characterized by feed-back-loops and crosstalk that alter the therapeutic efficiency. Hence, combinations of drug treatments such as drug codevelopment approaches may provide a better response to a certain cancer therapy and finally decrease tumor drug resistance. Therefore, it is necessary to gain novel biomarkers for tracking the success of a treatment and performing a pre-selection of patients for a distinct treatment concept (Yap et al 2013). Besides these intrinsic mutations, a small group of distinct cancers is induced by infections through viruses such as human papilloma virus that encodes genes favoring tumorigenesis via the action of oncogenes or inactivation of tumor suppressor genes (Munger and Howley 2002).
Tumorigenic mutations can affect signaling pathways that are activated in physiological tissue growth stimulations through the growth factor receptor tyrosine kinases (RTKs) such as epidermal growth factor receptor (EGFR), the small GTPase Ras, the cytoplasmic tyrosine kinase Src and the phosphoinositide 3-kinase (PI3K) (figure 6). Developmental signaling pathways such as Wnt, Hedgehog (Hh), Hippo, and Notch can also be affected including their downstream targets such as cell cycle effectors (the cyclins). In addition, many tumor suppressors can function as negative regulators in signal transduction pathways such as the adenomatous polyposis coli protein (APC) impairing Wnt signaling pathway and the phosphatase PTEN hindering the PI3K-Akt signaling pathway (figure 6).
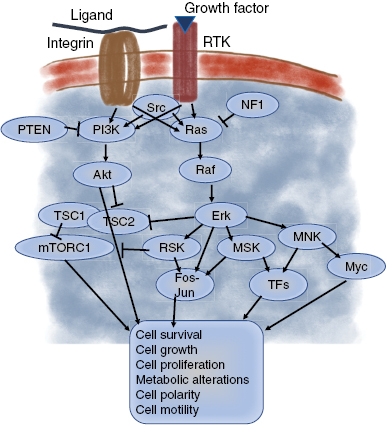
Figure 6. Receptor-tyrosin growth factor and integrin signaling pathways. The signaling can be through PI3K-Akt or Ras-Erk signal transduction pathways and finally causes a multiple set of functions such as cell survival, growth, proliferation polarity and motility.
Download figure:
Standard image High-resolution image1.3.2. PI3K-Akt-mTOR signaling pathway in cancer and mechanotransduction.
The PI3K-AKT-mTOR signaling pathway is known to be a major regulator of cellular physiology under normal conditions and is challenged under cancerous conditions such as human squamous cell carcinomas (Janus et al 2017), brain cancer (Mantamadiotis 2017), prostate cancer (Crumbaker et al 2017), acute myeloid leukemia and multiple myeloma (Piddock et al 2017). The PI3K-AKT-mTOR regulates cellular division by controlling the cell cycle, cellular metabolism, angiogenic processes, survival of cells, cellular motility, chemoresistance, intracellular membrane traffic and genomic instability (Mabuchi et al 2015, Marat and Hauke 2016). In more detail the class I of PI3K deals with the proliferation of cells, immunological reactions, the signaling of insulin and inflammatory processes (Mabuchi et al 2015). Under pathological conditions PI3K is often mutated in human cancers such as ovarian cancer (Gyori et al 2017, Aziz et al 2018). The class II of PI3K determines the membrane trafficking and the class III of PI3K supports the process of autophagy (Dobbin and Landen 2013). More precisely, PI3K can convert PIP-2 to PIP-3 that enables AKT and PDK1 to co-localize in close proximity to the cell membrane. Due to close neighborhood, PDK1 and also mTOR can phosphorylate the serin/threonine kinase Akt (synonymously termed protein kinase B) at Thr-308 and at Ser-473, respectively. The phosphorylated Akt can then directly activate mTORC1 or contrarily impair the TSC1/2 complex and thereby block the activation of mTORC1 (Cheaib et al 2015, Mabuchi et al 2015). However, Akt can also act together with Proviral-integration site for Moloney-murine leukemia virus proteins (PIMs) such as PIM1, PIM2 and PIM3, which are grouped to a family of serine/threonine protein kinases and are regualted by Ca2+/calmodulin protein kinases (Blanco-Aparicio and Carnero 2013). Hence, PIM and Akt regulate cellular growth and translation by employing mechanisms that partially overlap and thereby they phosphorylate multiple substrates that regulate mTORC1 (Warfel and Kraft 2015). In many cancer types such as prostate and endometrial cancers melanoma and glioblastoma (Cantley and Neel 1999, Rivas et al 2018), Akt fulfills a major function in cancer progression and hence the PI3K-Akt signaling pathway has been studied with major effort (Manning and Cantley 2007). Finally, Akt1 has been established as an oncogene (Carpten et al 2007). Indeed, this signaling pathway regulates the proliferation of cancer cells (Skeen et al 2006, Korkaya et al 2009) and contributes to the overall cancer phenotype (Nogueira et al 2008), which also seems to include the mechanical phenotype of the cancer cells. In line with this, Akt has been reported to be activated in several human cancer such as prostate and endometrial cancers melanoma and glioblastoma (Bhaskar and Hay 2007). Akt is also hyper-activated that can be regulated directly by its over-expression or mutation of Akt or even indirectly through alterations of PTEN, which overcomes apoptosis and partly induces the progression of the cell-cycle (Kandel et al 2002) All of which are major hallmarks of cancer (Hanahan and Weinberg 2011, Hambardzumyan et al 2008).
In glioblastoma (GBM) cells, the PI3K-AKT-mTOR signal transduction pathway represents a main transformed phenotype and is associated with the loss of tumor suppressor PTEN (O'Neil et al 2009, Populo et al 2013). The mechanistic target of rapamycin (synonymously termed mTOR) kinase can be located in two cellular multiprotein complexes, which are named mTORC1 and mTORC2 and possess each a distinct subunit composition, specific substrates and distinct functional mechanisms. Thus, targeting of the mTOR pathway has been suggested to be efficient in GBM therapy. In contrast, it has turned out that none of these two complexes is totally impaired by the allosteric mTOR inhibitor such as rapamycin or analogs. Thus, the ATP-competitive inhibitor PP242 seems to be highly suitable to inhibitor both mTORC1/2 and subsequently decrease GBM growth and cancer cell dissemination (Populo et al 2013). Indeed, GBM cells treated with PP242 showed a significant decreased activation of mTORC1 and mTORC2. Moreover, the insulin facilitated activation of mTORC1 and mTORC2 has been reported to be abolished upon pretreatment with PP242. This phenomenon is due to the fact that PP242 leads only to a moderate activation of the extracellular regulated kinase (ERK1/2). Indeed, the cell proliferation of GBM cells was reduced by PP242 and further decreased by combined treatment with rapamycin. Subsequently, PP242 impairs the motility of GBM cells due to altered cellular behavior, whereas cytoskeletal remodeling is not regarded to be affected. These findings revealed that PP242 seems to have a therapeutic effect in GBM growth and its dissemination into the surrounding tissue. Moreover, the targeting if this pathway may also change the mechanical properties of the cells and secondly also of their microenvironment (Rubashkin et al 2014). Hence, this needs to be included in future investigations. In fibrosarcoma cells such as HT1080, it has been shown that Akt increased the invasion by enhancing cellular motility and elevating the production of the matrix metalloproteinase-9 (MMP-9) that is strongly dependent on Akt activity and the ability to translocate to the plasma membrane (Kim et al 2001). Indeed, the extracellular matrix has been found to be restructured and mechanically stiffened during the malignant progression of cancer (Gehler et al 2009, Levental et al 2009, Egeblad et al 2010, Lopez et al 2011, Plodinec et al 2012). Increased cross-linking of collagen causes a stiffening of the extracellular matrix environment in vivo and in vitro and in turn elevates the phosphorylation of focal adhesion kinase (FAK) at amino acid 397 that and supports the progression of the mammary tumor. However, when the cross-linking of the extracellular matrix can be prevented and the tension of the tissue causes decreased FAK activity, the invasion of cancer cells and subsequently cancer metastasis can be impaired (Levental et al 2009, Pickup et al 2013). As the extracellular matrix mechanical properties determine the activation state of FAK (Provenzano et al 2009), the FAK-ERK signaling pathway also plays a role and possibly also impacts mechanotransduction processes in cancer cells.
1.3.3. Crosstalk between the Ras-ERK and PI3K-Akt signaling pathways and impact on mechanotransduction.
The Ras-extracellular-regulated kinase (ERK) signaling pathway (termed also RAS–RAF–MEK–ERK signaling pathway and receptor tyrosine kinase (RTG)-Ras signaling pathway) has been determined to be hyperactivated in a broad variety of tumors that even carry the most frequently activating mutations in the KRAS, NRAS and BRAF genes. Moreover, these pathways target molecules involved in cellular mechanics and may hence contribute to the invasive phenotype of cancer cells. In concrete terms, stress fibers themselves can serve as a platform for tension-induced activation of biochemical signals. The MAP kinase, ERK, becomes activated on stress fibers as a function of myosin II. However, when myosin II is inhibited in cells, uniaxial stretching of the cell adhesion substrates causes the restoration of ERK activation to stress fibers (Hirata et al 2015).
Many of the mutated genes of cancer types target components of the PI3K-Akt and Ras-ERK signaling pathways. As response to growth factors and ligand facilitated activation of cell-matrix adhesion receptors such as integrins, both signal transduction pathways are usually activated, whereas genetic alterations cause constitutive signal transduction activation that is even present in the absence of external stimuli such as growth factors. The constitutive active PI3K-Akt pathway is obtained on the one hand by amplification or activating mutations that target several PI3K-Akt-pathway proteins such as the type I PI3K isoform PIK3CA (p110a), Akt, and the adaptor protein PIK3R1 to activate them and on the other hand by deletion or inactivating mutations that target in the phosphatases, which hydrolyze PI3K products such as phosphatidylinositol 3,4,5-trisphosphate (p1p3), PTEN and INPP4B tumor suppressors to inhibit them. Moreover, further downstream in the PI3K-Akt pathway, mutations in the tumor suppressors TSC1 and TSC2 lead to hyperactivation of the PI3K-Akt pathway through increased mTORC1 signaling (Laplante and Sabatini 2012), which is a major target protein therein. Similar to the PI3K-Akt pathway, the Ras-ERK pathway can be constitutively activated by two mechanisms: one mechanism is based on mutations in Ras or its target protein Raf and the other mechanism is based on the inactivation of GTPase-activating proteins (GAPs) such as NF1 (Cichowski and Jacks 2001), DAB2IP (Min et al 2010), and RASAL2 (McLaughlin et al 2013) that facilitate the GTP hydrolysis, when GTP is bound to Ras. Another down-stream target protein of the Ras-ERK signaling pathway is Myc that is also a component of many other signaling pathways and is amplified or overexpressed in distinct cancer types (figure 6). Besides its binding the promotor regions of genes, Myc can affect gene transcription of genes without Myc binding sites in their promotors, as it prolongs the transcriptional activity of the polymerase II beyond the targeted gene. Thus, Myc can act primarily as a universal amplifier of expressed genes rather than as an initiator of de novo transcription (Rahl et al 2010, Lin et al 2012, Nie et al 2012). In summary, oncogenic mutations, amplification or gene fusions of RTKs such as EGFR, ErbB2, fibroblast growth factor receptor (FGFR), and platelet-derived growth factor receptor (PDGFR) facilitate constitutive active Ras-ERK and PI3K-Akt signaling pathways. These altered intracellular signaling may change the cellular mechanical properties and subsequently the invasive phenotype. A similar oncogenic activation of the two signaling pathways has been found by mutations in G-protein-coupled receptors (GPCRs). Hence, substances in the tumor microenvironment activate biochemically the progression of cancer by stimulation of these receptors. Simultaneously, the matrix mechanical properties of connective tissue surrounding cancer cells are altered and they can in turn interact with cancer cells that migrate out of the primary tumor and support the formation of secondary tumors to metastasize. The motility and invasiveness of cancer cells can be further induced by the matrix mechanical properties that cause for example a contraction of the stress fibers within cancer cells. This mechanical stimulation causes then the activation of the mitogen-activated protein kinase-extracellular signal-regulated kinase (MAPK-ERK) signaling pathway that facilitates the activity of transcription factors, which control the genes that are required for cell proliferation and differentiation (DuFort et al 2011, Aguilar-Cuenca et al 2014, Fedorchak et al 2014, Feller et al 2015).
In conclusion, the identification of dysregulated synthesis of growth factors is considered to be responsible for many cancer types. Through the dysregulated synthesis of growth factors on cells expressing appropriate amounts of the growth factor receptors can cause an autocrine loop-based mechanism that decreases the expression of these receptors on the cell's biomembrane surface. As an alternative, the growth factors can be secreted directly in the matrix environment of tumors, where they are bound to extracellular matrix components that can be released by aggressive and invasive cancer cells (Bonnans et al 2014). The autocrine loop can be performed the cleavage and release of anchored soluble growth factors driven by surface expressed a disintegrin and metalloproteinase (ADAM) proteases that are themselves activated by oncogenic signaling pathways (Turner et al 2009). An alternative pathway for the autocrine loop is a paracrine stimulation, which means that neighboring cells synthesize growth factors. Under both conditions, the both the Ras-ERK and PI3K-Akt signaling pathways are enhanced activated.
It has been shown that a mechanical stress stimulates cells such as chondrocytes (Whitney et al 2012). In more detail, ultrasound has been used to mechanically exert stress on chondrocytes. Thereby, intracellular signaling through mechanotransduction processes are activated that transfer extracellular mechanical stress into gene regulatory mechanisms. How this is precisely performed requires more investigation. However, it is revealed that the ultrasound mechanical probing of chondrocytes causes the phosphorylation of FAK, CrkII, and Erk, Src, p130 Crk-associated substrate (p130Cas), that are all key players in Ras-ERK and PI3K-Akt signaling pathways. In addition, it has been found that the impairment of integrin receptors, Src, and MAPK/Erk kinase (MEK) by using pharmacological drugs decreased the effect of the ultrasound facilitated increased phosphorylation of Erk, which leads to the suggestion that integrins and Src act upstream of Erk (Whitney et al 2012). Moreover, these results show that mechanical stimulation through ultrasound is sensed by integrin receptors and transduced through the MAPK/Erk pathway (Whitney et al 2012). These results are in line with the finding that expression levels of the integrin subunits α5 and β1 in chondrocytes are elevated after exposure to a continuous ultrasound stimulation with 5.0 MHz (0.14 mW cm−2) (Hasanova et al 2011). Besides the Ras-ERK and PI3K-Akt signaling pathways, the TGF-beta signaling pathways may also have an impact on mechanotransduction processes in cells and on the microenvironment of cancer cells.
1.3.4. TGF-beta signaling pathways and their impact on the cancer surrounding matrix mechanical properties.
Mechanical stress such as from a primary tumor on TGF-β1 can switch it from an insoluble molecule to a soluble. When myofibroblasts are required for the generation of stiff fibrotic tissue around tumors and when this rigid extracellular matrix is critical for the differentiation of myofibroblasts, what is the initial event? It has been shown that the contraction of myofibroblasts facilitates the activation of latent TGF-β1 (Wipff et al 2007). In more detail, the unbinding of TGF-β1 from their extracellular matrix enables the chondrocytes to bind the growth factor that directly turns cellular mechanical forces into biochemical signals. As TGF-β1 is seen as a major regulatory factor in physiological tissue repair and in pathological development of fibrosis that has been discussed to play a role in malignant cancer progression (Mierke et al 2018), it represents a candidate for regulating the invasive phenotype of cancer cells. When TGF-β1 is unbound from its storage, it facilitates the inflammatory response that leads to increased production of extracellular matrix components, elevated synthesis of tissue inhibitors of metalloproteinases, reduced synthesis matrix degrading proteases synthesis, which subsequently causes the differentiation of myofibroblasts (Hinz et al 2007, ten Dijke et al 2007). In general, TGF-β1 signaling provides tissue homeostasis through the regulation of the proliferation of various cell types such as epithelial cells, endothelial cells, immune cells, and fibroblasts (ten Dijke et al 2007, Wakefield and Stuelten 2007, Jenkins 2008). Hence, an inhibition of TGF-β1 in order to treat fibrosis is not recommendable as it will be uncontrollable due to diverse side effects (Varga and Pasche 2008).
There are several mechanisms that cause the activation of latent TGF-β1 and thereby may support a cell-specific inhibition of TGF-β1 in order to reduce the side effects. For example, latent TGF-β1 can be activated by dissociation from the latency associated peptide (LAP), which is co-synthesized and associated to TGF-β1 (Jenkins 2008). Commonly, the cell types can secrete TGF-β1 that is a part of a large latent complex containing TGF-β1, LAP and the latent TGF-β1 binding protein (LTBP-1) (Jenkins 2008). In more detail, LTBP-1 belongs to the fibrillin family of extracellular matrix proteins. It can bind several other extracellular matrix components such as fibronectin, vitronectin and fibrillin-1 that ensures a continuous reservoir of latent TGF-β1 in the matrix microenvironment (Jenkins 2008). The latent TGF-β1 is activated by dissociation from LAP that is evoked by diverse mechanisms due to cell type and the physiological conditions. The activation of TGF-β1 can be trigged in several ways such as by proteolytic cleavage, interaction with thrombospondin 1 and interaction with mannose-6-phosphate receptor (Jenkins 2008). In addition, cell-matrix adhesion receptors such as integrins facilitate the connection between the cell through extracellular transmembrane components coupling the cell to the extracellular matrix via focal adhesions and thereby regulate the activation of latent TGF-β1 (Sheppard et al 2005, Wipff and Hinz 2008). There exist at least two mechanisms for the activation of growth factors through integrins. The first mechanism of TGF-β1 activation depends on proteolytic cleavage and is hence sensitive to protease inhibitors. Moreover, integrins such as αvβ6 are supposed to be a common binding partner for latent TGF-β1 and proteases that activate latent TGF-β1 during the onset of lung tissue fibrosis (Annes et al 2004, Jenkins et al 2006). Moreover, knockout of αvβ6 integrin leads to a phenotype in mice that is similar to the phenotype of the TGF-β1 knockout (Yang et al 2007). In line with this, in β6 knockout mice, the incubation of their lungs with bleomycin cannot induce fibrosis (Sheppard et al 2005).
In contrast, the second mechanism is not dependent on proteolytic reaction and instead requires the exertion of cellular traction forces, which are transmitted through integrins towards the latent complex (Sheppard et al 2005, Jenkins 2008, Wipff and Hinz 2008). However, it has solely been suggested that cell-generated forces function in the αvβ6 integrin-driven latent TGF-β1 activation, as simply the incubation of purified αvβ6 integrin with latent TGF-β1 cannot release the active TGF-β1. Additionally, it has been shown that firstly the disruption of actin filament bundles using cytochalasin D and secondly truncation of the cytoplasmic tail of the αvβ6 integrin, which couples the integrin to the cell`s cytoskeleton, both impair the activation of latent TGF-β1 (Sheppard et al 2005). Subsequently it has been found that β6 integrin-transfected fibroblasts that overexpress either constitutively active or dominant-negative forms of the small guanosine triphosphatase RhoA support that the contractile cytoskeleton plays a major role in the activation of latent TGF-β1 (Jenkins et al 2006). In more detail, the activation of RhoA is a crucial for the actin-myosin contraction and hence the level of active RhoA correlates with the level of active TGF-β1 (Jenkins et al 2006), which is an example for cellular mechanical properties that provide the progression of diseases such as fibrosis or cancer. During the process of fibrosis in epithelialized tissues such as kidney and lung tissues, αvβ6 integrin-dependent activation of latent TGF-β1 seem to contribute to the generation of myofibroblasts through the transition of epithelial to mesenchymal phenotypes of cells (Kim et al 2006). This transition is also dependent on mechanical properties such as cellular jamming (Sadati et al 2013, Oswald et al 2017, Mierke et al 2018). Contrarily, during the progression of pulmonary fibrosis towards end stage of the fibrotic lung disease, the αvβ6 integrin-negative myofibroblasts persistently express and activate the latent TGF-β1. Myofibroblasts facilitate the progression of fibrosis in various organs that have even no pronounced epithelium and display no αvβ6 integrin expression.
The mechanical stress dependent activation of latent TGF-β1 has been shown to be a function of the contractility and stiffness of the extracellular matrix. By increasing the contraction of myofibroblasts by the stimulation with thrombin, endothelin-1 or angio-tensin-II evokes the release of active TGF-β1 from the extracellular matrix microenvironment (Wipff et al 2007). Based on these results, a model has been developed for the direct integrin-dependent activation of latent TGF-β1 in contractile myofibroblasts. In the first step, integrins of the myofibroblasts provide a connection between the extracellular LAP content in the stored complex and contractile stress fibers inside the cytoplasm of the cells. In the second step, the stress fiber-based forces exert a pulling force on the latent complex through the connections to the integrins. In the third step, the connection of LTBP-1 to the extracellular matrix tissue withstands mechanically the pulling on the complex, which finally causes the opening of the complex to release active TGF-β1.
Mechanical properties of the microenvironment play a role in TGF-β1 activation. The physical connection between TGF-β1 and matrix environment is mediated through the LTBP-1 within the complex. In more detail, mutant LTBP-1 that consists of solely the LAP-binding domain and the extracellular matrix-binding hinge region can fulfil the function of full-length LTBP-1 regarding the integrin-facilitated activation of latent TGF-β1 (Annes et al 2004). In turn deletion of one of these two regions impairs the function of LTBP-1 (Annes et al 2004). However, the activation of latent TGF-β1 by the contraction of myofibroblasts depends on the mechanical properties of the matrix microenvironment. More precisely, when the microenvironment is soft (a Young's modulus around 5 kPa), the contraction dependent mechanism is not employed (Wipff et al 2007). This threshold is even lower than the minimal stiffness that is needed to assemble α-SMA into stress fibers (16 kPa) (Goffin et al 2006). It seems to be likely that the matrix stiffness increases with the remodeling of myofibroblasts during the initiation and progression of fibrosis (Hinz 2006). Moreover, it can be hypothesized that the stress-dependent activation of latent of TGF-β1 is critical for the development of fibrosis and its progression, as the adaption of myofibroblasts by their differentiation towards a remodeled and hence contraction-stiffened microenvironment is thereby facilitated.
In turn, the mechanical properties of cells play a role in TGF-β1 activation. Another requirement for the activation of latent TGF-β1 through the contraction of myofibroblasts is that the intracellular force is transferred to the large latent complex by integrins. All integrins activating the latent TGF-β1 (directly or indirectly by proteolysis induction) interact with LAP of this complex (Sheppard et al 2005). In line with this, the impairment or the deletion of the Arg-Gly-Asp (RGD) motif, which is an integrin binding sequence, in LAP abrogates the activation of latent TGF-β1 cells such as epithelial cells similarly to the TGF-β1 knockout phenotype (Yang et al 2007). In myofibroblasts the inhibition of RGD in LAP also abolishes the activation of latent TGF-β1. Although myofibroblasts display no αvβ6 integrins, it has been shown that other LAP binding candidates such as the integrin αvβ5, and to some extent also β1 and αvβ3 integrins can be impaired by using function-inhibiting antibodies directed them, which diminished their ability to facilitate the activation of latent TGF-β1 by contraction-dependent force exertion (Wipff et al 2007). Moreover, integrins αvβ5 and αvβ3 are reported to be upregulated in systemic sclerosis fibroblasts and their impairment by deletion or inhibition diminishes the latent TGF-β1–based differentiation of myofibroblasts (Asano et al 2006). Finally, the TGF-β1 signaling pathway is an example for the mechanical impact of the extracellular matrix on regulating functions of cells embedded in the connective tissue.
1.3.5. Notch signaling is linked via YAP/TAZ to cell mechanics.
Yes associated protein/PDZ-binding protein (YAP/TAZ) regulates the size of organs in embryonic development by employing the induction of the amplification of progenitors within several tissues such as the epidermis (Pan 2010, Schlegelmilch et al 2011, Zhang et al 2011, Ramos and Camargo 2012, Low et al 2014). Moreover, YAP/TAZ are reported to be crucial transducers of mechanical signals in cells (Dupont et al 2011, Wada et al 2011, Aragona et al 2013). Indeed, cells, in which YAP/TAZ is active, can sense the rigidity of the extracellular matrix microenvironment: on a stiff substrate the cells spread and display a tensed cytoskeleton, whereas on a softer substrate the cells detach or display only small adhesive areas (Halder et al 2012). Hence the mechanical regulation of YAP/TAZ in epidermal progenitor cells seems to be a mechanism that can be affected by the structure and the matrix mechanical phenotype of the tissue microenvironment. How is the mechanical regulation of YAP/TAZ mediated on short length scale interactions such as the communication of neighboring cells? How does this impact the cells fate? Within the epidermis, the paradigm of such a communication is the Notch signal transduction pathway. In more detail, the activation of Notch is crucial to support the differentiated state in suprabasal cells, whereas in basal cells, the Notch signaling needs to be impaired (Blanpain et al 2006, Rangarajan et al 2001, Nowell and Radtke 2013). The contrasting functions of YAP/TAZ and Notch signaling in terms of epidermal cell fate have not been reported earlier. In fact, mechanical signals have been found to employ YAP/TAZ to regulate the Notch signal transduction pathway (Totaro et al 2017). At the transcriptional level, the YAP/TAZ can control the expression of Notch inhibitors such as the epidermal somatic stem cell factor DLL1 that diminishes the Notch signaling 'in cis', which thereby protects the undifferentiated cell state (Lowell et al 2000, del Alamo et al 2011, Palmer and Deng 2015). In summary, the YAP/TAZ mechanotransduction processes connect the regulation of Notch signaling with structural and physical signals such as the coordination of mechanosensitive pathways, cell-cell interactions and maintenance of the somatic stem cell state.
1.3.6. Wnt signaling pathway and its contribution to mechanotransductive forces.
The well-known Wnt signaling pathway facilitates the regulation of a broad variety of cellular processes such as survival, proliferation, differentiation, polarity, migration and wound repair (MacDonald et al 2009, Tsaousi et al 2011). The Wnt pathway is highly complex, as there are 19 Wnt ligands, ten Frizzled receptors and multiple coregulatory proteins. The canonical and non-canonical pathways have been found to respond to mechanical forces in cell such as mesenchymal stem cells (Kuo et al 2015) and osteocytes (Hu et al 2015) and function in the maturation of vessels and during tissue repair in zebrafish (Li et al 2014). During the formation of the lymphatic valve the oscillatory shear stress caused the enhanced activation of beta-catenin and leads to the expression of beta-catenin regulated genes that facilitate directly the valve formation (Cha et al 2016). For example, in endothelial cells of developing cardiac valves it has been demonstrated that Krüppel-like Factor 2 (KLF-2) regulates the expression of Wnt-9a due to shear stress alterations (Goddard et al 2017). In contrast, when endothelial cells are exerted to unidirectional, laminar shear stress, the non-canonical pathway involving Wnt-5a and Wnt-11 has been determined to impair the axial polarization of the cells and diminish their sensitivity to flow, which is independent of downstream effects of KLF-2/KLF-4 signaling (Franco et al 2016). Hence, it can be concluded that lymphatic, valvular and vascular endothelium respond differently towards shear stress, as the Wnt-signaling pathway is regulated in different ways.
It has been reported that the Wnt signaling can be altered in cells such as adult endothelial cells, when an external flow is applied that has been detected in multiple, microarray studies, in which shear stress altered the expression of genes (Maimari et al 2016). Upon shear stress, several members of the canonical Wnt signaling pathway have been found to be altered in their expression such as Porc, Wnt-1, Frizzled-1, TCF/LEF, and cyclinD. Additionally, members of the non-canonical Wnt-5a/Ca2þ pathway such as Frizzled-1, PLC and PKC and of the non-canonical Planar Cell Polarity pathway such as Frizzled, Prickle, and RAC have been demonstrated to be altered. However, it still needs to be investigated how the amount of shear stress altered the members of these pathways.
Moreover, the prominent Wnt signaling pathway plays a central role in at least two physiological processes such as the development and homeostasis of tissues that is facilitated by the precise regulation of endogenous stem cells. In multiple cancer types, the aberrant Wnt signaling is associated with the onset of cancers and their malignant progression by reprogramming the cancer stem cells (CSCs). The CSCs are needed for the formation of the primary tumor and the relapse of the cancer disease, when by-passing drug treatment by development of resistance. Thus, the development of new therapeutic treatments that affect the Wnt signaling pathways seem to be highly needed in order to diminish CSCs and avoid the recurrence of the cancer disease. However, the mechanical issue of this signaling pathway has been purely developed and requires to be further explored. The treatments of excessive Wnt signaling should be safe enough for normal somatic stem cells that undergo the Wnt pathway signaling in order to fulfill their physiological functions such as tissue homeostasis and wound healing processes after tissue injury. However, they are still specific enough for the elimination of dysregulated Wnt pathways in CSCs in solid tumors.
In order to understand why it is important to target dysfunctions of the Wnt pathway, it is necessary to understand the major parts of this signaling pathway. Akt phosphorylates GSK3 and thereby inhibits its function that subsequently stabilizes beta-catenin, a Wnt target gene. Thereby beta-catenin is induced to translocate into the nucleus, where it acts together with T-cell factor/lymphoid enhancer-binding factor1 (TCF/LEF1) as a transcription factor (figure 7) (Haq et al 2003, Korkaya et al 2009, Ma et al 2013). In the nucleus beta-catenin induces the transcription of target genes such as Myc and cyclin D, which both regulate cell proliferation. Another beta-catenin interacting protein is Akt that can phosphorylate beta-catenin and thereby induce its dissociation from E-cadherin-dependent cell-cell adhesions. This mechanism enhances the available amount of beta-catenin proteins for translocation in the nucleus and transcriptional activity (Fang et al 2007). When beta-catenin is not translocated into the nucleus, it is degraded by GSK3 (Polakis 2001, Korkaya et al 2009).
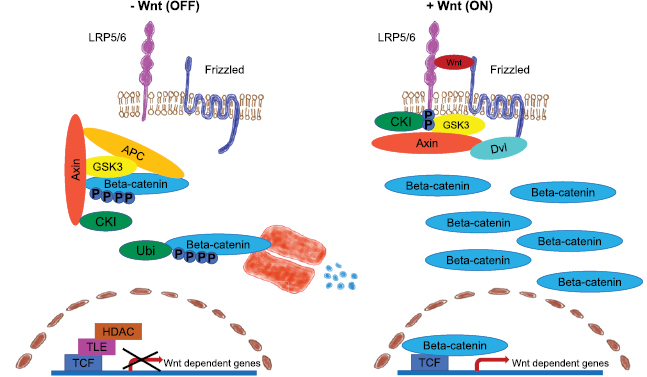
Figure 7. Wnt signaling pathway. (Left image) In the absence of wnt signaling, cytoplasmatic beta-catenin is in a complex with APC, Axin, GSK-3 and CKI. The cytoplasmatic beta-catenin is proteolytically degraded in the proteasome, when phosphorylated by CKI and GSK-3. Wnt target genes are prepressed. (Right image) Upon Wnt signaling, Frizzled and LRP5/6 form a complex. The recruitment of Dvl by Frizzled leads to phosphorylation of LRP5/6 by CKI and GSK-3 and Axin binding. The beta-catenin phosphorylation and degradation via Axin is impaired and beta-catenin translocates together with TCF in the nucleus and co-activates wnt target gene transcription.
Download figure:
Standard image High-resolution imageBesides the Wnt signaling pathway, the PI3K-Akt and Ras-ERK pathways facilitate cellular motility including migration and invasion via several downstream proteins that function as effectors (Cain and Ridley 2009). Firstly, Rho family GTPases such as RhoA, Rac1, Cdc42 and ARF6 are known to facilitate the regulation of cytoskeletal components such WAVE/WASP-family members, the actin branching protein actin-related protein 2/3 (Arp2/3) complex, actin interacting protein formins, the actomyosin contractile machinery, the kinase LIMK and cofilins (Raftopoulou and Hall 2004). Secondly cell matrix adhesion receptors such as integrins and associated intracellular focal adhesion proteins such as focal adhesion kinase (FAK), paxillin, vinculin or calpains regulate cellular motility (Mierke et al 2008a, Devreotes and Horwitz 2014). Thirdly, extracellular proteases that degrade extracellular matrix proteins promote the migration of cancer cells in confined space movements, as they enlarge pore-sizes and hence overcome the previous obstacle for cell migration. Moreover, they also decrease the number of adhesive sites within the surrounding matrix and thereby reduce cell-matrix adhesion strength, which enables cell migration. These cancer cell derived proteases can be secreted within the extracellular matrix and bind to these networks stably. Fourthly, the cell-cell adhesion sites affect pronouncedly the migratory capacity, as they provide stable protein interactions regulating cell-cell adhesion strength. Fifthly, transcription factors such as AP1 and Ets2 are activated and increase the expression of genes controlling the migration and polarity of cells such as matrix metalloproteinases (MMPs), plasminogen activator, cadherins, and actin regulators. These targeted molecules can affect also mechanical properties of the cells, when they alter structural components of the extracellular matrix microenvironment.
The outcome of alterations in certain genes that are a critical component of certain signaling pathways depends on the isoform type such as Akt1 that suppresses migration through inhibition of ERK, or phosphopalladin-induced actin bundling, whereas Akt2 promotes cell migration via regulation of integrin expression, which then performs an epithelial-mesenchymal transition (EMT) of the cells (figure 8) (Chin and Toker 2011). In a similar manner act some isoforms of ERK that target RSK: some isoforms facilitate cellular migration and invasion through increased transcription and activation of integrins, whereas others reduce cellular migration via remodeling of the actin cytoskeleton (Sulzmaier and Ramos 2013). This remodeling of the cell's actin cytoskeleton can also alter the mechanical properties of the cells and subsequently their invasive phenotype.
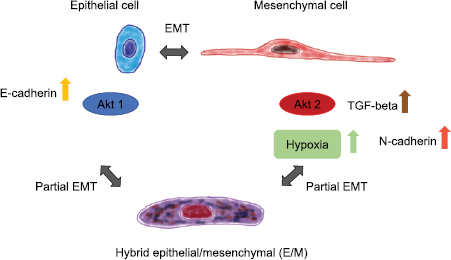
Figure 8. The epithelial to mesenchymal transition and partial transition is termed hybrid epithelial/mesenchymal (E/M) and represents an intermediated form of the two well-known classical phenotypes of cancer cells.
Download figure:
Standard image High-resolution imageThe knowledge of the pathology of cancer including the relevant signaling pathways that help to develop and manifest the metastatic phenotype in cancer cells, is essential for its diagnosis and treatment. However, it is still not yet well understood whether specific 'metastatic' genes can be identified that regulate the malignant progression of tumors. During the malignant progression of tumors, these 'metastatic' genes may be self-activated or activated by proteins such as activated oncogenes or inactivated tumor suppressor genes, which are themselves activated in earlier stages of oncogenesis. All these mechanisms contribute to the final metastatic phenotype of the tumor (Bernards and Weinberg 2002). Rho family of GTPases are found to be precise regulators of cellular motility and hence facilitate an invasive phenotype of cells (Jaffe and Hall 2002). Beside their function in cellular motility, the small Rho family members such as Rac, Cdc42 and RhoA are functional regulators of the actin cytoskeleton (Tapon and Hall 1997). As Rac is essential for lamellipodia-driven cell migration at the leading edge of the cell and therefore, it is regarded as the driving factor of cellular motility (Jaffe and Hall 2002), whereas Cdc42 does not seem to be required for the migration of cells. Moreover, Cdc42 has been observed to induce the polarity of cells and subsequently to determine directly the persistence of motion, which all have been analyzed mainly in 2D motility assays (Allen et al 1998, Nobes and Hall 1999). The GTPase Rho functions in the polymerization of F-actin stress fibers and is found to be colocalized in focal adhesions through its activation of the downstream effector mDia and ROCK I and ROCK II kinases (Amano et al 1997, Watanabe et al 1997). However, it is still not fully understood whether Rho is essential for the cellular motility (Allen et al 1998) by providing cellular contractility that is needed for certain modes of cell migration or whether it solely affects cellular motility indirectly by increasing cell adhesion strength through the up-regulation of F-actin stress fiber assembly (Cox et al 2001).
1.3.7. Role of RhoGTPases in mechanotransduction involved in cancer.
Among the members of the Rho family GTPases are activators and effectors of the cancer progression pathway. Under normal physiological conditions, cells utilize Rho family GTPases to perform cell migration and proliferation. In various cancer types, the levels of Rho protein family members such as Rho, Rac and CDC42, which are necessary for Ras transformation, are differentially expressed (figure 9). In several human cancer types, the expression of Ras is increased, which is provided in the first line by mutations blocking the hydrolysation of GTP and Rhod family members that are activated through alterations in upstream regulatory proteins. Rho family members have been revealed to be oncogenes by using in vitro cell culture assays. RhoGEFs contain DBl homology (DH) domains, which are closely associated with neighboring PH domains and other proteins that have the capacity to couple GEFs to upstream signal transduction pathways. In particular, Rac GEF Tiam1 possesses a Ras binding domain. In line with this, Tiam1-deficient mice exhibit resistance to Ras-facilitated activation of Rac and subsequently to Ras-triggered tumorigenesis (Lambert et al 2002, Malliri et al 2002). Moreover, pancreatic cancer cells grow anchorage-independent is facilitated by activated (G12V) K-Ras that requires the combined expression activated Ras and Tiam1. Hence, Tiam1 dependent activation of Rac is needed for the Ras transformation. Moreover, it has been shown that Rac can alter the mechanical properties of cancer cells (Fischer et al 2017) and fibroblasts (Kunschmann et al 2019).
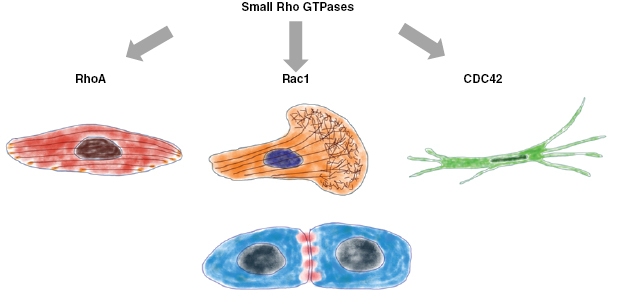
Figure 9. The main three small Rho GTPases are RhoA, Rac1 and CDC42. The precise regulation of these GTPases determines the phenotype of the cells and subsequently their migration mode.
Download figure:
Standard image High-resolution imageHow are integrin-based signal transduction processes coupled with growth factor facilitated signaling events to provide the proliferation of adherent cells? Among these mechanisms is one that involves the combined interaction of integrin and growth factor signaling pathways facilitated by targeting of Rac towards the cell membrane by the process of cell adhesion (del Pozo et al 2000). In particular, a mechanism is of high interest that involves alterations in the lipid rafts and caveoli at the plasma membrane through integrin signaling due to Rac activity (Ren and Schwartz 1998).
More than twenty members of the family of Rho GTPases are identified that have been shown to fulfill various cellular functions. The members of the Rho-family GTPases are small GTPases that have each a size in a range of 20–40 KDa. The most prominent family member is RhoA, which is highly related RhoB and RhoC. All these three GTPases have been subgrouped as RhoA subfamily and possess 88% amino acid identity. All of them can act as a regulator for common effector proteins such as Rho kinases (ROCK), which is involved in the regulation of cellular mechanical properties such as contractility, as it has been reported to play a role in activation of growth factors from the matrix microenvironment (Ridley 2015). Beside all similarities, RhoA, RhoB, and RhoC facilitate pronouncedly different effects on the migration and invasion of cancer cell migrations, which lead to the suggestion that they signal through different signal transduction pathways (Ridley 2015). In their active state, all three GTPases are bound to the cell membrane, whereas solely RhoB can additionally localize to endosomes (Ridley 2015). The various effects of the Rho family GTPases are based on the nume-rous interacting proteins. The assembly of actin stress fibers is regulated by RhoA through two classes of downstream targeted proteins, which can also affect the cellular mechanical properties and subsequently the invasive phenotype of cells.
Rho GTPases can be altered in their function by at least two classes of proteins. One class are the Diaphanous-related formins (DrFs), which are a subgroup of at least 15 identified formins, that facilitate the actin polymerization and microtubules formation. Another class are the ROCKs that facilitate the contraction of the actomyosin cytoskeleton through increased phosphorylation of the myosin light chain and hence increase the stability of actin filaments through the inactivation of cofilin, which acts as an actin filament severing protein. In particular, it has been shown that cancer cells possess at least two distinct migration modes, which mainly differ in their dependence on Rho facilitated signaling via ROCK. One migration mode is a protrusive migration that is based on lamellipodial driven motility facilitated by active Rac, which is associated with the decreased Rho-ROCK signaling. This migration mode has been detected in Src-transformed cells, in which cofilin is dephosphorylated. Moreover, in activated Ras transformed cells, diminished Rho-ROCK signaling is regulated by the activity of the MAPkinase ERK (Vial et al 2003). Following the signal transduction pathway, ERK provides the activation of the AP1 component Fra1 that then leads to an inactivation of integrin β1 facilitated signal transduction by Fra1 and causes subsequently the downregulation RhoA. Simultaneously, ERK facilitated signaling increases the expression of the urokinase plasminogen activator receptor (uPAR) that then causes and activation of Rac.
In contrast to this Rac-driven lamellipodial migration mode, the migratory capacity of certain cancer cell types depends on Rho-ROCK signal transduction processes (Sahai and Marshall 2003). Instead of a lamellipodial phenotype, these cells display a rounded so-called blebby morphology and possess a special polarized distribution of ezrin, when they invade into 3D extracellular matrices. Certain cancer cell types can employ both migration modes, the lamellipodial-based mesenchymal migration mode that utilizes proteolytic activity and the blebbing-driven mode that is proteolytic activity independent but relies on Rho-ROCK-dependent signal transduction processes. Based on these findings, the impairment of cellular motility requires both the inhibition of matrix metalloproteases and ROCK. Based on these results an efficient and promising therapeutic strategy that abolishes the metastatic capacity of cancer seems to be a simultaneously treatment of matrix metalloproteinases and ROCK.
The DrFs direct the assembly of actin filaments, which are polarized to straight actin filaments at their barbed end, as well as the assembly of microtubules and hence they are regarded as active scaffold mediators (Kühn and Geyer 2014). DrFs can be activated by small Rho GTPases. The DrFs are characterized by their autoinhibition function (resting state) that is mediated by the intramolecular interaction between the C-terminal Diaphanous autoregulatory domain (DAD) and its N-terminal recognition domain (termed FH3) (Watanabe et al 1999, Alberts 2001, Li and Higgs 2003, Lammers et al 2005, Nezami et al 2006), which subsequently switches the central actin polymerization domain from an active to an inactive state. The autoinhibition is reversible by interaction with a GTP-bound Rho, Rac or Cdc42 GTPase that leads to displacement of the DAD domain from the N-terminal FH3 recognition site and turns the DrF active for actin filament polymerization.
The autoinhibition seems to be activated by the interaction of DrFs with active Rho GTPases such as Rho, Rac and Cdc42. The GTPase binding domain (GBP) is located in the N-terminal region, upstream of FH3. All formins contain multiple domains, possess a weight of 140 kDa and are grouped together due to the presence of a formin homology 2 (FH2) domain. As the flanking regions of the FH2 domain of DrFs vary among different formins, they may explain the different cellular functions and regulatory processes of the formins. In particular, the FH2 domain interacts with G- and F-actin molecules and hence facilitates actin molecule nucleation, actin polymerization to filaments and their elongation by inhibition of the binding of actin filament capping proteins (Zigmond et al 2003, Moseley et al 2004, Goode et al 2007). The actin polymerization is increased by a proline-rich FH1 domain that interacts with profiling to assemble G-actin molecules (Romero et al 2004, Kovar et al 2006).
Other scaffold proteins are the class of WAVE/Scar proteins such as WAVE-1, -2, and -3 in mammals that facilitate Rho family GTPases driven alterations of actin cytoskeleton that subsequently can impact the mechanical phenotype of the cells. In particular, the WAVE/Scar proteins connect Rho family GTPases to the Arp2/3 protein complex that promotes the branching of actin filaments through the nucleation of actin filament assembly as a new side branch of existing filaments. The activation of WAVE-1 is induced by Rac, whereas the Rac-driven signaling is impaired by the WRP protein that is a WAVE-1-associated GTPase-activating protein. In order to investigate the function and properties of WAVE-1, WAVE-1 knockout mice have been generated (Soderling et al 2003). WAVE-1 knockout mice are viable indicating that WAVE-1 is not essential for generating mice in general, but these mice exhibit a broad variety of sensoric and motoric defects and possess even learning deficits, which are highly similar to those deficits that are described for haploinsufficiency of WRP in humans. In conclusion, alterations in the Rac facilitated assembly of actin filaments seem to contribute to the involved mechanism of mental retardation.
The expression of activated Rho family GTPases has a broad impact on gene expression and function and hence functional proteomics have been applied to characterize gene expression alterations in cell expressing mutants of activated RhoA, Ccd42 and Rac1 using 2D-Page combined by MALDI-qTOF peptide mass sequencing (Kabuyama et al 2006). They found that firstly, the expressions of several distinct genes are regulated by a single specific GTPase and secondly, the expressions of other genes are regulated by two or more GTPases. In total, 22 proteins have been identified and among these were 19 novel targets of Rho GTPases. One of these new targets is the protein-tyrosine phosphatase 1B (PTP1B), which is post-translationally modified by RhoA and thereby inhibited in its function.
Simultaneously, p130Cas can be phosphorylated at its tyrosine by PTP1B, which is facilitated by RhoA. p130Cas is critical for the regulation of the turnover of focal adhesions, which is dissimilar to the Rho kinase effector pathway and p130Cas has been found to provide cellular mechanical properties (Sawada et al 2006). These results indicate that PTP1B seems to be a novel effector of the RhoA driven signaling pathway, as it regulates the phosphorylation levels of p130Cas independently of RhoA signaling processes. Subsequently, the phosphorylation of p130Cas induces cell migration and invasion that seem to depend on altered mechanical properties possibly via the phosphorylation of Rac1.
1.3.7.1. Impact of CDC42 on cancer and cell mechanics.
The actin cytoskeleton of mammalian cells predicts the cell's shape and is involved in the cell's motility, cytokinesis, phagocytosis and the transport of substances within the cell. Since its involvement in cellular processes, the precise regulation of the actin cytoskeleton is critical for many basic cellular functions. Rho and Rac are two members of the Ras-related conserved superfamily of small GTPases that regulate the actin polymerization to assemble stress fibers and lamellipodia, respectively (Nobes and Hall 1995). Cdc42 is also a member of the same superfamily and builds the formation of an actin-based structure termed filopodia, which represents finger-like extrusions of the cell's membrane that contain parallel aligned actin filaments. Additionally, Rho regulates the assembly of focal adhesions and subsequently focal adhesion complexes. Independently of Rho, Rac and CDC42 facilitate the formation of multicellular focal complexes underneath the cell's membrane containing vinculin, paxillin and focal adhesion kinase (figure 9). In general, activation of Cdc42 causes the activation of Rac and subsequently, the activation of Rho indicating a coordinated control of cellular motility by the Rho family of GTPases. Cdc42 is activated similar to other Rho family of small GTPases members through the binding of GTP and is inactivated by binding of GDP (Jaffe and Hall 2005). There exist proteins for the activation and inactivation of Cdc42 such as guanine nucleotide exchange factors (GEFs) that trigger the hydrolysis of GTP to GTP by catalysis (Schmidt and Hall 2002). In particular Cdc42 has the ability to regulate the polarization of actin, the dynamic remodeling of microtubules and gene expression (Jaffe and Hall 2005, Otani et al 2006). Hence, it can be hypothesized that Cdc42 can alter the mechanical properties of cells regulating cellular functions. As these various cellular remodeling processes are controlled by Cdc42, it acts as a regulator for membrane trafficking of vesicles, cell-cell adhesion, cell-cycle rates, motility and polarity of cells (Jaffe and Hall 2005, Harris and Tepass 2010, Citi et al 2011). Coordinated regulation of the actin and the microtubule cytoskeleton is performed by the binding of Cdc42's to the binding Wiskott-Aldrich syndrome protein (WASP) that thereby induces its activation (Aspenström 1997, Tian et al 2000). Moreover, the Cdc42 driven polymerization of actin is facilitated by activation of WASP and N-WASP (Rohatgi et al 1999, Hall 2005). The assembly of microtubules is facilitated by interaction of Cdc42 with CIP4 that causes deformation of the membrane (Aspenström 2009).
The knock-down of Cdc42 leads to an intact basic actin machinery, but cannot polarize the Golgi Apparatus and hence regulate the coordination of the motility system. The loss of Cdc42 impairs the interaction between three main cellular systems such as (i) the assembly of actin via formin FML2 and the actin branching protein Arp2/3, (ii) the localization of active myosin II and (iii) the integrin-driven cellular adhesion dynamics.
1.3.7.2. Effect of Rac1 on cell migration and barrier function.
Two main functions of Rac1 are known: firstly, it enhances the migration of cells (single and collective cell migration), which is mainly studied on 2D flat substrates and secondly, it provides cell-cell adhesion-based barrier function of cell monolayers such as endothelial blood or lymph vessel linings or cell clusters such as spheroids or organs (figure 9).
The small GTPase Rac1 acts intracellularly in the signal transduction pathways and therefore it is able to switch between two states: on inactive state, in which GDP is bound and an active state, in which GTP is bound. Both states underlie a precise regulation by many downstream effector pathways that determine a broad spectrum of cellular functions (Van Aelst and D'Souza-Schorey 1997, Hall 1998, Bishop and Hall 2000, Zheng 2001, Etienne-Manneville and Hall 2002).
1.3.7.2.1. Effect of Rac1 on cancer cell migration.
Main results on the effect of Rac1 in cellular motility are obtained from 2D migrations studies and hence in 2D systems, in which the underlying mechanisms have been intensively investigated. Previous studies showed that Rac1 is a key signaling molecule that regulates the actin cytoskeletal organization in mammalian cells (Hall 1998). Rac1 is known to facilitate the formation of protrusive structures such as lamellipodia and causes the ruffling of cell membranes upon stimulation with serum and Platelet derived growth factor 2 (PDGF2) (Ridley et al 1992, Ridley 2011) that is facilitated by p21-activated kinase 1 (PAK1)-dependent and -independent signaling processes (Lamarche et al 1996, Sells et al 1997, Sells et al 1999). However, another Rho family kinase RhoA facilitates the actin stress fiber formation and focal adhesion assembly that is partly driven by ROCK (Ridley and Hall 1992, Pawlak and Hellfman 2002, Wilkinson et al 2005). Hence, Rac1 acts upstream of RhoA in the process of actin cytoskeleton reorganization (Ridley et al 1992, Sander et al 1999, Wilkinson et al 2005, Ridley 2011). Additionally, Rac1 functions in cell-matrix adhesion and cell migration (Nobes and Hall 1995, Allen et al 1997, Rottner et al 1999, Arthur et al 2002, Kaverina et al 2002) and has been found to switch from random Brownian motion to directionally persistent motion of cells (Pankov et al 2005). Moreover, the Rac1 facilitated cellular adhesion through integrins seems to be a critical component for avoiding cell anoikis, which is crucial for maintaining cancer progression (Coniglio et al 2001, Zugasti et al 2001). Anoikis is a specific kind of a programmed cell death that is caused by detachment of anchorage-dependent cells from their surrounding extracellular matrix environment. All these functions of Rac1 indicate that this protein seems to be a candidate for a central regulator of cell mechanical functions such as stiffness (Kunschmann et al 2019).
1.3.7.2.2. Physiological and pathological functions of Rac1.
The physiological of function of Rac1 has been explored by using Rac1 null mice and cell lines generated from mouse embryonic fibroblasts. The majority of biological studies on the function of the Rac1 GTPase were performed with cells that overexpress constitutively active and/or dominant-negative Rac1 mutants. Numerous key discoveries have been made among which is finding that Rac1 is essential for the formation of lamellipodia formation and hence cell-matrix adhesion (figure 9). However, these approaches possess several limitations when it is taken into account that there exists an abundant signaling crosstalk between Rac1 and other Rho GTPases, which may vary between cell clones that are frequently used in these assays (Bishop and Hall 2000, Zheng 2001, Etienne-Manneville and Hall 2002). Besides clonal variability, the overexpression of one protein may also cause adaptions of interacting proteins and lead to an unbalanced situation evoking possibly a phenotype. In more detail, on the one hand, a dominant-negative Rac1 mutant sequesters the upstream GEFs that include more than 80 members in the human genome and among them are many factors that serve various RhoGTPases (Zheng 2001). As dominant-negative Rac1 has no catalytic activity, an excessively high level of the dominant-negative mutant protein is necessary for effectively sequestering GEFs in order to impair endogenous Rac1 activity, which in turn is likely to impact the functions of variant Rho GTPases (Bebreceni et al 2004).
On the other hand, the overexpression of the constitutively Rac1 mutant (Rac1 in a GTP-bound state) can possibly cause an activation a numerous Rac1 effectors, which are also effectors of other Rho GTPases such as PAKs, IQGAPs and IRSp53 (Bishop and Hall 2000) that may confusedly lead to Rac1-irrelevant functional phenotypes (Sarner et al 2000, Aoki et al 2004). As the balanced GTP binding/GTP hydrolysis cycle of Rac1 needs to be precisely balanced for an effective signal transduction process (Cerione 2004), overexpression of the Rac1 mutants in cells can cause artifacts insofar that the small GTPase is blocked in one conformational state leading to an arrest in a specific intracellular location. Moreover, due to the fact that Rac1 activity and its induced cellular functions can be altered by genomic alterations during the process of Rac1 mutant in vitro cell cloning, passage and selection procedures, it is possible that the functional phenotype of the Rac1 mutant expression is artificially dependent of clonal history rather than on the effect of the mutation. Hence, genetic approaches in primary cells are preferred such as conditional gene-targeted Rac1 loxp/loxp mice for the generation of Rac1 knockout mouse embryonic fibroblasts (Guo et al 2006).
1.3.7.2.3. Rac1-dependent cell migration is altered by Strip1 and STRIPAK.
Physiological processes such as morphogenesis and the formation of organs during the embryonic development require the precise regulation of the migration of cells in the mesoderm tissue section. There exist many in vitro studies of cell migration that report that the regulation of cellular motility is manifested in the cell's cytoskeleton (Theriot and Mitchison 1992, Pollard and Borisy 2003, Mierke et al 2011a, b). However, the migration mechanism of cells within the mesoderm tissue section is still not yet well understood in vivo. An exception is a study that investigates the identification and characterization of a mutation in the striatin-interacting protein 1 (Strip1), which is known to impair the mesoderm migration after finishing the gastrulation step, when a successful transition from a mesenchymal to epithelial phenotype (MET) has been performed (Bazzi et al 2017). Besides gastrulation, the following migration of the mesoderm is needed for proper generation of the entire body and the patterning of tissues within the mammalian embryo (Rivera-Pérez and Hadjantonakis 2014). The epiblast consists of a single pseudostratified epithelial layer and upon gastrulation start, the cells of the primitive streak (the posterior epiblast region) need to perform the epithelial-to-mesenchymal transition (EMT). At the primitive streak fibroblast growth factor (FGF) signaling induces the increased expression of the transcription factor SNAIL that represses then the transcription of the cell-cell adhesion molecule E-cadherin, which enables the transition of the epithelial to mesenchymal phenotype to facilitate the mesoderm migration (Burdsal et al 1993, Ciruna and Rossant 2001). In many solid cancers the expression of E-cadherin is elevated, whereas the expression of E-cadherin in migratory and invasive cancer cells is downregulated (Frixen et al 1991). Moreover, the migration capacity of mesoderm cells is regulated by the actin cytoskeleton reorganization and hence the migration is driven by the WAVE complex and the small GTPAse Rac1 (Rakeman and Anderson 2006, Migeotte et al 2011). In in vitro experiments striatin-interacting phosphatases and kinases (STRIPAK) complexes, which organize striatin in their core center, have been identified to be involved in mammalian cell migration (Madsen et al 2015). Moreover, striatins belong to calmodulin-binding WD-repeat proteins, as their strong expression has been reported initially in the striatum of the brain (Castets et al 1996). In more detail, striatin assemblies a complex consisting of the catalytic and regulator subunits of the protein phosphatase 2A (PP2A) (Moreno et al 2000) that is part of a much larger multiprotein complex composed of the kinases STK24 (synonymously termed MST3), STK25, and STK26 (synonymously termed MST4), which all together form the highly conserved STRIPAK complexes (Goudreault et al 2009, Hwang and Pallas 2014).
STRIPAK complexes are proposed to facilitate PP2A activity, which depends on the activity of the STRIPAK kinases that in turn serve as substrates for the phosphatase PP2A (Gordon et al 2011, Hwang and Pallas 2014, Shi et al 2016). Mouse Strip1 consists of 837 aa and is a core component of STRIPAK. Null mutants of Strip1 lead to developmental arrest in midgestation involving disruptions in mesoderm organization that finally cause a male function in anterior extension of the axial mesoderm. The migration failure impairs also cell spreading, focal adhesion formation, actin cytoskeleton formation and subsequently decreases migration speed (Bazzi et al 2017). In summary, the STRIPAK complexes are required for cellular motility and the overall morphogenesis of tissues in vivo. STRIPAK complexes are regulators of several signaling processes such as Hippo, MAP (Mitogen-activated protein kinase) and the restructuring of nuclear receptors and the actin cytoskeleton (Hwang and Pallas 2014, Shi et al 2016).
Moreover, the absence of Strip1 shortens the anterior-posterior axis and results in the direct connection of the head to the tail without a trunk that is similar to the phenotype caused by the absence of Rac1 (Guo et al 2006). As Strip1 depletion leads to a similar phenotype as Rac1 depletion, it has been suggested that Strip1 functions in Rac1-driven cellular motility (Bazzi et al 2017). This Rac1-driven cell migration is more based on collective cell migration rather than on single cell migration, in which Rac1 also plays an important role. Indeed, it has been shown that the Strip1 phenotype causes abnormal migration of two cell populations that elongate the anterior-posterior axis in axial and paraxial directions of the mesoderm (Shi et al 2016). In addition, in cell culture studies, it has been revealed that Strip1 is required for the actin cytoskeleton and focal adhesion organization, which are both essential for physiological mesoderm cell migration and finally the body axis formation (Shi et al 2016). Besides the physiological functions of STRIPAK complexes such as cell growth, proliferation, cell death (apoptosis), metabolism and immune system regulation, it also acts in pathological processes such as tumorigenesis. In particular, dysregulation of STRIPAK has been found to be associated with cancer. However, it remains to be explored whether STRIPAK affects the mechanical properties of cancer cells, when regulating their invasive phenotype.
1.3.7.2.4. Rac1 function on the microenvironment of cancer cells such as endothelial 'migration barriers'.
As the endothelium provides the inner lining of blood and lymphoid vessels, it is critical for the regulation of the exchange of fluids, biomacromolecules and cells such as leukocytes between the vessel's surrounding tissues and the vessel's lumen (figure 10). The maintenance of the endothelial barrier function needs to be sustained and hence protected from endothelial cell driven nitric oxide (NO) production that is known to impair the integrity of the endothelial barrier by induction of leakage within the endothelial monolayer lining (Predescu et al 2005, Hatakeyama et al 2006), as it often occurs in cancer. The NO production is facilitated by the activation of enzymes such as the nitric oxide synthase (NOS) that can be inhibited naturally by methylarginases such as guanidine-methylated arginine analogue L-n(g)-mono-methyl-arginine (L-NMMA) or asymmetric dimethylarginine (ADMA) (Vallance and Leiper 2004). ADMA has been revealed as a cardiovascular risk factor and increased plasma levels of ADMA correlated the dysfunction of the endothelium and subsequently with vascular leakage (Vallance and Leiper 2004, Cooke 2005). However, the role of methylarginine in vivo in the regulation of endothelial barrier function in the malignant progression of cancer is not yet clear. In general, methylarginines impair the synthesis of NO. In vivo a decrease of NO levels increases the leakage of microvessels in the pulmonary circulatory system of eNOS deficient mice (eNOS–/–) treated with the NOS inhibitor L-NAME (N(G)-nitro-l-arginine methyl ester) (Predescu et al 2005) and similarly in the isolated lung of rabbits after NOS inhibition (Mundy and Dorrington 2000). However, the mechanisms are not yet revealed. Hence NOS inhibitors seem to alter directly the barrier function of the endothelial vessel lining in vivo (Predescu et al 2005) that can support the malignant progression of cancer. In addition, these NOS inhibitors may cause indirect effects such as increased neutrophil adhesion (Rumbaut et al 2000). Besides inhibition of NO production, ADMA seems to uncouple NOS, which leads to superoxide production (Cardounel et al 2005). In several model systems, the oxidative stress can enhance pulmonary hypertension, which breaks-down the endothelial barrier function by either increasing (van Wetering et al 2002) or decreasing ROS levels (Wojciak-Stothard et al 2005). As it has been shown that ADMA alters the activity of the actin dynamics regulators of such as the RhoA, Rac1 and Cdc42 differently in pulmonary artery endothelial cells (Wojciak-Stothard et al 2007), it may determine the overall function of the endothelial barrier including the strength of the barrier. Endothelial permeability is increased through RhoA activation that causes enhanced actomyosin contractility in endothelial cells, which then loses cell-cell adhesions and forms intercellular gaps between adjacent endothelial cells (van Nieuw Amerongen and van Hinsbergh 2002). Contrary to RhoA, Rac1 activity is necessary for the assembly, maintenance and recovery of endothelial cell-cell adherence junctions (Waschke et al 2004, Wojciak-Stothard et al 2005, Baumer et al 2009). Moreover, Cdc42 induces the recovery of cell-cell junction after treatment with thrombin in human pulmonary arterial endothelial cells (Kouklis et al 2004). Similarly, RhoB regulates endothelial barrier recovery through the inhibition of Rac1 (Marcos-Ramiro et al 2016). The downstream effectors of NO such as the cAMP-activated protein kinase A (PKA) and the cGMP-activated protein kinase G (PKG) can facilitate effects of methylarginines. The latter have both shown to be involved in the regulation endothelial permeability, however, their effect on the endothelial barrier is either protective or destructive, which depends of the individual vascular bed (van Nieuw Amerongen and van Hinsbergh 2002). PKA and PKG can phosphorylate the vasodilator-stimulated phosphoprotein (VASP) that couples the cell-cell adherence junctions to integrins to the actin cytoskeleton (Krause et al 2003). Moreover, in genetically altered mice, a critical function of VASP in providing vascular integrity has been identified (Furman et al 2007), whereas the VASP phosphorylation that maintains junctional integrity is still not well understood. The PKA facilitated phosphorylation of VASP is suggested to increase the adhesion strength of cell-cell adhesion (Comerford et al 2002), whereas it has been demonstrated that VASP interacts with the cortical actin cytoskeleton assembly and thereby reduces the cell-cell adhesion strength (Benz et al 2008). PKG facilitated phosphorylation of VASP increased the endothelial cytoskeletal reorganization and enhanced angiogenesis (Chen et al 2008), whereas the function on the endothelial barrier is elusive. In addition, the localization and activity of VASP in endothelial cells is highly associated with the activation of Rac1 activation and the integrity of endothelial (Schlegel et al 2008).
Figure 10. The endothelial barrier function. The endothelium lines the innermost site of blood or lymphoid vessels to provide vessel integrity, stability and restricts the transfer of substances or cells.
Download figure:
Standard image High-resolution image1.3.7.3. RhoA and its role in motility.
RhoA has a function in providing mechanical properties of cells such as cellular contractility. Viscoelastic recoil of individual stress fibers after laser cutting has demonstrated that it is partially slowed by inhibition of Rho-associated kinase and effectively eliminated by immediate inhibition of myosin light chain kinase (Kumar et al 2006). Based on Rho activity-dependent biosensors it has been shown that RhoA is activated at the cell's leading edge of migrating cells (Pertz et al 2006, Kurokawa and Matsuda 2005), which is in line with studies that show RhoA functions in the ruffling of the membrane and the formation of lamellae (Nishiyama et al 1994, Fukata et al 1999, O'Connor et al 2000, Kurokawa and Matsuda 2005). Moreover, RhoA is associated with the blebbing of the cell membrane, which is utilized in amoeboid-like motility (Charras and Paluch 2008). Several studies reported a role for RhoA in cell migration and propose that Rac and RhoA are located in different spaces: activated Rac is found at the cell's leading edge, whereas activated RhoA is located at the cell's trailing edge (Spiering and Hodgson 2011).
Upon the availability of FRET-based Rho GTPase activity biosensor, activated RhoA has been detected at the cell's leading edge of migrating cells together with active Rac1 and active Cdc42 in a spatial and temporal different manner: RhoA activation is followed by activation of Rac and Cdc42 (El-Sibai et al 2008, Machacek et al 2009, O'Connor and Chen 2013). These studies are consistent with previous studies detecting RhoA at the cell's leading edge, where it supports cell migration (Nishiyama et al 1994, Fukata et al 1999, O'Connor et al 2000, Narumiya et al 2009, O'Connor et al 2012, Kurokawa and Matsuda 2005). A main question is still unanswered that is how can RhoA act on the one hand in providing stress fibers and on the other hand in facilitating the formation of lamellae? Can simply the decision on a single effector molecule regulate both membrane ruffling and stress fiber polymerization? Indeed, these two processes are facilitated by Rho effectors ROCK and the mammalian homolog of Drosophila diaphanous (mDia) (Narumiya et al 2009). Moreover, it has been shown that in the absence the Rac1, RhoA induces the formation of lamellae after the binding of laminin to its receptor, the integrin α6β4, in colon cancer cells during haptotactic migration (O'Connor et al 2000). Using RhoA biosensors, RhoA activity has been shown to be increased at the cell's leading edge of fibroblasts. Besides the location of active RhoA in the front of the cells, it has also been detected in the rear of HeLa cervical cancer cells, when cells migrate randomly on 2D collagen coated surfaces. In addition, due to growth factor stimulation RhoA activity persists within membrane ruffles (Kurokawa and Matsuda 2005). How is the activation of different small Rho GTPases such as RhoA, Rac1 and Cdc42 regulated in time upon stimulation? The activation of RhoA is correlated with protrusion exertion and is locally restricted to the outermost 2 µm layer of the cell's leading edge, which is followed by the activation of Rac1 and Cdc42 (Machacek et al 2009). Hence, the timely regulated expressions and the precisely (but solely slight) special separation of Rac and Rho activities provide an explanation for answering the question of how integrin- and Rac-activated GAP activities can be present together with RhoA at the cell's front. Moreover, it is pointed out that a precise regulation of RhoA activity at the cell's front is required for the protrusion of the cell's membrane.
1.3.7.4. Effect of cytoskeletal components such as Arp2/3 (downstream target of Rac1) or filamin A (regulating Rac1 activity) on cellular mechanics and migration.
1.3.7.4.1. Effect of the actin cross-linker Arp2/3 (a downstream target of Rac1) on migration.
In the protrusive migration of cells, lamellipodia, which are flat, sheet-like membrane perturberances, play a crucial role together with branched actin structures (Yamaguchi and Condeelis 2007, Vinzenz et al 2012). The Arp2/3 is a major component of lamellipodial structures and acts as a nucleator for actin network polymerization by facilitating the branching of actin filaments (Firat-Karalar and Welch 2011). Actin nucleation factors such as Arp2/3 play a major role in enhancing the polymerization of actin polymerization, as they facilitate the addition of monomeric actin units to actin filaments either to their filament ends or at new branching points (Le Clainche et al 2003). All of which can have a major impact on the mechanical properties of the cells. Moreover, the Arp2/3 complex is a highly conserved complex that consists of seven protein subunits. Among this complex are two proteins such as actin-related proteins 2 and 3 (Actr2 and Actr3, respectively) that are structurally similar to actin. Additionally, this complex consists of five subunits such as Arpc1, Arpc2, Arpc3, Arpc4 and Arpc5 (Welch et al 1997, Robinson et al 2001). In more detail, there exists two isoforms of Arp1 in humans: Arpc1A and Arpc1B. At the branching site on the actin filament, Arpc2 and Arpc3 are associated with the pointed end of the new daughter actin filament, whereas Arpc2 and Arpc4 interact with the mother actin filament. However, the precise functions of β-propeller protein Arpc1 as well as Arpc3 and ARPC5 are still not yet well understood (Goley and Welch 2006).
Several knockdown approaches have shown that the disruption of the Arp2/3 complex in mouse embryonic fibroblasts impairs the formation of lamellipodia and subsequently also the migration of cells (Bailly et al 2001, Rogers et al 2003, Steffen et al 2006), whereas there has been observed no effect on the lamellipodial structure (Di Nardo et al 2005). However, it has been seen that the Arp2/3 complex is indeed crucial for the formation of lamellipodia. Although the disruption of Arp2/3 complex cannot fully impair the migration of cells, it determines the directionality of cell migration that impairs fibroblasts in sensing alterations of the extracellular matrix, which causes a coherent cell movement along the gradients (Suraneni et al 2012, Wu et al 2012). Arp2/3 complex-free (depleted) cells build instead of lamellipodia, filopodial structures in order to facilitate cell migration and invasion. The importance of the Arp2/3 complex in the formation of lamellipodia and subsequently cell migration is based on the finding that the correct localization of Actr2 mRNA at the cellular protrusions forces the cells to move in a certain direction (Liao et al 2011). Arpc1A and Arpc1B have been found to be possible target genes for 7q22 amplification (targeting cyclin kinase 6) in pancreatic cancer, as the migratory capacity of cancer cells can be decreased by siRNA mediated silencing of Arpc1A and Arpc1B (Laurila et al 2009). Moreover, a systematic study on the role of Arp2/3 complex in pancreatic cancer have been performed through the analysis of the expression levels of all Arp2/3 complex subunits and the effect of silencing of each of the complex members for the migration of pancreatic cancer cells. As the Arp2/3 complex silencing causes usually a decreased migration of cells, the migration is still not fully impaired. The residual migration can provide an altered migration mode, as the migration mode has been switched from a chemotaxis-driven directional migration mode to a more random migration mode that resembles the migration of Arp2/3 complex-depleted mouse embryonic fibroblasts (Suraneni et al 2012, Wu et al 2012).
1.3.7.4.2. Filamin A (a regulator of Rac1 activity) and its effect on migration mode.
In 1975, Filamin a (FLNa) has been identified as first actin filament crosslinking protein in human non-muscle cells (Hartwig and Stossel 1975). FLN a consists of two 280 kDa subunits and self-assembles in order to build a 160 nm long semi-flexible strand (Nakamura et al 2011). The dimerized molecule exhibits a V-shaped structure. Human FLNa belongs to a family of three proteins that are named filamin b (FLNb) and filamin c (FLNc) that are produced by their individual genes. FLNs function as scaffolding proteins and thereby interact with more than ninety other binding proteins such as transmembrane receptors, channels or intracellular signaling associated proteins and transcription factors, which play a role in cell adhesion and motility. Mutations of FLNa cause a wide range of cellular and tissue anomalies (Stossel et al 2001, Feng and Walsh 2004, Robertson 2005, Popowicz et al 2006, Zhou et al 2010). Therefore, it can affect also the mechanical properties of cells and subsequently affect their invasive phenotype.
Each FLN subunit has an N-terminal region that is termed spectrin-related actin-binding domain (srABD). The srABD is followed by a 24 repeats beta pleated sheet unit (repeats are termed immunoglobulin-like domains). The repeats 1–15 built the rod 1, repeats 16–23 form rod 2 and repeat 24 contains the self-association domain (Gorlin et al 1990, Pudas et al 2005). The three domains are separated by two calpain-sensitive 'hinges' (Stossel et al 2001, van der Flier and Sonnenberg 2001), which introduces additional flexibility into the molecule. The increased flexibility enables the molecules to link and stabilize orthogonal networks containing F-actin branches with a large angle (Hartwig et al 1980). All FLNs are important and highly conserved actin-binding proteins that perform essential roles in cell and organ development, in the assembly of tissues and morphogenesis and also in mechanosensing involved processes (Yamazaki et al 2002, Kim and McCulloch 2011, Razinia et al 2012).
Among the three human FLNs, FLNa is the most widely and abundant expressed similarly expressed is FLNb, whereas FLNc expression is mainly restricted to striated muscle cells (Stossel et al 2001, van der Flier and Sonnenberg 2001). All three FLNs possess overlapping and total independent functions and activation modes (Kesner et al 2010). Moreover, missense mutations or truncations in each of the three FLNs belong to a wide range of congenital anomalies such as malformations of the body's skeletal structure and of tissues such as cells of the skeletal muscle, brain or the cardiovascular system (Robertson et al 2003, Zhou et al 2007, Zhou et al 2010, Nakamura et al 2011). These alterations are proposed to affect the interactions with specific FLN-binding proteins and subsequently lead to disorders in cytoskeletal and signaling networks, which underlie the pathologies, however, the molecular determents are still elusive. Due to FLNs crosslinking activity of the actin cytoskeleton, FLNs can function as mechanosensory proteins that even regulate the cellular responses to different matrix mechanical properties such as lateral matrix densities. Thus, they are involved in the regulation in cell motility and facilitate impairment of cell-matrix adhesion receptors such as integrins.
The individual loss of either FLNa or FLNb has been reported to have only a minor effect on cell migration, whereas the knockdown of both FLNa and FLNb, or proteolysis of all three FLNs, inhibits cellular motility (Baldassarre et al 2009). The effect is based primarily on a defect in the initiation of cellular motility rather on the maintenance of the migration speed. FLN-deficient cells have significant spreading defects. The rescue of this phenotype by re-expression of a full length FLNa, can restore cell spreading and impair the induction of migration, whereas the re-expression of mutated FLNa, which lacks the immunoglobulin domains 19–21, cannot rescue the phenotype (Baldassarre et al 2009).
Although FLNs function as essential actin-binding proteins facilitating cell adhesion, the dimerization domains are not necessary to rescue the FLNa KD phenotype (Gorlin et al 1990, Pudas et al 2005). Besides diseases facilitated by alterations in FLN genes of the germline, FLN mutations have been found in human breast and colon cancers (Nakamura et al 2011). FLNs facilitate as actin binding proteins the coupling of the actin cytoskeleton to cell-matrix adhesion receptors and subsequently, the extracellular matrix, which indicates that they fulfill a critical role in cellular motility. However, the focus on FLNs is on interventions of the cell motility and dynamics of the cytoskeleton and membrane, whereas the remodeling of the extracellular matrix is rather ignored. Using FLNa-deficient melanoma cells, a crucial function of filamins in cell migration has been observed (Cunningham et al 1992). In addition, FLN deficiency seem to cause impaired neuronal migration (Nagano et al 2002). Other knockout mice studies (Feng et al 2006, Hart et al 2006) and studies employing small interference RNA (siRNA) revealed the depletion of only one FLN isoform reduces cell migration, whereas the depletion of two or more FLNs also abolished the initiation of migration and spreading of cells (Heuze et al 2008, Baldassarre et al 2009, Lynch et al 2011). However, FLNa expression has been found to be negatively associated with migration and invasion of breast cancer cells and subsequently cancer metastasis in targeted organs (Xu et al 2010, Caruso and Stemmer 2011). Using shRNA facilitated knockdown of FLN in HT1080 human fibrosarcoma cells, it has been found that a decrease in FLNa down-regulates the expression and secretion of tissue inhibitor of metalloproteinase 2 (TIMP-2) and thereby increases the MMP activity through the activation of MMP2, which then provides an increased degradation of the extracellular matrix nearby the cell to enhance its migration and invasion in these 3D scaffold confinements (Baldassarre et al 2012). However, the mechanism is not yet fully clear. It can be hypothesized that FLN directly causes the secretion of TIMP-2. In line with this, the migration on 2D environments is not impaired. The effects of FLN on proteolytic extracellular matrix remodeling represents a possible mechanism for the reduced FLNa levels in the process of cancer metastasis. In more detail, the actin binding domain of FLNa and IgFLNa domains 1–15 are essential to impair the degradation of the extracellular matrix, whereas the dimerization and ligand binding of integrins is not necessary (figure 11). Similar results are obtained for FLNb (Baldassarre et al 2012). The remodeling of the extracellular matrix also affects the structure and the matrix mechanical phenotype.
Figure 11. Filamin A (FLNa) dimerizes and crosslinks actin filaments that form network-like structures. In particular, FLNa binds via its srABD domain to the actin filaments. The srABD domain is followed by the rod 1 and 2 domains and finally repeat 24 contains the dimerization domain.
Download figure:
Standard image High-resolution image1.4. Mechanical properties of cells facilitate cell migration
The mechanical properties of cells have been investigated more pronouncedly in the last decade and have become a focus of biophysical and soft matter physics research. However, the main questions are still not yet fully answered: Are aggressive and invasive cancer cells soft or stiff? Are highly migratory and invasive cells soft or stiff? What impact has the heterogeneity of cells and different cell types or cancer types on mechanical stiffness? What role has a 3D microenvironment on cellular stiffness? There exist several studies about the cellular mechanical properties of cells which are not conducted in a comparable manner by addressing the effect of cell adhesion on the mechanical properties such as cellular stiffness and contractility. Some of the stiffness measurements are performed independently of cell adhesion by forcing the cells to adapt a rounded morphology during the measurement in an aqueous buffered solution using an optical cell stretcher (Guck et al 2005, Meinhövel et al 2018) or a suspended microchannel resonator, inculing a cell sorting function (Shaw Bagnall et al 2016). Moreover, the mechanical phenotype of cells can be determined adhesion-independent with real-time cytometry (Xavier et al 2016). Other stiffness measurements are performed with atomic force microscopy (AFM), where mostly adherent cancer cells are measured (Lekka et al 2016) and in certain cases also suspended cells such as breast cancer cells (Fischer et al 2017). As the oncogenic transformation of cells creates a specific phenotype that is characterized by excessive cellular growth, decreased cellular differentiation and altered interaction with adjacent cells and the surrounding extracellular matrix, alterations in their cytoskeletal organization and subsequently cellular mechanical properties (Mierke et al 2008b, Hanahan and Weinberg 2011, Jinka et al 2012, Mierke 2014). It has been reported by several studies that structural alterations of the cytoskeleton usually cause a larger deformability of single cells such as cancer cells of bladder, prostate, thyroid, ovarian or breast tissues (Lekka et al 1999, Guck et al 2005, Faria et al 2008, Prabhune et al 2012, Xu et al 2012, Lekka et al 2012a). The mechanical alterations of cancer cells are found to be correlated with either a decrease in actin filaments levels or an increased disorganization of microtubules (Yamaguchi and Condeelis 2007, Pachenari et al 2014) that all is a result of a cytoskeletal scaffold of lower density compared to normal cells. Therefore, the ability of single cells to deform themselves has been studied with several biophysical techniques such as AFM, magnetic tweezers, magnetic twisting cytometry or optical cell stretching. These investigations of cellular mechanical properties are based on the assumption that the cellular deformability is altered due the development and the progression of a certain disease such as cancer. Indeed, certain diseases such as muscular dystrophies and laminopathies have been found to be associated with genetic mutations affecting structural and molecular alterations within single cells (Pasternak et al 1995, Schäpe et al 2009, Puttini et al 2009, Banerjee et al 2013, van Zwieten et al 2014). In addition, both increased and decreased deformability have been reported in various cancer types. However, there is still the agreement that mechanical properties have the potential of a non-labelled biomarker for the detection of cancer progression. The identification of such a biomarker, which is based on the mechanical properties of individual cancer cells, has supported the development and invention of novel and powerful techniques for analyzing the mechanical properties and their effect on cellular structures and functions. Among these biophysical techniques is the AFM (Alessandrini and Faci 2005) and indeed the first studies reported differences in mechanical properties of cancer cells that are suitable to identify them (Lekka et al 1999). In particular, the cellular deformability of malignant human bladder cancer cells was even one order of magnitude larger than control cells of non-malignant cancers. Additionally, these AFM results have been supported by optical cell stretcher measurements showing that cancer cells differ in their entire deformability due to their malignant potential and hence invasiveness. Compared to AFM measurements, the optical cell stretcher measurements are a high throughput technique. Three human cell lines have been initially analyzed: the non-tumorigenic breast epithelial MCF10 cells, non-metastatic breast epithelial cancer MCF7 cells and MCF7 cells that have been transformed with phorbol ester to obtain a metastatic cell line. As expected, the deformability of invasive cells such as transformed MCF7 breast cells was increased compared to non-transformed MCF7 cells and non-metastatic MCF10 cells (Guck et al 2005). Other measurements of single cell elasticity also demonstrated that cancer cells including bladder, prostate, thyroid, ovarian or breast cancer cells possess a significantly higher deformability compared to normal healthy cells. Moreover, the deformability of cancer cells increased during the malignant progression of cancer (Faria et al 2008, Xu et al 2012, Lekka et al 2012b).
Using AFM based deformability measurements a Young's modulus (elastic modulus) of the cells can be determined which enables us to compare the elastic properties quantitatively. The values of the Young' modulus of cells such as vascular smooth muscle cells, fibroblasts, bladder cells, red blood cells, platelets and epithelial cells, which contain measurements of normal healthy cells and cancer cells that are, when cultured in vitro, in the range of 1–100 kPa (Radmacher 2002). The mechanical stability of cells is based on its cytoskeletal structures such as the three main filament types: actin filaments, intermediate filaments and microtubules. In order to reveal which of the three components act as a major contributor for the mechanical response of cells after stress exertion, the influence of each component on the stiffness can be tested by impairing its stability through the addition of an inhibitory drug (Rotsch et al 2000, Wakatsuki et al 2000, Lu et al 2008, Seltmann et al 2013, Ramos et al 2014). Cytochalasin D is still the most common cytoskeletal drug used in these assays, although its specificity is relatively broad. It has been shown that cytochalasin D depolymerizes actin filaments, which is also mirrored in the elasticity measurement of cells using AFM (Lekka 2016). In melanoma cells, it has been demonstrated that cytochalasin D treatment leads to actin filament depolymerization and an increase in cellular deformability indicated by a decrease in the Young's modulus (Rotsch et al 2000, Wakatsuki et al 2000, Ramos et al 2014). For a long time, solely the organization of actin filaments inside the cells have been considered, as the presence of actin stress fibers such as bundles of single actin filaments provide pronouncedly the elasticity of cells (Gavara and Chadwick 2016) such as bladder cancer cells (Lekka et al 2012b). Besides the drug cytochalasin D, which causes many side effects, latrunculin A can be used as an inhibitor for actin polymerization in order to investigate whether cellular stiffness (Young's modulus) can be attributed precisely to the actin cytoskeleton (Fischer et al 2017). Additionally, some studies found besides the organization of actin filaments that the total actin content contributes to the cellular stiffness (Chiou et al 2013, Efremov et al 2015). In conclusion, as the actin density is more associated to alterations in cellular stiffness (or elasticity) that are evoked by the oncogenetic transformation of cells, it seems to play a more prominent role than the purely structural organization of the actin filaments within the cell's cytoplasm. Moreover, several studies showed that the mechanical properties of colon cancer cells are determined by the ratio of actin filaments to microtubules (Pachenari et al 2014). As the stabilization or destabilization of cytoskeletal filaments depends on the individual cell type, the chemical component, the cell-cycle stage, cellular stimulation by cytokines and the type of filaments and the impact on cellular deformability can be an increase in deformability (softening of the cell) or a decrease in deformability (stiffening of the cell).
The analysis of mechanical properties in living cells using AFM requires the comparison of cancer cells with normal, healthy cells that serve as control. For several cancer cell types such as thyroid (Prabhune et al 2012), breast (Lekka et al 2012a, Coceano et al 2016), prostate (Faria et al 2008, Lekka et al 2012b), bladder (Lekka et al 1999) and kidney (Rebelo et al 2013) cancer types have mechanical properties been determined and correlated with the disease stage. Thus, the single cell deformability analysis seems to be a suitable tool for establishing the Young's modulus (cellular stiffness) as a reliable and quantitatively accessible biomarker for cancer-dependent alterations. The mechanical biomarker 'stiffness' should be related to the studies investigating the classical medical histological grading of cancers such as ovarian cancer (Xu et al 2012). Similar to other cancer types, the Young's modulus of non-malignant immortalized ovarian surface epithelial cells is 2.472 ± 2.048 kPa, whereas the Young's moduli of two cancer cell types that are derived from the same ovarian cancer cell lines such as HEY (0.884 ± 0.529 kPa and HEY A8 (0.494 ± 0.222 kPa) are dramatically decreased (Xu et al 2012). Moreover, the migration and invasion rates of both HEY and HEY A8 cancer cells are higher than those of the immortalized ovarian surface epithelial cells (control), which suggests that the cellular stiffness is inversely correlated with the motility and subsequently the metastatic capacity.
The finding of softer single cancer cells compared to their normal counterparts, seems to contradict the finding of cancer are sensed by palpation as a solid and stiff mass of cells within tissues. In line with this, the Young's modulus of tissues of solid primary tumors is often higher than that of normal tissues (Levental et al 2007, Rebelo et al 2013, Lekka 2016). The differences may be attributed to the diverse methods employed for analysis of the Young's modulus that for tissue samples in the macroscopic length scale (a sample of mm or cm size) and hence the elasticity is based on the overall mechanical response of the entire sample volume without a distinct separation on constituents such as single cells or extracellular matrix components. Therefore, the increased deformability of cancer cells that have been detected using the AFM technique can be associated with their ability to display enhanced migration and invasion such as seen for ovarian cancer cells (Xu et al 2012). Whether this relation is also eminent in other cancer cell types needs to be analyzed. However, the increased stiffening of primary tumors detected by macroscopic measurements revealed that the extracellular matrix components seem to play a crucial role in providing mechanical properties of tissues and subsequently, the malignant progression of cancer (Krouskop et al 1998). Indeed, in measurements of tissue stiffness using AFM is has been detected that there exist broad distributions of the Young's modulus (Plodinec et al 2012, Lekka et al 2012b, Zhou et al 2013). In particular, for such heterogeneous tissue samples, smaller Young's modulus values can be attributed to the elasticity of single cells, whereas larger values can be attributed to extracellular matrix components such as collagen fibers building dense deposits in several cancer types (Paszek et al 2005, Plodinec et al 2012).
As the role of FLNa on cytoskeletal remodeling, focal adhesion based mechano-coupling and cell motility has been described extensively before, FLNa has been supposed to affect cellular mechanical properties such as stiffness. Indeed, FLNa crosslinked actin filament networks contain several aspects of cellular mechanical properties: Under low shear stresses these networks act as weak elastic solids, as their mechanical behavior is based on the flexible property of FLNa-actin filament crosslinks. However, large shear stresses cannot be supported and cause a strain-stiffening of these networks that exhibit then a non-linear behavior (Gardel et al 2006, Kasza et al 2009a, 2009b). The observed mechanical properties of these cellular networks are provided by the unique structure of FLNa's and how this structure interacts with actin filaments to build orthogonal actin filament branches. Through their high avidity binding towards actin filaments, which is provided by dimerization and multiple binding to the actin filaments via the FLNa ABD and rod1 domains, a strain-stiffening on the FLNa-actin filament cross-linked networks is evoked (Nakamura et al 2007).
Traction stresses in human metastatic breast, prostate and lung cancer cell lines were deteremined by traction force microscopy and metastatic cancer cells were found exhibit increased tractions than non-metastatic counterparts (Kraning-Rush et al 2012). In line with these results this relation was confirmed in the isogenic MCF10AT series of breast cancer cells (Kraning-Rush et al 2012). Moreover, increased matrix stiffness and collagen density further facilitate increased traction forces, and within all variation of matrix properties tested metastatic cells generated higher forces than non-metastatic cells.
1.5. What type of microenvironment such as tumor mass or stroma do cancer cells sense during the process of metastasis?
1.5.1. Cancer cells in the tumor mass (cell cluster).
Primary solid tumors such as carcinomas create during their excessive growth progression an environment for cancer cells that is characterized by rapid establishment of an oxygen gradient, which decreases towards the tumor's center of mass (figure 12). This dramatic change of the cell's growth conditions requires the adaption of cancer cells to the tumor conditions. As the reprogramming of the energy metabolism has been described as one of the two new hallmarks of cancer (Hanahan and Weinberg 2011), cancer cells inside the primary tumor mass experience altered conditions such as enhanced or decreased activation and expression of molecules. The deregulated proliferation such as chronic and uncontrolled growth of cells is essential for ongoing neoplastic diseases such as cancer. Besides the involvement of proliferative signaling pathways, the energy metabolism needs to be adapted to provide enough metabolites for cell growth and division. Normal cells grow by processing firstly glucose under aerobic conditions such as producing pyruvate through the glycolysis cycle, which operates in the cytoplasm of the cells and secondly carbon dioxide is produced in the mitochondria.
Figure 12. Physical anomalies of the tumor stroma are interrelated such as altered extracellular matrix stiffness, increased solid and interstitial pressure, hypoxia and abnormal (chaotic) vascular system within the primary tumor causing increased permeability for cells and substances. Another factor is the presence of small Rho GTPases such as the RhoA, which facilitate the growth factor regulation of the migration modes by supporting the lobopodial migration mode or the blebbing migration mode. The matrix mechanical properties such as elasticity (stiffness) can favor a distinct migration mode such lamellipodial (non-linear matrix elastic behavior) or lobopodial (linear matrix elastic behavior) migration modes.
Download figure:
Standard image High-resolution imageIn contrast, cells within the solid tumor mass are forced to grow under anaerobic conditions similar to cells such as bacteria utilizing fermentation processes such as anaerobic glycolysis. Thus cancer cells still fully support and prefer the process of glycolysis, but impair the pyruvate degradation in the oxygen-requiring mitochondria. The characteristic feature of cancer cell's energy metabolism is that cancer cells can restrict their glucose metabolism to glycolysis has been described initially by Otto Warburg and termed thereafter aerobic glycolysis (Warburg 1930, 1956a, 1956b). The switch to the aerobic glycolysis of cancer cells has been established and is known as a novel hallmark of cancer termed the preprogramming of energy metabolism, which seems to be at the first glance unexpected, as the cancer cells have chosen an approximately 18-fold lower efficiency of ATP production compared to ATP generated by an oxidative phosphorylation based on the citrate acid cycle inside the mitochondria. How can the cancer cells overcome the lower efficiency of the glycolysis-based ATP generation? Indeed, it has been shown that cancer cells can overcome the inefficiency by upregulation of glucose transporters such as GLUT1 leading to an enhanced glucose import into the cytoplasm (DeBerardinis et al 2008, Hsu and Sabatini 2008, Jones and Thompson 2009). Moreover, increased glucose levels in the cytoplasm have been detected in numerous human cancer types, which have been obtained through increased glucose uptake. The uptake of glucose of these cancer cells has been measured via the uptake of a reporter, which is the radiolabeled analog of glucose such as 18F-fluorodeoxyglucose (FDG), using positron emission tomography (PET) (Haberkorn et al 1991, Vesselle et al 2000).
The glycolytic energy generation is known to be correlated with increased activated oncogenes such as RAS and MYC and also connected with mutations in tumor suppressor genes such as TP53 (DeBerardinis et al 2008, Jones and Thompson 2009). These tumor suppressor gene alterations in cancer cells have supported those mutations with increased capacity for cell proliferation, suppression of cytostatic controls and impairment of apoptosis, which are all three hallmarks of the malignant progression of cancer (Hanahan and Weinberg 2011). In turn, under hypoxic conditions the dependence on glycolysis is further promoted and hence enhanced in several tumors. In more detail, the hypoxia response utilizes a multifactorial signaling cascade increasing the amount of glucose transporters and many enzymes of the glycolysis cycle (DeBerardinis et al 2008, Jones and Thompson 2009, Semenza 2010a). Finally, the Ras oncogene and hypoxia can individually elevate the HIF1alpha and HIF2alpha transcriptions factors, which then increase the glycolytic production of ATP (Kroemer and Pouyssegur 2008, Semenza 2010a, 2010b). However, it is still not yet understood why cancer cells switch from the mitochondrial oxidative phosphorylation dependent ATP energy production to the inefficient glycolytic ATP energy production. One hypothesis has been first raised by Potter (1958) and is further expanded by Vander Heiden et al (2009). The hypothesis states that enhanced glycolysis provides glycolytic intermediates which can themselves act in numerous biosynthetic signaling pathways such as the generation of nucleosides and amino acids. In turn, the generation of such products induces the biosynthesis of biomacromolecules and entire organelles, which are essential for the formation of new cells. Hence, the Warburg-dependent metabolism is proposed to be employed in several fast proliferating tissues such as embryonic tissues, which also promotes its role in large-scale and fast biosynthesis programs that are active in enhanced proliferating cells.
In certain tumors, two subpopulation of cancer cells have been identified that can be distinguished by their generation of energy. The first subpopulation uses the Warburg-dependent metabolism (also termed Warburg-effect) that can be detected by the secretion of lactate and the second subpopulation uses the lactate as a substrate for their main energy generation by utilizing parts of the citrate acid cycle (Semenza 2008, Feron 2009, Kennedy and Dewhirst 2010). Thus, both subpopulations act together in symbiosis, as the first subpopulation of hypoxic cancer cells depends on glucose for energy generation and produces lactate that is exocytosed as waste and endocytosed by the second subpopulation and then used for the highly efficient oxygenated energy production. However, this mechanism needs to be verified for different cancer types, but it is not a unique feature of primary tumor growth, as muscles utilize the same mechanism for energy production (Semenza 2008, Feron 2009, Kennedy and Dewhirst 2010). Moreover, it has been revealed that oxygenation can range from normoxia to hypoxia in solid tumors, but underlies continuous and varying fluctuations in time and space (Hardee et al 2009), which is based on the disordered nature of neovasculature within the solid tumor mass depending on assembly and disassembly and the dynamically changing its architecture. The altered energy metabolism has been identified as a characteristic feature of cancer cells and established as another basic and properly also core hallmark of cancer, as the aerobic glycolysis can be seen as another phenotype of cancer cells evoked by oncogenes that facilitate cell proliferation (Hanahan and Weinberg 2011). Support for determining energy metabolism as a hallmark of cancer comes from the finding that activating (gain-of-function) mutations in enzymes of the citrate acid cycle such as the isocitrate dehydrogenase 1/2 (IDH) have been detected in gliomas and specific other human tumors (Yen et al 2010). The mutations of IDH can be selected as certain cell clones that still possess reproducibly an altered energy metabolism. However, it needs be answered whether the hallmark of an altered energy metabolism is independent of other core hallmarks such as decreased genome stability and increased angiogenesis and invasion that can also be caused by elevated oxygenation and stability of HIF-1 transcriptions factors, respectively (Reitman and Yan 2010). Hence the hallmark of altered energy metabolism is regarded as an emerging hallmark, which shows still its importance in cancer progression, but leaves the question of independency of other core hallmarks unanswered. The different metabolism can additionally affect the mechanical properties of cancer cells. The less oxygenation of the core of the primary tumors may additionally cause a stiffening of the core by sclerosis formation by dying cancer cells.
1.5.2. Heterogeneity of cancer cells in tumors and the microenvironment.
Inside solid tumors exist an enormous heterogeneity, which is also reflected in the heterogeneity of circulating tumor cells (CTCs) and subsequently in multiple spread single new tumor seeds. A good example is prostate cancer, which represents a heterogeneous tumor mass that has been built by different cancer cell clones (Andreoiu and Cheng 2010, Cooper et al 2015). Metastases formation of prostate cancer cells is usually driven by polyclonal seeding of several diverse cancer cell clones (Gundem et al 2015), however, opposite approaches exist that propose that conserved so-called driver lesions that facilitate metastases occurrence in targeted organs or tissues of individual organisms (Kumar et al 2016). The knowledge of these driver lesions seems to be promising in selecting distinct treatments on the basis of predicted molecular vulnerabilities in the patient for a fast and positive treatment response. Hence these personalized treatments predict the susceptibility of the individuals to specific therapeutics, whereas those individuals with malignancies without the alteration will not respond to this treatment. The personalized treatment approach is supported by studies that report the existence of an extensive diversity in genomic aberrations and treatment responses even in cancers of the same histological classification (Perou et al 2000, Tannock et al 2004, Frattini et al 2013, Sato et al 2013, Verhaak et al 2013).
The individualized treatment strategies are based on distinct molecular biomarkers. Since genetic instability in cancer cells exists, the monoclonality of these cells can be vanished by becoming genetic heterogeneous during the progression of cancer (Zhang et al 2016a, b). The heterogeneity can exist between the primary tumor and secondary metastases, between various metastatic lesions and within the primary or secondary solid tumors. Since the tumor heterogeneity is based on diverse protein functions, the diagnosis and treatment are no longer precise and efficient. In order to detect and monitor tumor heterogeneity CTCs can be easily analyzed to identify the mutations and expression alterations in the individual patient (Zhang et al 2016a).
1.5.3. Environmental changes in solid tumors range from normoxia to hypoxia and impact cancer cell mechanics and motility.
A major feature of solid tumors of epithelial origin are hypoxic (Wilson and Hay 2011), which is a consequence of inefficient and dysfunctional vascularization of tumors (figure 12). Moreover, tumor endothelial vessels are often leaky, consist of cancer cells within the vessel lining that mimic endothelial cells by adapting their gene expression pattern such as VE-cadherin expression, chaotic and heterogeneous organization of the vessels. In addition, the branching of the vessels occurs irregularly and vessels contain abnormal shunting that alters the blood flow causing a heterogeneous blood flow (Carmeliet and Jain 2011), which then establishes endogenous hypoxia gradients in the entire tumor including extremely low partial pressure levels of oxygen (pO2) in distinct regions of the primary tumor mass (Li et al 2007). In turn, the primary tumor can adjust their metabolism to the reduced oxygen levels, which establishes a cell-layered metabolite gradient in the tissue. In summary, the vascular heterogeneity of the vessels evokes extracellular oxygen gradients (or pO2), which then changes the intravascular pO2 and subsequently alters the metabolic consumption rate of cancer cells and stromal cells within the entire tissue. Finally, the combination of all events causes pO2 and metabolite gradients inside the tissue, which in turn have a major effect on the behavior of cancer cells. Moreover, also the mechanical properties of cancer cells may be altered by the environmental conditions such as oxygenation and metabolite concentrations.
1.5.4. Alterations of the 'normal' pH gradient in the tumor mass may impact cancer cell mechanics and motility.
The intracellular pH (pHi of 7.2) of differentiated epithelial cells is slightly lower than the extracellular pH (pHe of 7.4). Within a primary tumor the pH can alter dramatically, as it depends on the pO2 content and the availability of metabolites that converts a normal pH gradient either to a higher pHi of 7.4 (Webb et al 2011) or a lower phe of 6.5–7.0 (Stuewe et al 2007, Gallagher et al 2008, Hashim et al 2011). Alterations in the pH inside of cancer cells are based on the increased expression and/or activity of ion pumps and ion transporters such as H+ ATPases (Martinez-Zaguilan et al 1993),  –
– exchanger NHE1 of the SLC9A family (McLean et al 2000), and monocarboxylate–
exchanger NHE1 of the SLC9A family (McLean et al 2000), and monocarboxylate– efflux cotransporters MCT1 and MCT4 or the SLC16A family (Pinheiro et al 2008, Chiche et al 2012) that regulate the efflux of
efflux cotransporters MCT1 and MCT4 or the SLC16A family (Pinheiro et al 2008, Chiche et al 2012) that regulate the efflux of  , which causes the high pHe. During hypoxia, the activity of the carbonic anhydrases CAIX and CAXII is increased and hence enhances the hydration of extracellular CO2 to
, which causes the high pHe. During hypoxia, the activity of the carbonic anhydrases CAIX and CAXII is increased and hence enhances the hydration of extracellular CO2 to  and
and  (Chiche et al 2009, Swietach et al 2009). Subsequently, the decreased blood flow velocity and the loss of oxygen at the early stages of solid primary tumors can switch the metabolism from a citrate acid cycle based to a glycolysis-based metabolism (Gatenby and Gillies 2004). In particular, the enhanced flux of carbons, which is a result of the glycolytic metabolism that leads to an acidification of the extracellular pH around the primary tumors, which is even increased in luminal structures (Barathova et al 2008). Similar to pO2 and metabolite gradients, primary tumors create pH gradients, which in turn affect the behavior of cancer cells and also leads to the broad heterogeneity of tumors including their mechanical properties that evoke alterations in cellular migration.
(Chiche et al 2009, Swietach et al 2009). Subsequently, the decreased blood flow velocity and the loss of oxygen at the early stages of solid primary tumors can switch the metabolism from a citrate acid cycle based to a glycolysis-based metabolism (Gatenby and Gillies 2004). In particular, the enhanced flux of carbons, which is a result of the glycolytic metabolism that leads to an acidification of the extracellular pH around the primary tumors, which is even increased in luminal structures (Barathova et al 2008). Similar to pO2 and metabolite gradients, primary tumors create pH gradients, which in turn affect the behavior of cancer cells and also leads to the broad heterogeneity of tumors including their mechanical properties that evoke alterations in cellular migration.
1.5.5. Electric field alterations in the tumor mass may alter cancer cell mechanics and motility.
In cell and tissue homeostasis the electrical signaling has a fundamental role. In particular, the potential of the cell membrane is based on the unequal distribution of ions over the cell membrane, which leads subsequently to differences in voltage between the cytoplasm and the extracellular microenvironment of the cell (Yang and Brackenbury 2013). Within epithelial tissues exist natural electrical fields that are kept constant through the spatial organization of ion pumps. In particular, the apical part of the cell possesses highly enriched in  channels and Cl− transporters, whereas the basolateral part of the cells consists of
channels and Cl− transporters, whereas the basolateral part of the cells consists of  –K+ ATPases (McCaig et al 2009). These ionic gradients cause an extracellular ionic current flow that subsequently creates endogenous voltage gradients. As normal healthy epithelial cells are highly polarized, the membrane potential of cancer cells seems to be often depolarized that leads to a decrease in transepithelial potential difference (James et al 1956, Faupel et al 1997). Moreover, depolarized cancer cells exhibit enhanced intracellular
–K+ ATPases (McCaig et al 2009). These ionic gradients cause an extracellular ionic current flow that subsequently creates endogenous voltage gradients. As normal healthy epithelial cells are highly polarized, the membrane potential of cancer cells seems to be often depolarized that leads to a decrease in transepithelial potential difference (James et al 1956, Faupel et al 1997). Moreover, depolarized cancer cells exhibit enhanced intracellular  levels, stable K+ levels, and Cl− as well as Ca2+ influx (Yang and Brackenbury 2013). The depolymerization of the primary tumor creates an extracellular voltage gradient between normal healthy cells and cancer cells (Cuzick et al 1998), which can vary largely within one single primary tumor and differ widely between different tumor types. In rat prostate glands, a steady direct current electrical field of 500 mV mm−1 measured across the luminal wall can be detected and the transepithelial electrical potential difference of −210 mV has been measured, which leads to a negatively charged lumen (Szatkowski et al 2000, Djamgoz et al 2001). However, in the mammary duct, the transepithelial electrical potential is +30 mV that leads to a positively charged duct lumen (Pu et al 2007). Electric field alterations between normal and cancerous tissues may cause a galvano-based cell migration mode due to the electric field changes that are based on Ca2+ ion concentration differences. These differences in Ca2+ ion concentration may also have an impact on cellular mechanical properties that regulate the migration and invasion of cancer cells.
levels, stable K+ levels, and Cl− as well as Ca2+ influx (Yang and Brackenbury 2013). The depolymerization of the primary tumor creates an extracellular voltage gradient between normal healthy cells and cancer cells (Cuzick et al 1998), which can vary largely within one single primary tumor and differ widely between different tumor types. In rat prostate glands, a steady direct current electrical field of 500 mV mm−1 measured across the luminal wall can be detected and the transepithelial electrical potential difference of −210 mV has been measured, which leads to a negatively charged lumen (Szatkowski et al 2000, Djamgoz et al 2001). However, in the mammary duct, the transepithelial electrical potential is +30 mV that leads to a positively charged duct lumen (Pu et al 2007). Electric field alterations between normal and cancerous tissues may cause a galvano-based cell migration mode due to the electric field changes that are based on Ca2+ ion concentration differences. These differences in Ca2+ ion concentration may also have an impact on cellular mechanical properties that regulate the migration and invasion of cancer cells.
1.6. During cancer progression distinct cancer cells leave the primary tumor
Commonly (regardless of the solid cancer type), the process of cancer metastasis from a biophysical point of view starts with the dissemination of cancer cells to surrounding healthy tissues and organs, which are located far beyond the initial tumor origin site and supports the initiation of secondary new tumors (termed metastases). This single event can cause death in cancer patients. At the time point of the first cancer diagnosis, many patients exhibit clinically detectable metastases and micrometastases that cannot easily be detected. The most life-threatening step in cancer disease is the occurrence of metastasis, which is a process that follows a clear linear propagation (termed metastatic cascade) with interruptions, when the cancer cells stay dormant in specific niches. The process of metastasis increases the complexity of cancer and turns it into a multiplex disease. During the progression of the metastatic cascade, alterations in cell-cell and cell-matrix adhesion receptors occur likely and are highly important in malignant cancer progression (Martin and Jiang 2009). The process of metastasis involved at least major steps such as the spreading from the primary tumor, the invasion into the surrounding tissue, intravasation into vessels and possibly extravasation out of the vessels into the targeted tissue sites. The increased cell-cell adhesion strength causes the dissociation of malignant cancer cells to migrate out of the primary tumor mass. In parallel the cell-matrix adhesion changes enable the cancer cells to migrate into the surrounding tumor stroma.
The invasion steps also utilize proteolytic degradation of the basement membrane and the extracellular matrix and in addition, the cell surface receptor expression is adapted to cell migration and invasion. When the primary tumor reaches a size larger than 2 mm in its diameter, the ingrowth of vessels into the tumor is induced, which transports the waste away from the tumor and supports it with oxygen (Brooks 1996). As the metastatic cascade mainly depends on loss of adhesion strength or even the depletion of adhesion receptors, the ability to change the expression of distinct cell-cell adhesion molecules such as E-cadherin have been the classical model for the initiation of cancer dissemination from the primary tumor and hence the malignant progression of cancer. In order to determine the onset of the malignant progression of cancer, alterations in the mechanical properties of cancer cells may help to predict their migratory capacity and subsequently the invasive phenotype of cancer cells. Moreover, these cellular mechanical property alterations may determine the mode of cellular migration that is also affected by the environmental cues such as matrix mechanical properties.
At the periphery of tumors or spheroids cells undergo a continuous transition from a round morphology in the spheroid core to a highly aligned elongated morphology, which is driven by both β1-integrin-based cell-matrix adhesion and myosin II/ROCK-based cell contractility (Valencia et al 2015). This isotropic to anisotropic transition of the cells is related to a shift in cell migration, such as a slow and unpolarized movement at the spheroid core is turned into a fast, polarized and persistent movement at the spheroid periphery (Valencia et al 2015). The conversion of the migratory phenotypes can be facilitated by the extracellular matrix environment that exerts anisotropic contractile stresses on the cells at the periphery (Valencia et al 2015).
When investigating the tissue level, a wetting approach can be employed (Wallmeyer et al 2018). Cell can be seen as a liquid drop on a surface, which has three interfaces carrying mechanical tension. When an interfacial force equilibrium is to be maintained during the quasi-static spreading process, the wetting physics can be a useful tool for predicting the change in contact angle over time. Although experimental values vary considerably, the model permits us the scaling of all measured contact angle dynamics to a single master curve that describes the collective cell movement. Fundamental and complex developmental mechanisms, as they occur at the onset of embryogenesis can be explained by just three main parameters: firstly, the offset tension strength, which indicates the interfacial tension strength compared to other force-generating mechanisms, secondly, the stress (tension) ratio between the different interfaces and thirdly, the stress variation rate, which defines the time scale of the whole process (Wallmeyer et al 2018).
2. General physical migration models depend on the microenvironmental cues
There exists a high plasticity in the migration modes (synonymously termed migration types) utilized by cancer cells depending on the cancer cell type, cancer cell mechanical properties, cell adhesion molecules, capability of matrix degradation, the microenvironmental cues such as matrix composition, pore/mesh size and cross-linking state or matrix mechanical properties such as stiffness (Wolf et al 2013, Kassianidou et al 2017, Mierke et al 2017, Paul et al 2017, Malandrino et al 2018). A specific cell employs a certain migration mode and can switch between migration modes, which is termed transition. The switch is mediated by mechanical cues such as external stress or by up- and down-regulation of characteristic migration mode specific molecules (Thiery 2002, Wolf et al 2003, 2013, Mierke 2014, Paul et al 2016, Mierke 2017).
2.1. Introduction in migration modes of cancer cells
Migration modes have been initially identified by describing them as morphology based different types of migration strategies. However, a precisely defined functional description is still not yet fully available. The current knowledge is that there exist at least two basic migration modes: a protrusion-based mesenchymal migration mode, in which the cells are polarized and a blebbing driven migration mode that exhibits a rounded cellular morphology without any protrusions. As it is mostly the case, there is an intermediate migration mode that involves both the exertion of protrusions such as lobopodial structures and the blebbing of the cell's membrane. In this lobopodial migration mode, the cells are still polarized. Besides these migration modes, there exists compartmentalized pressure-based migration modes and a migration mode that is driven by osmosis. In the following, all migration modes will be described in more detail.
2.2. Protrusive migration mode
The migration and invasion of cells in three-dimensional (3D) extracellular matrices is on the one hand a prerequisite for tissue assembly, homeostasis and regeneration, immune cell trafficking upon cell injury such as wounds and on the other hand cellular motility plays a role in numerous diseases such as cancer. During the malignant progression of cancer disease, the process of metastasis depends on the migration of single cancer cells that spread out of the primary tumor and breach through endothelial vessel linings. In more detail, the migration of cancer cells through the extracellular matrix protein network building connective tissue is a cyclic process that involves several steps: firstly the actin polymerization-dependent protrusion of pseudopods at the leading edge; secondly the integrin-facilitated adhesion to the extracellular microenvironment; thirdly the degradation of the nearby extracellular matrix proteins evoked by its cleavage through membrane exposed proteases such as matrix-metalloproteinases; fourthly the actomyosin-driven contraction of the cell's body, enhancing longitudinal tension; and fifthly the retraction of the cell's rear part in the direction of the cell migration front leading to a translocation of the whole cell (Doyle et al 2012). These five steps describe only one special migration mode that can be chosen by aggressive and metastatic cancer cells of epithelial origin. Hence, this distinct mode of migration is named the protrusive migration mode (figure 13) (Maruthamuthu and Gardel 2014). Moreover, it has been shown that cells with low adhesion site formation rates prefer to move perpendicular to adhesive stripes, whereas cells with high adhesion site formation rates prefer to move parallel to adhesive substrate stripes. Hence, effects of different substrate pattern geometry and the actin polymerization strength seems to determine on the directionality of cell crawling behavior (Mizuhara et al 2017).
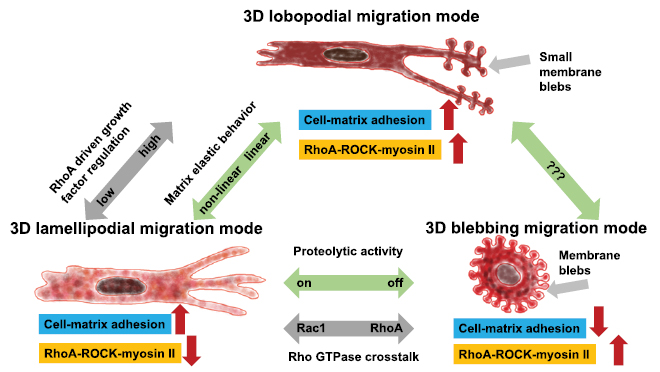
Figure 13. In 3D migration and invasion of cells into confined extracellular matrix microenvironments, the migration modes can be switched from protrusive migration modes to blebbing and hence amoeboid migration modes, when the cells are impaired in proteolytic degradation of the matrix.
Download figure:
Standard image High-resolution image2.3. Cellular membrane blebbing occurrence
2.3.1. Blebbing-driven programmed cell death (apoptosis) or blebbing-driven mode of migration?
There is exists no common agreement on the blebbing-driven migration mode and on its importance in cell migration and invasion of fibroblasts and cancer cells. However, it is proposed that during the blebbing mode of migration, a cell has connections parallel to surrounding collagen fibers in a 3D extracellular matrix confinement balancing itself inside the collagen fiber network and keeping its entire cell body in weak contact with the collagen fiber bundles (figure 13). The blebbing mode of migration can be either the driving factor of cell migration and invasion or a side effect of migration caused by the continuous secretion of vesicles during cell migration. Another possibility for the occurrence of membrane blebbing during cell migration is the event of a sudden programmed cell death (termed apoptosis). During this process, the cells exert blebs on their cell surface and finally rupture into pieces by themselves. The induction of apoptosis during migration seems to be likely, as confinements of the microenvironment may cause nuclear ruptures and affect cell transcriptional gene expression that finally lead to apoptosis. These nuclear ruptures can appear, when the confinement such as the pore size is under the limit of the nuclear deformability. Some of these nuclear ruptures can be repaired and others are more severe and hence lead to apoptosis. It has been reported that these nuclear ruptures can also be associated by nuclear blebs. For the occurrence of blebs on the cell surface arises the main question of how the blebbing mode of migration can be distinguished from the blebbing events during apoptosis? An answer may be that the blebbing during migration is highly reversible and the blebs appear to be smaller.
2.4. Blebbing migration mode
Besides the well-known protrusive migration mode, there seems to be other modes of cancer cell migration, such as the blebbing migration mode. In this blebbing mode, the cells extend and retract suddenly membrane blebs, in which the membrane has no contact to the actin cytoskeleton (Laser-Azogui et al 2014). When cells use this type of migration mode, it not yet well understood and still subject of ongoing research. Moreover, it is not clear whether the protrusive and the blebbing migration modes are clearly distinguishable from each other or whether mixtures exist that involve both phenomena, the formation of protrusions and the rapid extension and retraction of blebs on these protrusions. Which migration mode is preferred by a specific type of cancer cell and how the switch of migration modes is affected by the specific biomimetic environmental conditions is still elusive. Morover, the blebbing movement can be used to induce hydraulic fractioning of cells and tissues, which is termed interfacial fracture and can be described by Griffith's theory (Arroyo and Trepat 2017). A force is applied to a deformable material, such as a cell or tissue. More precisely, the mechanical work on the cell or tissue, such as the applied force times the resulting displacement, is saved in it as elastic energy. In order to relax, the cell or tissue can crack and hence suddenly release the stored elastic energy. The new free surface within the material requires interfacial energy. Based on Griffith's theory, the fracture is a competition between external work, such as hydraulic pressure in fluid-filled cracks, elastic energy release and interfacial energy.
2.5. Lobopodial migration mode
In addition to these two migration modes (protrusive migration mode and blebbing migration mode), there is another migration mode described, which is named the lobopodial migration mode that can be seen as an intermediate state that is located between the blebbing and the protrusive migration mode phenotypes, in which both migration modes can be simultaneously employed (figure 13). This lobopodial mode has not been observed in cancer cells, however, so far it has only been detected in fibroblasts. Thus, we suggest that cancer cells also choose this lobopodial migration mode under certain circumstances. Hence, further effort is needed to analyze whether and how cancer cells are able to use this fibroblastoid, lobopodial migration mode.
As it has been described above, the capability of cancers to metastasize depends on the cancer cell's ability to break through the basic tumor mass and basement membrane, migrate out of the primary tumor and invade connective tissue, adhere, and possibly breach through and transmigrate through a barrier such as basal membrane and the endothelium of blood and lymphoid vessels. Alternatively, cancer cells can enter the vascular system directly in the primary tumor as many tumor-endothelial vessels are leaky or consist of cancer cells mimicking endothelial cells.
2.6. Compression–based migration mode depends on osmosis
In vivo, cells mostly migrate into 3D extracellular matrices and invade further through them. Thereby, cells experience 3D longitudinal tracks within the extracellular matrix with bordering 2D interfaces that are similar to channels. These channels are assembled by the connective tissue and basement membranes of various tissues such as muscle, nerve and epithelium (Friedl and Alexander 2011). These 3D longitudinal tracks or channels are generated in between adjacent bundled collagen fibers inside fibrillary, interstitial tissues. In addition, cells can migrate in vivo through 3D channels (Alexander et al 2008). The cross-sectional areas (Wolf et al 2009) of those channels can range in vivo range from 10 to >300 µm2, which indicates that cells migrate under varying physical confinement pressures. Moreover, it has been suggested that physical confinement itself may have the capacity to impact the migration of cells and induce a switch the migration mode (Pathak and Kumar 2012, Balzer et al 2012, Konstantopoulos et al 2013, Stroka et al 2013). In addition, it has also been shown that physical confinement such as bi-axial restriction alters the cell-cycle of sarcoma cells by reducing cell division (Moriarty and Stroka 2018).
However, cell migration and invasion through physically confined spaces such as channels even exists in the absence of the hallmarks of 2D migration on flat substrates such as the polymerization of actin and the myosin II-based contractility of the cells. Thus, another migration mechanism seems to be involved that depends on the permeation of water into the cell. The permeation of water is based on both active and passive ion transport mechanisms under spatial confinement. This migration mode is termed the osmotic engine model and represents a compression-driven migration mode that is regulated by the permeation of water, which is based on an osmotic process, caused by the semipermeable cell membrane and cellular polarization (figure 14) (Stroka et al 2014). In more detail, the osmotic engine migration mode is driven by the cell-volume regulation, which is provided by fluxes of ions and water into and out of the cell. A polarized cell inside a narrow channel confinement can create a spatial gradient in the distribution of ion channels and pumps such as  /
/ pumps and aquaporins such as aquaporin 5 over the cell membrane, which evokes a net inflow of water and ions at the cell's leading edge and a net outflow of water and ions at the cell's trailing edge (Stroka et al 2014). The influx of water at the front leads to a displacement of the entire cell, as the water flow based by difference of the osmotic pressure differences across the cell membrane (Lang et al 1998). In addition, external osmotic shock at the leading or trailing edges of the cells is supposed to impact cell migration strongly. In 2D cell migration assays, ion channels and aquaporins have been shown to support migration (Schwab et al 2007, Papadopoulos et al 2008). However, the underlying molecular mechanism are not yet clear. As cytoskeletal components have been reported to facilitate the activity of ion channels (Grunnet et al 2002, Dreval et al 2005, Mazzochi et al 2006), the regulation of the cell's volume via these ion pumps demands an intact cytoskeleton.
pumps and aquaporins such as aquaporin 5 over the cell membrane, which evokes a net inflow of water and ions at the cell's leading edge and a net outflow of water and ions at the cell's trailing edge (Stroka et al 2014). The influx of water at the front leads to a displacement of the entire cell, as the water flow based by difference of the osmotic pressure differences across the cell membrane (Lang et al 1998). In addition, external osmotic shock at the leading or trailing edges of the cells is supposed to impact cell migration strongly. In 2D cell migration assays, ion channels and aquaporins have been shown to support migration (Schwab et al 2007, Papadopoulos et al 2008). However, the underlying molecular mechanism are not yet clear. As cytoskeletal components have been reported to facilitate the activity of ion channels (Grunnet et al 2002, Dreval et al 2005, Mazzochi et al 2006), the regulation of the cell's volume via these ion pumps demands an intact cytoskeleton.
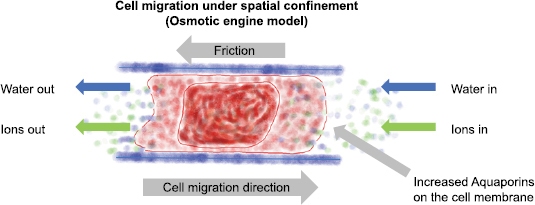
Figure 14. Schematic drawing of the osmotic engine model that is based on osmotic water permeation through the cellular membrane at the cell's leading and trailing edges during migration under spatial confinement. Cell migration is based on the water permeation across the membrane. In particular, the water flows in at the cell's leading edge that causes a cell extension at the leading edge and water flows out at the cell's rear that causes a retraction of the rear. Subsequently, the cell body is translocated forward in the direction of movement.
Download figure:
Standard image High-resolution image2.7. Nuclear piston-migration mode
The investigation of cellular motility in 3D extracellular matrix environments has revealed a multitude of cell migration mechanisms (Friedl and Wolf 2010, Petrie and Yamada 2012, Charras and Sahai 2014). Various cell types possess the capacity to switch between two or even more distinct migration mechanisms, the so-called migration modes, in response to environmental or cellular alterations (Wolf et al 2003, Petrie et al 2012, Liu et al 2015, Madsen et al 2015, Ruprecht et al 2015). Hence, it needs to be deciphered how the plasticity affects the migration modes in order to gain a comprehensive understanding of both normal (physiological) and metastatic (pathological) 3D cell motility. In the absence of proteolytic activity, the cells can switch to other migration modes. It is clear that primary fibroblasts and cancer cells have the ability to switch between distinct migration modes, whereas it is not clearly understood whether these two cell types switch between the same modes or whether their migratory plasticity is solely regulated by highly similar mechanisms.
Recently a novel mode of cell migration has been found that is termed the nuclear piston mode. The nuclear piston mode is actually a forward movement of cells that is based on protrusions of the leading edge and hence cellular polarity (figure 15) (Petrie et al 2017). It has been reported that primary human fibroblasts have the ability to utilize their bulky nucleus as a piston that switches from low to high pressure protrusions in response to the nearby surrounding 3D extracellular matrix scaffold constrains. In a 3D environment, migrating cancer cells have the capacity to alter their mode of migration due to 3D matrix alterations such as collagen concentration, composition or increased crosslinked collagen fibers. However, it is not yet fully clear, whether cancer cells switch between migrations modes that are characterized by high- or low-pressure protrusions similarly as human primary fibroblasts perform their switch. Therefore, the intracellular pressure in front of the nucleus and also behind it, needs to be determined in both cell types, when the cells migrate in 3D confinements. For polarized HT1080 fibrosarcoma cells (metastatic cancer cells), the nuclear piston mechanism is usually inactive, when the cells migrate through a 3D extracellular matrix, whereas it is activated in elongated and polarized cancer cells when the MMP activity is impaired. Hence, dissimilar to human primary fibroblasts, the nuclear piston mode of migration is not found to be active in HT1080 fibrosarcoma cells in the presence of proteolytic activity (Petrie et al 2017). In the absence of proteolytic activity, the nuclear piston mode of migration can be detected, as compartmentalized pressure is observed that is required for the movement of the piston (represented by the bulky nucleus) (Petrie et al 2017). In order to produce a compartmentalized pressure, the nucleoskeleton-cytoskeleton linker protein nesprin 3, actomyosin contractility and integrin-facilitated cell-matrix adhesion need to be present. These features of the nuclear piston migration mode are consistent with lobopodia-driven migration of fibroblasts. However, the activation of the nuclear piston migration mode causes a slower movement of HT1080 fibrosarcoma cells in 3D environments (Petrie et al 2017). Finally, these results show the nuclear piston migration mode can be rescued, when the proteolytic activity is impaired during the migration of polarized cancer cells through 3D microenvironments, which can be identified by analyzing the pressure alterations in different cell compartments at similar levels as in healthy human primary fibroblasts (Petrie et al 2017). In addition, human fibroblasts can alter their migration mode from a low-pressure lamellipodia-driven migration mode to a high-pressure lobopodial protrusion-driven migration mode, when the 3D extracellular matrix is highly crosslinked similarly to mammalian dermis or cell-derived matrices (Petrie et al 2012). In lobopodial primary fibroblasts, actomyosin-based contractility of the cells can pull the nucleus forward similarly as a piston in a cylinder, which enhances the hydraulic pressure of the cytoplasmic compartment in front of the nucleus in the direction of motion (Petrie et al 2014). The compartmentalized pressure seems to be responsible for the migration of cells, as the pressure in the cell's front compartment can push the lobopodial membrane forward in the direction of motion rather than that the actin polymerization driven 'Brownian ratchet' causes the forward movement of the cells. The latter Brownian ratchet mechanism is utilized during the lamellipodial protrusive migration modes.
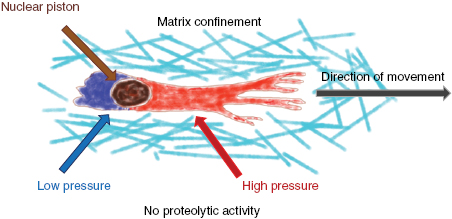
Figure 15. The nuclear piston migration mode. For 3D migration under physical (spatial) confinement, cell possess a non-uniform intracellular pressure: In front of the nucleus and behind the cell's leading edge, the compartment pressure is higher compared to the compartment pressure in the cell's tailing edge (behind the nucleus). The cells move in a certain direction by utilizing the nucleus as a piston the increased the pressure in the cell's leading edge compartment.
Download figure:
Standard image High-resolution image2.8. Amoeboid migration modes
Besides the piston migration modes, non-adherent fibroblasts employ a third migration mode in a 3D microenvironment, that is termed A1 amoeboid (figure 16) (Liu et al 2015). The third type of 3D migration can be investigated when primary fibroblasts are placed between two surfaces, which are treated chemically in order to prevent integrin facilitated cellular adhesion to the surfaces and are then compressed. These fibroblasts undergo then the third type of 3D migration that is termed 'fibroblast amoeboid motility' or A1 motility (Liu et al 2015). As proposed, the cytoplasmic pressure together with a weakening in the cell-matrix adhesions between the cell membrane and the underlying actomyosin cytoskeleton causes the dynamic exertion of membrane blebs, which protrude the cell membrane independently of the formation of lamellipodia (Charras et al 2006). In nearly all cell types, but with the exception of normal epithelial cells, a subpopulation of spontaneously polarizing cells can adapt an amoeboid-like morphology and employ a fast migration mode. Two of these fast migration modes have indeed been identified. The first fast migration mode is termed A1 and similar to the migration of normal human dermal fibroblasts: the cells exhibit a round shape and possess a small leading edge. The second migration mode is termed A2, in which the cells exhibit an elongated ellipsoid morphology of the entire cell body which exerts a large uropod (figure 16) and hence is similar to migrating neutrophil granulocytes. The migration modes can be distinguished by their altered migration speed: A2 cells migrate at significantly faster migration speeds (5.3 ± 1.5 µm min−1) compared to the migration speeds of A1 cells (1.7 ± 0.4 µm min−1) (Liu et al 2015). These migration modes are both faster compared to the relatively low migration speeds of mesenchymal cells (0.234 ± 0.09 µm min−1) (Liu et al 2015). Which of the two fast migration modes such as A1 and A2 is chosen for cell migration depends on the cell type, and within a distinct cell type, the two modes can both exist with varying percentages. Moreover, there can even exist a preference for switching to a distinct migration mode (Lämmermann and Sixt 2009, Liu et al 2015, Callan-Jones and Voituriez 2016). The choice of the A1 or A2 migration mode may be based on minor molecular differences between the cell types or within subpopulations of one cell type.
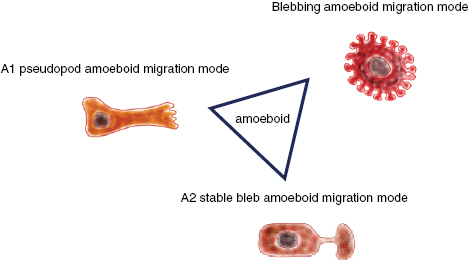
Figure 16. Amoeboid migration modes of cells. Cells can utilize different migration modes such as the blebbing (multiple blebs) migration mode, A1 (pseudopod) or A2 (stable bleb) amoeboid migration modes that are distinguishable in their specific morphology and shape. Under various conditions cells can switch between migration modes. Cell can migrate with a stable bleb (A2) in front through a matrix confinement or they can migrate with a pseudopod that points in the direction of movement (A1). Cells can move by utilizing a blebbing based migration mode. The migration modes are influenced by environmental factors such as adhesion strength, physical confinement strength or by intracellular parameters such as physical fluctuations in cortical contractility, which leads to polarized cells.
Download figure:
Standard image High-resolution imageThe amoeboid metazoan cell migration has been detected as a characteristic feature of single cells such as neutrophils and dendritic cells, when they move and alter rapidly their cell shape due to exertion of small membrane extensions compared to the movement of adherent fibroblasts (Lämmermann and Sixt 2009). In contrast to the protrusive migration mode of mesenchymal cells, amoeboid cancer cells are morphological defined as round cells that can exert membrane blebs at the cell's leading edge (Sahai and Marshall 2003, Wolf et al 2003). There exist two amoeboid and hence adhesion-independent migration modes that are distinguishable on the degree of retrograde actomyosin flow inside the cells. In particular, amoeboid fibroblasts (termed A1) have a protrusion at their leading edge with a distinct pattern of actomyosin flow, whereas the A2 termed mode of amoeboid migration displays a different flow pattern. A fast mode migration is termed A1 and an even faster and more conserved contractile migration mode is termed A2. Among A2 cells are the Walker 256 carcinosarcoma cells, which have the capacity to form blebs during cell migration, and leukocytes, which exert a large stable bleb at the cell's leading edge. During the adhesion-independent amoeboid fibroblast migration the myosin II can flow fast to the cell's rear by utilizing cell's leading-edge protrusions and finally it manages to be probably accumulated and uniformly distributed all over the cell's actin cortex. Moreover, myosin II can be fully dispensed for employment in fast and rapid cellular amoeboid movement in these cells (Liu et al 2015). The driving force of the A2 integrin-independent migration mode is based on a global retrograde flow of actin and myosin II in the central region of the cell, whereas the driving force of the A1 integrin-independent migration mode is based on a retrograde flow that is locally restricted in the cell's leading edge.
As the speed of primary fibroblasts migrating and invading in fibrillar 3D extracellular matrices is found to be reduced after the inhibition of integrins (Petrie et al 2012b), it seems to be likely that this amoeboid fibroblast myosin II independent mechanism is not sufficient to migrate efficiently in more structurally complex fibrillar 3D extracellular collagen or fibroblast-derived matrix scaffolds. A low adhesion-based migration mode of 3D fibroblast migration is supposed to be based on low traction forces (Bergert et al 2015), however, the connection to intracellular pressure and protrusion exertion still needs to be figured out. In addition, whether pressure or decreased adhesion strength are crucial for the formation of various bleb types during the amoeboid migration mode of cancer cells needs to be determined. In conclusion, there remains still the question unanswered whether the classical round amoeboid migration mode of cancer cells is mechanistically similar to the adhesion-independent A1 and A2 amoeboid migration modes.
2.9. Actin flows are characteristic features of mesenchymal and amoeboid migration
It has been hypothesized that common features, such as actin flows (Theriot and Mitchison 1991, Svitkina et al 1997, Hawkins et al 2011, Poincloux et al 2011, Heuze et al 2013, Paluch and Raz 2013), which have been identified as a hallmark of mesenchymal migration (Theriot and Mitchison 1991, Svitkina et al 1997) contribute in general to the migration of cells. Moreover, the actin flows have been identified to play a role in amoeboid migration (Callan-Jones and Voituriez 2016). Hence, it can be hypothesized that common elementary mechanisms drive the different migration modes of cells, although at the first glance these migration modes, such as mesenchymal or amoeboid migration, seem to be largely divergent. To fill be gap between the molecular scale of cellular motility and the mesoscopic scale, the cell behaviors during migration are described commonly by statistical physics (Jülicher et al 2007, Prost et al 2015). Indeed, a basic physical property of the acto-myosin system, such as the spontaneously flow at the cell scale (Voituriez et al 2005, 2006, Salbreux et al 2009, 2012), is the driving factor of both mesenchymal and amoeboid migration modes and additionally facilitates the polarity of the cell. The path (or trajectories) on which a cell moves through the microenvironment are supposed to depend on the actin flow based polarity of the cell (Callan-Jones and Voituriez 2016). Basically, the acto-myosin system converts the chemical energy, such as the hydrolysis of ATP, into mechanical energy. Thereby, two mechanisms are employed: the first mechanism is based on the actin filament polymerization, which leads to protrusive forces, and the second mechanisms is the myosin (commonly myosin II) driven active cross-linking of actin filaments, which causes the contraction of cell structures, such as actin stress fibers. Although on the molecular scale these processes are identified, on the mesoscopic scale principles from condensed matter physics, such as active gel theory, have been employed to determine the impact on motility (Kruse et al 2004, 2005, Jülicher et al 2007). Indeed, analysis of mesenchymal migration mode has been performed largely in the prototypical keratocytes that migrate in flat adhesive substrates (Theriot and Mitchison 1991, Svitkina et al 1997, Keren et al 2008, Wilson et al 2010), and hence several physical models have been successfully employed (Jülicher et al 2007, Callan-Jones et al 2008, Keren et al 2008, Blanch-Mercader and Casademunt 2013, Danuser et al 2013).
Since the amoeboid phenotype with a broad uropod and a smoothly rounded cell front appears to be conserved among cell lines in non-adhesive, confined microenvironments, it is referred to as a stable bleb phenotype in relation to that found in zebrafish embryonic cells (Ruprecht et al 2015). This stable bleb phenotype is driven by the retrograde flow of cortical actin and the contractile rear of the cell. Since the transmission of force towards substrate is independent of adhesion molecules, it is hence based exclusively on friction (Byun et al 2013). The major characteristics of the stable bleb phenotype are strongly preserved and can be modeled with physical theories, based essentially on the active gel theory (Hawkins et al 2011, Callan-Jones and Voituriez 2013, Recho et al 2013, Bergert et al 2015, Liu et al 2015, Ruprecht et al 2015), and can be reproduced in reconstituted artificial systems (Abu Shah et al 2014).
The advection by actin flow leads to the establishment of polarity in early Caenorhabditis elegans embryos (Munro et al 2004, Goehring et al 2011), and seems to be crucial for the mechanisms of pattern formation in acto-myosin systems (Goehring et al 2011, Howard et al 2011, Goehring and Grill 2013, Maiuri et al 2015). In migrating cells, such as bone marrow dendritic cells, actin retrograde flows are supposed to influence the concentration profiles of actin-interacting molecules: faster retrograde actin flows are said to lead to steeper concentration gradients, with higher concentrations at the rear (posterior) and lower at the front (anterior) (Renkawitz et al 2009, Maiuri et al 2015). The concentration gradients of actin interacting proteins may be particularly valid to polarity cues, leading to the conclusion that an increase in the retrograde flow velocity of actin should stabilize cell polarity by increasing the asymmetry of concentration profiles of polarity cues (Maiuri et al 2015).
Accordingly, advection by actin flows can trigger a positive feedback loop that should be sufficient to polarize the cell and facilitate migration (Hawkins et al 2011, Callan-Jones and Voituriez 2013, Recho et al 2013, Ruprecht et al 2015, Liu et al 2015). The candidates for the feedback loop are myosin II or the actin polymerization regulators Arpin (Dang et al 2013, Gorelik and Gautreau 2015), whose rearward accumulation has been demonstrated to be pronouncedly improved due to faster actin flows. In turn, steeper gradients of myosin II increase actin retrograde flows and hence enhance cell polarity. This mechanism can be described by active gel theory, which, for general reasons, predicts that an acto-myosin cortex, when sufficiently contractile, is unstable and spontaneously acquires a polarized state with retrograde actin flows and accumulation of actin myosin at the rear at the mesoscopic scale (Hawkins et al 2011, Callan-Jones and Voituriez 2013, Recho et al 2013, Lui et al 2015, Ruprecht et al 2015). Cell-to-cell differences are identified by analyzing a single parameter to adjust the balance between the formation of polarity and intracellular noise (Alonso et al 2018).
3. Physical role of the cell's nucleus in cancer cell migration
The motility of cells through 3D tissue microenvironments is based on a balance between cellular deformability and physical tissue restrictions. The rates of cell migration are regulated additionally by the ability of migrating and invasive cells to degrade their surrounding confinements such as the extracellular matrix by utilizing matrix degrading enzymes such as matrix metalloproteinases (MMPs). Besides the matrix degradation capacity of cells, the migration speed and efficiency rely on the integrin and actomyosin based mechanocoupling of the cells with their environment. However, it is still elusive how these parameters are coordinated and cooperate in confined space. When the MMP-degradable collagen fiber matrixes or non-degradable substrates with defined and non-varying porosity are utilized, the limits of cellular motility can be quantitatively analyzed under distinct physical constraints. The migration capacity of cells has been reported to be dependent linearly on pore-size and deformability of their nuclei, when the MMP-dependent degradation of the extracellular matrix is inhibited. In the absence of matrix degradation capacity, cells are fully impaired in migration, when the pore-size reaches 10% of the nuclear cross section such as 7 µm2 for cancer cells, 4 µm2 for T-lymphocytes and 2 µm2 for neutrophil granulocytes (Wolf et al 2013). Residual migration under these strong constraints is firstly only possible in the presence of MMP-driven matrix degradation that causes a diminishment of restrictions by enlargement of the pore-sizes and secondly is based on the integrin-based actomyosin dependent force generation pushing the nucleus through the confined space. The restrictions of cell migration through interstitial tissues are matrix scaffold porosity, deformability of the nucleus, extracellular matrix degradation capacity and mechano-coupling between the cell's cytoskeleton and the external matrix surroundings (Mierke et al 2008c, Wolf et al 2013).
3.1. Nuclear deformability and the impact of integrins (connection between nucleus, cytoskeleton and microenvironment)
The nuclear deformability plays a prominent role in confined space migration. It has been suggested that integrins are crucial for alterations in nuclear components that cause the repositioning of the nucleus and its mechanical properties during the migration of cancer cells. How can integrins as integral membrane proteins couple the cell membrane to the cell's nucleus? How can integrins regulate the cellular motility to enable cancer cells to invade and subsequently infiltrate the surrounding tissue? The knowledge how cell receptors and the nucleus are functionally connected will provide a novel target for therapeutic targets in the malignant progression of cancer (Madrazo et al 2017).
In normal eukaryotic cells, the nucleus envelopes the genome and protects it from the cytoplasm. In contrast to normal, healthy cells, cancer cells display an aberrant nuclear morphology, which is characterized by nuclear invaginations, irregular nuclear shapes and volumes, aberrant chromatin densities in the nucleoplasm, nuclear bodies and multilobulation of the nucleus (Zink et al 2004, Lever and Sheer 2010, De Las Heras and Schirmer 2014). All of which may affect the deformability and hence squeezeability of the nucleus that is the largest obstacle inside the cells for cell migration and invasion through narrow confinements. Thus, the disorganization of the nucleus of cancer cells can serve as a tool for diagnosis and prognosis of cancer and helps to define the grade of cancer malignancy (Bussolati et al 2008). However, defects in the components of the nucleus have not yet been shown to cause cancer, but they are related to pathological features in humans (De Sandre-Giovannoli et al 2003, Padiath et al 2006). Thus, it can be suggested that a disruption of the nuclear organization of cancer cells is evoked by cancer transformation and the malignant progression of the disease.
The cell nucleus is covered by a nuclear envelope (NE) that consists of the outer and inner nuclear membranes and contains nuclear pore complexes. Underneath the NE is the nuclear lamina network that consists mainly of several lamins and lamin-associated membrane proteins coupling the lamins to the chromatin and cell's cytoskeleton (Martins et al 2012). Lamins belong to the group of nuclear intermediate filaments that are subgrouped into A-type and B-type lamins. They regulate the nuclear architecture and function in gene expression and the overall chromatin organization (Dechat et al 2008, Kaminski et al 2014). The nuclear lamina network is composed to several components coupling the nuclear lamina to the chromatin and the cell's cytoskeleton. The coupling is facilitated by the LINC (Linker of nucleoskeleton and cytoskeleton complex), which is assembled by nesprin and SUN proteins, titin, emerin and all-spectrin. In more detail, Nesprins and SUN proteins belong to NE transmembrane proteins and structurally connect to actin (nesprin-1 and -2), plectin (nesprin-3) dynein and kinesin (nesprin-4 and KASH5). In addition, lamin has other interacting proteins such as lamin B receptor (LBR) and LAP2 that are found to be connected with chromatin rich regions of the nucleus (Kaminski et al 2014). Moreover, other proteins fulfill important roles in both the cytoskeleton and the nucleoskeleton. Among these are actin and actin associated proteins such as Wiskott-Aldrich syndrome protein (WASP), FAK, actinin, Arp proteins, myosin and ERM (Ezrin, Radixin, Moesin) that can be located in the nucleus and in the cytoplasm and hence shuttle between the nucleus and the cytoplasm. In particular, the nuclear fraction of these proteins provides a linkage to the epigenetic machinery of the cell, the nuclear lamina scaffold and even facilitate nuclear functions (Lim et al 2008, Di Cristofano et al 2010, Falahzadeh et al 2015, Sadhukzadeh et al 2015, Sathe et al 2016).
Chromatin inside the nucleus consists of the deoxyribonucleic acid (DNA) that has polymerized to DNA strands and DNA associated proteins such as histones. The structure of chromatin is highly conserved and precisely regulated in order to provide the regulation of gene expression, coordinate cell-cycle progression, avoid and protect DNA from damage, support cell development and differentiation (Kouzarides 2007). Chromatin is found to be condensed and hence relaxed at distinct nuclear regions regulated by epigenetic alterations. These epigenetic alterations are characterized by posttranslational modifications of the DNA (methylation), histone proteins (methylation, acetylation, ubiquitination, SUMOylation) and non-coding RNA sequences, which all contribute to the regulation of the chromatin structure, function and the entire nuclear architecture (Kouzarides 2007). Many epigenetic alterations have been found in cancer cells that cause genomic instability through an aberrant expression of distinct genes during the malignant progression of cancer or upon cancer relapse after therapeutic treatment. Based on the detected aberrant nuclear architecture of cancer cells, nuclear alterations are proposed to be a hallmark of cancer and hence they may serve as a target for novel therapeutic drugs (Dawson et al 2012, You and Jones 2012).
How is the cell's nucleus crucial for cell migration under confinements? As the nucleus is positioned by the cell's cytoskeleton such as actin stress fiber that span over the nucleus, the interplay between the cytoskeleton and the nucleus is eminent for the fundamental process of cell migration through structurally restricted extracellular matrix networks. Hence, the ability to alter the nuclear shape, determine the position of nucleus by translation and rotation and subsequently to deform the bulky nucleus is critical for the migration of normal healthy cells and cancer cells.
3.2. Nuclear shape and nuclear motion such as translocation and rotation
Based on cellular tensegrity, the cell nucleus moves in a coordinated manner with the entire cell body (Crisp et al 2006). The cellular tensegrity concept highlights the importance of the intracellular filament network connecting the two cell poles and all organelles (Crisp et al 2006). In more detail, the cell nucleus seems to be able to rotate for the alignment of its shape to the axis of cell migration and behind the centrosome of a polarizing and migrating cell. The nuclear rotation is regulated by microtubules and the actomyosin contractility of the cell and hence the rotational step represents a promoting factor for the cell polarization and subsequently for cell movement through structural confinements (Starr and Fridolfsson 2010). Thereby, cytoskeletal rearrangements can facilitate the cell's polarity, provide the cell's leading and trailing edges and increase internal cytoskeletal and nucleoskeletal dynamics. More precisely, migrating cells possess three stress fiber subtypes, such as dorsal, transverse arcs, and ventral stress fibers, which differ in their origin, location and their extracellular matrix linkage (Lee et al 2018). Moreover, the LINC complex represents a nuclear node that can provide the transmission of forces between the cytoskeleton and the nucleus and therefore, it represents a crucial regulator for the positioning and reorientation of the nucleus and the attachment of the centrosome to nucleus (figure 17) (Wilhelmsen et al 2006). Nuclear translocation is supposed to be critical for the migration of mesenchymal stem cells (Swift et al 2013). The nuclear location has to be coordinated with the cell body migration through the actomyosin cytoskeleton that can apply either pulling or pushing forces on the cell nucleus such as a piston. Thus, the piston-based mechanism plays a key role for a lobopodial migration mode of fibroblasts and fibrosarcoma cells (Petrie et al 2017). Myosin IIA has been found to cooperate with the intermediate filament vimentin on the regulation of the contractility of myosin at the cell's leading edge of breast cancer cells by pulling on the nucleus (Thomas et al 2015). In particular myosin IIB can be detected closely around the nucleus in a perinuclear actin circle and at the cell's trailing edge that helps to facilitate the pushing of the nucleus from the cell's rear through the extracellular matrix confinements in the direction of the cell's leading edge (Thomas et al 2015). Besides actomyosin based contractility, microtubules and their associated proteins dynein and kinesin additionally promote the nuclear rotation and pull on the nucleus during cell migration to move the nucleus forward to the cell's leading edge, as it has been reported in myoblasts and neurons (Umeshima et al 2007, Cadot et al 2012, Wilson et al 2015). In summary, the LINC complexes are required for the controlling and balancing the maximum tension point for the nucleus of migrating cells such as fibroblasts (Alam et al 2015). Additionally, integrins are localized at the cell's leading edge of migrating cells and regulate the actomyosin dependent contractility through the Rho/ROCK signaling axis (Wu et al 2014a). However, the functional connections between the polarization of the cytoskeleton, LINC complex transmitted internal forces and the force applied to the nucleus need to be identified in order to reveal how cohesive forces manage precisely the control of the positioning of the nucleus in cancer cells. Indeed, when the nucleus is spatially confined by an actin capping structure of a polarized cells with filopodial extrusions (figure 18(A)), the nuclear shape is adapted to the polarization of the entire cell that seems to be based on actomyosin contractility. The mouse embryonic fibroblasts were cultured on laminin-coated flat substrates in culture medium containing a buffer control solvent. After 12 h, the actin filaments were stained with Alexa Fluor 548 Phalloidin, the nuclei with Hoechst 33342 and the cell membrane with Vybrant DiD (figure 18(A)). The fibroblasts were treated with a myosin II inhibitor blebbistatin (100 µM) for 5 h then stained (figure 18(B)). However, when myosin II is inhibited by 100 µM blebbistatin, the cell seems to be less polarized and the nucleus adapts a rounded, also non-polarized shape (figure 18(B)). Besides the nucleus, the actin cytoskeleton is altered by blebbistatin treatment, as the actin capping fibers cannot be seen and less pronounced actin filaments are detectable (figure 18(B)).
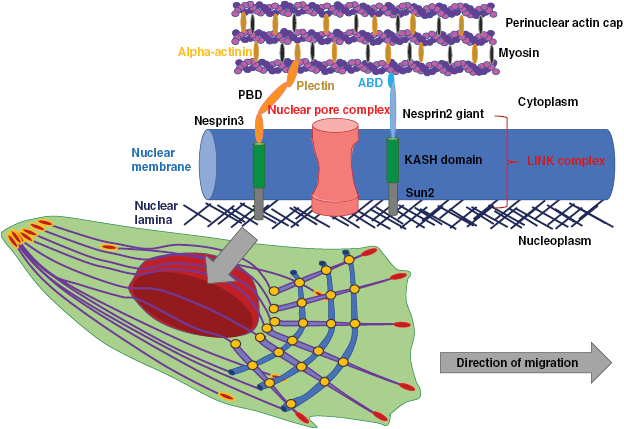
Figure 17. The cytoskeleton is connected to the nucleoskeleton via Sun2, Kash and nesprin-2 that form the LINC complex. The actin filaments span over the nucleus and build the actin cap structure, which is connected to the nuclear lamina. The actin cap can rotate and translocate the nucleus in migrating and invading cells.
Download figure:
Standard image High-resolution image
Figure 18. Confocal laser scanning images of mouse embryonic fibroblasts show after stimulation with 100 µM of the myosin II inhibitor blebbistatin that the actin cytoskeleton, cell polarization, cellular surface roughness and membrane lining (extrusions and tension) seem to be altered. Alexa Fluor 546 Phalloidin has been used to stain actin filaments, the nucleus has been stained with Hoechst 33342 dye and the membrane with DMI.
Download figure:
Standard image High-resolution image3.3. Nuclear mechanical properties such as deformability
The nuclear deformability is crucial for the migration of cancer cells in physical confinements (Caswell et al 2008). During the migration of cells, the nucleus becomes more flexible and hence is more sensitive to applied forces applied from the cytoskeleton. Nuclear deformability is characterized by various nuclear components such as lamins, chromatin and nucleoplasm. Hence, the nuclear lamina contributes mainly to the nuclear stiffness of human lung carcinoma, glioblastoma and mesenchymal stem cells (Swift et al 2013, Harada et al 2014). The ratio of lamin A to lamin B expression is supposed to control nuclear mechanical properties such as nuclear stiffness (Harada et al 2014). The lamin A expression is necessary for controlled movement of the nucleus, a decrease of E-Cadherin, an increase of the CSC phenotype and increases cell migratory capacity in human colorectal cancer cells (Willis et al 2008). The overexpression of lamin A is associated with the PI3K/AKT signal transduction pathway and subsequently with the malignant transition of pancreatic cells (Kong et al 2012). Contrarily, the migration and invasion of melanoma and neuroblastoma cells under confinements assumes decreased levels of lamin A that decreases the nuclear stiffness and hence increases the formability of the nucleus (Ribeiro et al 2014, Nardella et al 2015). There exist also differences of lamin A expression inside of tumor xenographs, as at the periphery of xenografts detected lamin A level are low compared to the tumor core center. This indicates that the cells at the leading edges of the tumor exhibit an invasive phenotype (Harada et al 2014). The contradictory findings on the function of lamin A levels can be explained by the utilization of different migration modes and other factors controlling the nuclear stiffness and subsequently migration efficiency. In line with this, defective lamin A expression has been shown to be linked to the structure of chromatin, the expression of the LINC complex and the localization as well as the existence of cohesive nuclear-cytoskeletal linkages in fibroblast and osteosarcoma cells (Haque et al 2006, Liang et al 2011). The lack of other nucleoskeletal components such as emerin can regulate the shape and the mechanical properties of the nucleus in human thyroid cancer cells (Kinsella et al 2013).
It is common knowledge that the migration and invasion of cells is connected with epigenetic alterations such as DNA methylation and histone modifications. Indeed, these epigenetic modifications are related to heterochromatin such as H4K20me1, H3K27me3 and H3K9me3 and are upregulated in several malignant breast, ovarian and melanoma cancer cells (Gerlitz et al 2007, 2010, Yokoyama et al 2013). Alterations of the histone 1 mobility are correlated with the migratory capacity of cells (Gerlitz et al 2007). The compaction of chromatin seems to be coupled with cytoskeleton to promote nuclear positioning, nuclear reshaping and provide mechanical nucleocytoskeletal connections (Chalut et al 2012, Guilluy et al 2014). Epigenetic alterations may affect directly mechanical properties of cell nucleus and they are found to occur in specific genomic regions that are coupled to lamins and possibly also other NE components such as lamina-associated domains (Shaklai et al 2007). These results are supported by the detection of novel non-genomic functions that are supposed to driven by epigenetic alterations (Bustin and Misteli 2016). Moreover, using the MTA (5'-deoxy-5'-methylthioadenosine) inhibitor to impair the methyltransferase decreased the condensation of chromatin, reduced proliferation and diminished invasion of human bladder cancer cells (Zhang et al 2013).
In gastric cancer cells, the usage the DNA methyltransferase inhibitor decitabine reduces the migration of cells, which indicates that this inhibitor can abolish the dissemination of cancer cells and hence be used as a therapeutic treatment (Shin et al 2012). It has been shown that progression of cancer evokes chromatin plasticity that in turn regulates the invasiveness and proliferation rates of melanoma cells (Maizels et al 2017). High heterochromatin levels have been found to elevate the stiffness of the nucleus and hence can abolish the migration of cancer cells, however, they can regulate the reshaping of the nucleus and control effective cytoskeletal driven forces during the translocation of the nucleus. Moreover, normal and cancer cells can both cause NE rupture and DNA damage, when they migrate and invade through highly spatial confined matrices (Denais et al 2016, Raab et al 2016). Both events need the induction of the DNA damage repair machinery and to restore the NE integrity by using the endosomal sorting complexes required for transport-III (ESCRT-III) machinery and subsequently both lead to genomic instability in cancer cells. Subsequently, these processes show how the chromatin structure can compromise the migration of cancer cells. Additionally, it has been analyzed precisely how osteosarcoma cells manage to migrate through confined spaces and thereby regulate the compaction of chromatin compaction and impaired mobility of DNA repair molecules as well as nucleases (Irianto et al 2016).
In conclusion, besides internal nuclear components such as lamins, LINC complexes and chromatin, the cytoplasmic perinuclear cytoskeleton has been shown to possibly control the nuclear deformability when the cells migrate under confined conditions. These confinements can induce the intracellular actin network to relocate several components such as the actin interacting proteins Arp2/3 complex and fascin around the nucleus in a circular manner and hence provide the nuclear deformation in immune and cancer cells (Jayo et al 2016, Thiam et al 2016).
3.4. Nuclear composition and intranuclear structures
The interaction between a cell and its extracellular matrix can evoke a remodeling of nucleoskeletal structures. Specific nuclear regions can influence the entire nuclear deformability and hence the ability of cells to invade and migrate under constricted conditions is impaired or promoted. The nucleolus possesses an ultrastructure that consists of multiple proteins and RNAs, which provide the precisely regulated functions of the nucleus such as gene transcription and possible posttranscriptional modifications such as splicing or alternative splicing (McStay 2016). Moreover, it has been demonstrated that the nucleolus is positioned in opposite to the microtubule organizing centers (MTOCs) in migrating Dictyostelium discoideum and the specific arrangement of the nucleoli is driven by microtubules (Sameshima et al 1991). Other structures of nucleus such as Cajal bodies are also redistributed by cell-extracellular matrix interactions. These Cajal bodies are highly mobile and have been shown to interplay with the nucleolus of HeLa cells (Platani et al 2000).
Besides the nuclear lamina and the chromatin structure, additional cytoskeletal components such as Rac1 can translocate into the nucleus and provide increased nuclear deformability that is required for proper cell migration and invasion through dense matrix confinements. In particular, Rac1 regulates the shape of the nucleus and the organization of chromatin and creates hence invasive phenotype of cancer cells such prostatic cancer cells (Navarro-Lerida et al 2015). In addition, besides Rac1, other components of the nucleus and the cytoskeleton such as WASP and FAK control the structure of chromatin structure. These two focal adhesion proteins can translocate from the cytoplasm into the cell nucleus. In the nucleus, they regulate the interaction with specific epigenetic remodeling proteins and alterations of histones such as the protein methyl CPG-binding protein 2 (Lim et al 2008, Sadhukhan et al 2014). Additionally, nuclear ERM proteins regulate the conformation of chromatin. In more detail, the specific phosphorylation of ezrin amino acid residues regulates its localization inside the nucleoplasm, which in turn seems to impact gene transcription, nuclear shape and mechanical properties of osteosarcoma cells (Di Cristofano et al 2010).
In conclusion, despite the model of cell tensegrity new experimental-based evidence has been archived that support the hypothesis that the nucleus possesses its own mechanical properties such as deformability. Nesprin-1 is known to regulate the mechanical response of isolated nuclei to external applied forces, as it phosphorylates emerin in order to alter its activation (Guilluy et al 2014). These data contribute to the suggestion that the nucleus is a mechanosensitive compartment that is independent of the cell's cytoplasm. However, the nucleus is connected through actin filaments and focal adhesions to cell-matrix receptors.
Integrins are well-known cell matrix adhesion receptors and crucial for the protrusive migration mode of cancer cells. A main question is not yet answered: What role play integrins in regulating nuclear modifications? Hence, the role of nuclear alterations and mechanical properties of the nucleus needs to be revealed to widen the knowledge of the molecular mechanisms regulating the migration and invasion of cancer cells and finally metastasis. When cells migrate, they can cause the relocalization of distinct cellular components such as MTOCs, mitochondria and the Golgi apparatus. It is common knowledge that integrins such as the alpha5 beta1 integrin can facilitate the repositioning of cellular organelles, when they regulate the activity of the protooncogene tyrosine-protein kinase Src and the myosin light chain kinase (MLCK), which then facilitates the Golgi apparatus distribution in fibroblasts (Lobert and Stenmark 2012). Besides the Golgi redistribution, another organelle redistribution such as the relocalization of mitochondria has been connected to Miro-1 and other mitochondrial regulators such as syntaphilin and kinesin KIF5B, which possess functions regulating cell invasion by linking mitochondria with the cytoskeleton in several normal and cancer cell types such as including fibroblasts, glioblastoma, prostate and breast cancer cells (Caino et al 2016).
As pronounced linkage between integrins, cytoskeleton and nucleus has been detected (Maniotis et al 1997), some later studies demonstrated how integrin dependent cell adhesion and signal transduction pathways guide the positioning and the entire properties of intranuclear components and prominent nucleus and cytoskeleton connecting structures during the migration of cells. In particular, external mechanical forces that are applied through integrins to the cell's cytoskeleton can redistribute the interaction and localization of Cajal Bodies (spherical subbodies in the nucleus of proliferating cells) components such as coilin and SMN in fibroblasts and human cervix cancer HeLa cells (Poh et al 2012). Moreover, the actin cytoskeleton and the nuclear lamina, are crucial for the transmission of force from the integrins to the Cajal bodies, whereas microtubules are not involved. These findings favor the hypothesis that an integral signal transduction network between integrins and nuclear structures exists (Poh et al 2012). In line with this, it has reported that the 3D extracellular matrix precisely governs the number of nucleoli and alterations of the nucleus in human breast cancer cells, which are dependent on β1 integrin facilitated signaling (Maya-Mendoza et al 2016).
Integrins are critical for the promotion of epigenetic alterations that hence may also provide the deformability of the nucleus. Indeed, there exists a precise interaction between integrins and integrin interacting proteins with histone methyltransferases. In particular, the depletion of these epigenetic components can reduce cell adhesion and the progression of the cell, as EZH2, which represents a key component of H3K27 methylation, controls the activity of cofilin and talin and hence facilitates the migration of lymphocytes and colon cancer cells (Ferraro et al 2014, Gunawan et al 2015). In addition, the knockdown of mDPY-30, RbBP5 and components of the H3K4 methyltransferase complex affects the endosomal recycling system and promotes the formation of protrusions and induces cell invasion (Xia et al 2010). Hence the endosomal recycling seems to be associated with the trafficking of integrins and the ability of cancer cells to recycle integrins and should further investigated in future studies.
In turn, cellular adhesion through integrins can cause epigenetic alterations that affect the nuclear behavior such as the expression of lamin A expression, heterochromatin levels and dynamics of telomeres (Makhija et al 2016). Other focal adhesion molecules serve as fundamental mechanotransductors and hence they transmit external forces into nuclear alterations such as expression of genes. The culture of cells within rather stiff extracellular matrices causes a translocation of YAP into the nucleus, which leads to the transcription of active SMAD factors in breast and squamous cancer cells (Dupont et al 2011). For the development of new mechano-based therapies, the interaction between integrins and their focal adhesion proteins as well as and the regulation of DNA repair molecules needs to be considered (Denais et al 2016). In conclusion, integrin-based cellular adhesion regulates the dynamics of chromatin and epigenetic alterations, which drive specifically the regulatory gene expression, nuclear mechanical properties such as deformability and subsequently cell migration and invasion.
As mentioned before, the cell nucleus needs to rotate and move according to the cell body movement and the dynamic contractile forces acting on the nucleus, which are evoked by the actomyosin cytoskeleton. Inhibition of cell matrix adhesion receptors such as alphav beta3 and beta1 integrins impairs the generation of the internal pressure that is utilized in the lobopodial migration mode of fibroblasts (Petrie et al 2014). In particular, this nuclear mechanism relies on the presence of nesprin-3 and laminA during the integrin-based cell adhesion (Petrie et al 2014). The beta3 integrin expression on the cell's surface is facilitated external stimuli and even the metastatic microenvironment such as the tumor stroma of several cancer cell types such as adenocarcinomas, breast cancers and squamous lung cancers (Felding-Habermann et al 2001, Page et al 2015). Hence, cancer cells seem to control the expression of beta3 integrins in order to switch their protrusive migration mode to a nuclear piston migration mode for 3D extracellular matrix invasion or to the canonical extracellular matrix degradation via MMPs.
In summary, integrins seem to fulfill cellular and nuclear governing functions during the process of cancer cell migration. As the current knowledge on the function of the nucleus on cell migration of normal and cancer cells have expanded, the role of the physical shape of the nucleus and its structure are likely to be crucial for distinct migration modes under confinements. Indeed, nuclear components such as lamins and histones regulate the translocation of the nuclei and nuclear mechanical properties such as deformability, which is needed when the cells squeeze through narrow spatial confinements.
Indeed, there is a functional linkage between integrins, nuclear components and alterations in nuclear deformability (figure 19). In more detail, integrins are directly found to be associated in the three main nuclear alterations such as the nuclear translocation, increased nuclear deformability and the functional linkages of other intranuclear structures such as the nucleolus to the cytoskeleton. However, there are still not yet answered questions: What are the underlying molecular mechanisms that facilitate the coupling of integrins to nuclear alterations? Do these couplings have to be exclusively nucleus-cytoskeletal connections? Are signaling pathways involved in these connections? How can alterations of the nuclear lamina, the chromatin conformation or the number of nucleoli determine the architecture of the nucleoskeletal structure and provide distinct mechanical phenotypes of cancer cells? All these points need to be addressed, when analyzing the migration of cancer cells under confinements.
Figure 19. Functional linkage between integrin cell surface receptors, focal adhesions, signal transduction pathways and the nucleus such as Cajal bodies and nucleolus.
Download figure:
Standard image High-resolution image3.5. Nuclear compartments and chromatin distribution
The contribution of the nuclear compartments and chromatin such as condensed and decondensed chromosomes to the nuclear mechanical properties such as deformability needs to be addressed. As the mechanical deformability of the cell's largest compartment, the nucleus, is a critical determinant for the efficiency of the migration of cancer cells, it may have a major impact on the overall cellular mechanical properties. An altered nuclear stiffness can be archived by adding trichostatin A (TSA), which leads to an increase in nuclear deformability. Moreover, by using 3D hydrogels, it has been revealed that cellular migration and invasion is impaired under confinements indicating a crucial role for the mechanical stiffness of the cell's nucleus in cellular motility.
3.6. Nuclear blebbing
During cell migration, it has been observed that under physical constraints cells such as fibroblasts and cancer cells migrate through a mechanism that involves the nuclear blebbing. These nuclear blebs can be exerted and retracted reversibly. However, little is known about the impact of the nuclear blebbing for cellular motility. In addition, it has to be figured out whether the nuclear blebbing has a precise function or whether it is simply a side effect of confined migration.
4. Extracellular matrix confinements affect cancer cell migration
The extracellular matrix of connective tissues fulfills several major functions such as providing the structural architecture and the mechanical phenotype of tissues. Moreover, the components of extracellular matrix can perform mechanical functions and alter matrix mechanical properties. The composition and structural characteristics of the extracellular matrix contribute to a broad variability of the tissues that fulfill different functions. However, in diseased tissues such as solid cancer, the extracellular matrix has been found to be highly complex and impacts the progression of the disease (Malandrino et al 2018, Mierke et al 2018). The composition and structure of the extracellular matrix surrounding primary solid tumors can be challenged by cancer cells and stromal cells, whereby the mechanical phenotype of the tumor microenvironment can be altered significantly. As the interaction of a solid tumor with the microenvironment is not only in one direction, the microenvironment of diseased tissue can also act in the opposite direction and further impact the solid tumor and hence tumor progression.
4.1. Can confinements induce a switch in migration modes?
The molecular and cell biological migration modes have been defined based on cellular morphology changes and experimental migration models based on flat 2D surfaces. Recently, the migration studies switched from purely 2D motility analysis to a more complex 3D migration mode. These 3D microenvironments represent confinements for cell migration, when their pore-sizes are much smaller than the cell's nuclear diameter. In addition, also the topology of surfaces plays a role for the initial adhesion of cancer cells to a distinct matrix in order to spread and migrate into the scaffold. However, different surface topologies can easily be tested in high throughput 2D cell culture assays, but still their effect has to be determined by using more reliable 3D microenvironments. Indeed, the results of the 2D migration are currently being checked and refined by using 3D migration models, which resemble the tissue more suitable compared to the rather simple 2D migration models.
4.2. Besides intrinsic determinants such as proteins and cell mechanics, extrinsic determinants, such as composition, structure, matrix mechanics, play a role for defining a special migration mode
However, what specifies a certain migration mode? How is the appearance or the switch between the different migration modes regulated? The answer is that it is still not yet well understood. The individual migration mode of certain cancer cells is proposed to play a central role for the regulation of the crossing of the basement membrane or the endothelial barrier and is also supposed to have an impact on their migration speed and directionality of the movement through biomimetic matrices such as collagen type I gels.
All signal transduction pathways, which are altered in cancer cells, are partly intrinsic determinants for the migration of cells. Intrinsic determinants for cellular migration and invasion are based on cellular mechanics properties that are provided by a distinct structure of the cell's cytoskeleton such as actin filaments, microtubules and intermediate filaments and their interacting proteins. Besides cytoskeletal components also the expression of distinct cell-matrix adhesion receptor such as alpha5 beta1 (Mierke et al 2011a) or alphav beta3 integrin (Mierke 2013) or the CD24 cell surface receptor (Mierke et al 2011b) has been found to be altered during the migration of cells such as fibroblasts and cancer cells. Intrinsic signals of cancer cells can influence the tumor microenvironment, as these signals drive extracellular matrix signals that regulate the migration and invasion of cancer cells through the extracellular matrix by inducing a structural remodeling of their surrounding stroma (Oudin et al 2016). How can cancer cells cause these extracellular matrix alterations in order to induce directed migration of cancer cells? A cancer cell mechanism is driven by the actin regulatory protein Mena that causes the remodeling of the extracellular matrix scaffold and provides fibronectin gradients that serve as paths for haptotactic migration and invasion. In addition to cell-matrix adhesion receptors, the coupling between these receptors and the actomyosin cytoskeleton seems to be crucial for cellular shapes and membrane surface topology. The coupling is facilitated by focal adhesion proteins such as vinculin, FAK, talin or paxillin. If the mechanocoupling protein vinculin is deficient in cells, it affects the membrane topology and mechanical properties such as stiffness and contractile force generation (Mierke et al 2008a) (figure 20). Representative electron microscopic images of F9 mouse embryonic carcinoma cells (vinculin wildtype), vinculin knock-out cells and vinculin knock-out cells stably transfected with the vinculin head (residues 1–821) or the vinculin tail (residues 811–1066), which were cultured on flat fibronectin-coated glass substrates, are presented in figure 20. Retransfection of either only the vinculin head or vinculin tail domain in the deficient cells displays altered surface topology, as the vinculin tail causes a smooth surface and vinculin head a rather rough surface similar to vinculin cells (figure 20). These results correspond to the altered mechanical properties of the cells: the vinculin tail domain can restore the contractile forces, whereas the vinculin head domain cannot (Mierke et al 2008a).

Figure 20. Scanning electron microscopic images of mouse embryonic epithelial cells, which are wildtype or vinculin deficient cells. In order to rescue the phenotype of vinculin deficient cells, these vinculin deficient cells have retransfected either with the vinculin head or the vinculin tail domain.
Download figure:
Standard image High-resolution image4.2.1. Dual role of the microenvironment in cancer cell migration
Additionally, the microenvironment of tumors plays a prominent role in promoting or opposing cancer cell migration (Wolf et al 2013, Cox and Erler 2014, Acerbi et al 2015, Oudin and Weaver 2016, Fischer et al 2017, Paul et al 2017, Mierke et al 2018).
How efficient a migrating cancer cell overcomes the different obstacles such as steric restrictions that are present in dense 3D biomimetic matrices is supposed to depend strongly on its mechanical properties. In more detail, how a cancer cell is strongly able to generate and transmit its protrusive forces seems to impact its migration mode and speed. Hence cellular forces and material properties of biomimetic matter or tissue such as pore-sizes or elastic modulus determine which migration mode is favored for a distinct cancer cell type or cancer cell subpopulation. Indeed, it has been reported that cancer cells with a certain cellular mechanical phenotype, such as increased contractile force transmission and generation, are capable to migrate more efficiently than less contractile cancer cells through 3D biomimetic networks such as dense collagen fiber matrices (Mierke et al 2008b, Mierke et al 2011a).
Nonetheless, what kind of migration mode is chosen when cancer cells have to squeeze through narrower pores or interendothelial junctions of vascular endothelial cell walls? In this special case of transendothelial migration and the transbarrier migration through the dense basal membrane of the vessels a blebbing migration mode seems to be more suitable than a protrusive migration mode. However, also an invadopodia-based protrusive migration mode may help to promote the transmigration of cancer cells. In preliminary results, it has been observed that membrane stiffness pronouncedly softens in primary human mamma and human cervix carcinoma cells, supporting in both cancer cell types the blebbing migration mode. Finally, we hypothesize that the mechanical properties of cells and biomimetic matter as well as the type of force generation of cancer cells determine the migration mode and regulates the switch between individual migration modes. The following questions still need to be answered: What are the mechanisms determining the individual migration mode of cancer cells? What role do microenvironmental properties of biomimetic matter such as the material mechanics and structure of the extracellular matrix play regarding the switch of the migration mode by the cancer cells? In order to investigate this, one needs to dissect the crosstalk between the migration modes of the cancer cells and the environmental confinements such as pore-size, overall cellular stiffness, plasma membrane surface tension and extracellular matrix rigidity, and the proteomics of the cellular adhesion machinery as well as the extracellular protein composition of the biomimetic matrix network.
To understand the mechanism how migrating cancer cells interact with their biomimetic environmental confinement and why individual cancer cells use a certain invasion mode such as blebbing or protrusive migration modes, the following major problems need to be addressed: What roles play the influence of cytoskeletal stiffness and cell contractility regarding the switch of the migration modes and cellular migration speed in biomimetic matrices? To what extent do mechanical properties favor either the protrusive or blebbing-based migration modes of cancer cells? How does the surface tension of the cell's membrane impact on the cellular migration behavior, such as protrusive-driven or blebbing-driven motility? What effect does the adhesion strength of cancer cells have on the preferred migration mode? Moreover, is the blebbing migration mode indeed preferred by small pore-sizes of the microenvironmental confinements and biomimetic matrices, such as the connective tissue matrix scaffold, whereas the protrusive migration mode is supported by large pore-sizes? Can the blebbing or protrusive migration modes of cancer cells impact the transmigration of cancer cells through barriers such as vascular endothelial cell linings and basal membranes of vessels?
Knowing the answers to these questions will contribute to the understanding of how cancer cells utilize a specific migration mode to migrate through the 3D microenvironment. Moreover, it can be determined what role the cellular mechanical properties and material properties of biomimetic microenvironments and connective tissue play. Moreover, this knowledge will contribute to reveal the individual impact of the mechanical properties of cancer cells and the mechanical properties of their microenvironments in supporting the migratory behavior and capacity of epithelial-derived carcinomas, during the malignant progression of cancers (metastasis). Moreover, the extracellular proteomic connection of cancer cells and their microenvironment that is caused by RTK shedding represents a novel mechanism through which cancer cells acquire drug resistance (Miller et al 2016).
4.2.2. Mechanical properties of tissues impact cancer cell invasion
The microenvironments of tumors are structurally and functionally altered by the emergence of a primary solid tumor (Levental et al 2009). Thus, the surrounding tumor environment is termed tumor stroma, which is characterized by increased matrix remodeling and enhanced matrix stiffness (Sinkus et al 2000, Butcher et al 2009). The stiffness of the tumor stroma in turn increases the growth of the primary solid tumor and even promotes the malignant progression of tumors such as the initiation of cancer cell migration (Lo et al 2000). Moreover, the extracellular matrix rigidity can lead to disordered tissue morphogenesis, when the tension in cells is elevated (Paszek et al 2005). A decrease in cellular tension can lead to a repression of the malignant phenotype of mammary epithelial cells and subsequently abolish the malignant behavior of cancer cells that behave then similar to normal, healthy cells in in vitro cell culture assays (Paszek et al 2005). The main questions still remain unanswered: What drives extracellular matrix stiffening in primary solid tumors? Can increased extracellular matrix tension cause malignant cancer progression?
In the stroma, the extracellular matrix scaffolding protein collagen is the most abundant and provides mainly the tensile strength of tissues (Kolacna et al 2007). The metabolism of collagen has been observed to be deregulated in cancer, as the expression of collagen is upregulated, collagen is deposited increasingly, the collagen fiber network possesses an altered organization and architecture, and increased MMP activity as well as enhanced turnover rates of collagen have been detected that all play a role in the malignant progression of cancer (Jodele et al 2006). The MMP-driven remodeling of collagen causes novel gaps and holes within the extracellular matrix, in which cells can then migrate. As the cleavage of the substrate such as the extracellular matrix leads to the production of fragments, these fragments have independent biological activity. Therefore, they can alter cellular adhesion to direct the architecture, determine the composition of tissues and can cause directly the activation or deactivation of distinct molecules or alter them indirectly through the modulation of the activity of signaling molecules (Page-McCaw et al 2007). Increased levels of MMPs are associated with poor cancer prognosis (Tetu et al 2006) and alterations in MMP activity have been observed to cause a change in the tumor phenotype (Zhang et al 2008), however, MMP inhibitors failed in clinical trials (Coussens et al 2002), which lead to the suggestion that other parameters also remodel the extracellular matrix and hence are responsible for the regulation of malignancy. The collagen type I has been seen as a barrier for cancer cell and primary tumor invasion, however, increased collagen expression lead to increased metastases formation (Ramaswamy et al 2003). In line with this, mammographic density that is based on increased levels of collagen type I, elevates the risk of breast cancer (Martin and Boyd 2008). In addition, the crosslinking of collagen fibers may accompany and subsequently cause tissue fibrosis (van der Slot-Verhoevena et al 2005), which in turn elevates the risk of malignant progression of cancer (Colpaert et al 2003). The covalent intra-and intermolecular crosslinking of collagen and collagen fibers is facilitated by the lysyl oxidase (LOX), which is a copper-dependent amine oxidase (Kagan and Li 2003) and can oxidatively deaminate distinct lysine and hydroxylysine amino acid residues located in the telopeptide domains (Yamauchi and Shiiba 2008). LOX is reported to be increased in distinct tumors (Erler et al 2009) and hence the active LOX can stiffen tissues in order to compromise their function (Pfeiffer et al 2005). In turn, the decrease of the LOX activity reduces the stiffness of tissues and impairs tissue fibrosis (Georges et al 2007). However, the connection between the mechanical properties of tissues such as collagen crosslinking, tissue fibrosis, tissue tension and cancer needs to be revealed with highest effort.
Inside tissues exists stiffness gradients (Oudin and Weaver 2016) and hence primary tumors are regarded as mechanically corrupted tissues as they consist of a usually high tumor solid stress in its core (tumor phenotype) that is even more pronounced when the tumor cell mass expands by increased proliferation and tumor growth. Subsequently, the growing and expanding tumor increases the interstitial pressure and simultaneously also the resistance to tumor growth exerted by the tumor surrounding stiffened and fibrotic extracellular matrix environment (Padera et al 2004, Paszek et al 2005, Willipinski-Stapelfeldt et al 2005). The stiffening of the extracellular matrix tissue can be observed in an elevated protein abundance of fibrillary proteins such as collagen type I and III, their reorganization and posttranslational modifications, which involves the LOX driven crosslinking of the tissue constituents (Erler and Weaver 2009, Levental et al 2009, Egeblad et al 2010, Cox et al 2016). There exists a broad heterogeneity of the extracellular matrix stiffness within primary tumors when performing AFM stiffness measurements on a microscale that is based on tumor stage and grade, whereas the stiffness on a macroscale measured by elastography or unconfined compression analysis revealed reliable an incremental increase of the tissue stiffness (Evans et al 2012, Plodinec et al 2012, Yi et al 2013, Fenner et al 2014). In particular, AFM indentation measurements have shown that the invasive front of human tumors possesses the highest stiffness (Acerbi et al 2015). Additionally, even the vasculature is a subject of stiffness heterogeneity, as endothelial cells within the core of the primary tumor are softer than the endothelial cells at the primary tumor periphery (Lopez et al 2011), which is supposed to be caused by altered pericyte attachment to endothelial vessels, which dramatically increases their stability and hence stiffness (Ribeiro and Okamoto 2015). Finally, these distinct regions of increased stiffness within the extracellular matrix create endogenous gradients of stiffness which in turn facilitate the migration and invasion of distinct cancer cells out of the primary tumor through a durotaxis-driven process (Lo et al 2000, Isenberg et al 2009, Plotnikov et al 2012). In line with this, the two most prominent organs for secondary metastases formation are the lung and the liver (Wynn 2008, Steeg 2016), which are both sensitive to develop chronic fibrosis due to age, certain diseases and obesity (Zeisberg and Kalluri 2013). Both the chronic fibrosis of the lung and the liver are characterized by stiffening of the tissues that is due to increased amounts of fibrillar-type collagens such as collagen I and III, tenascin C and fibronectin a s well as increased proteoglycan and LOX facilitated crosslinking of the tissue. All of which evokes a strong differential gradient of stiffness by which aggressive and invasive cancer cells are guided to targeted organs or tissues (Levental et al 2009, Chang et al 2017).
4.2.3. Physical and structural parameters of the microenvironment affect cell migration
A major parameter regulating cellular matrix migration and invasion are adhesive ligands. Cell culture experiments using precisely engineered microenvironments allow us to perform reliable high-throughput mechanistic studies by using well-defined migration models that contain clearly defined migration spaces. Using these migration models, individual factors such as the cross-sectional area or pore-size, which alter the efficiency of cell migration and invasion can be identified. In these migration models, mechanical properties of the extracellular matrix such as stiffness, adhesiveness, available ligand density and the presence of external gradients are decoupled and hence individual parameters be determined as most suitable in promoting or impairing cell migration (Irimia et al 2007, Pathak and Kumar 2012, Tong et al 2012, Paul et al 2017). However, not in all migration models are these factors fully decoupled and hence the interplay needs to be taken additionally into account.
When the tissue gaps exceed the critical cell size, the cell migration rates are reduced (Haston et al 1982, Harley et al 2008). This behavior of cells is based on a loss of most cell-matrix fiber interactions, as only very few fibers or even a single fiber remain to be coupled to the cell body. Hence, this type of migration has switched from a 3D to a 1D migration type (Doyle et al 2009). In contrast, when pores sizes are below the cell's diameter, the cells also slow down their migration speed and subsequently are trapped due to the physical steric hindrance (Haston et al 1982, Harley et al 2008). When the cells can produce and secrete matrix degrading enzymes such as matrix metalloproteinases, they can further migrate into and throughout the matrix by using an amoeboid migration mode (Wolf et al 2003, see above). Based on a response to extracellular matrix confinement, migrating and invading cells elongate to a spindle-like and polarized shape in order to stretch themselves and decrease their cellular diameter (figure 21), whereas the response to large pore sizes causes a non-polarized and rounded cell, which is a crucial hallmark of amoeboid migration (Friedl and Wolf 2009). In figure 21, a representative human breast MDA-MB-231 cancer cell (GFP transfected cell) migrated through a matrix confinement, where the pore-size or the cross-sectional area of the 3D collagen fiber matrix (stained with TAMRA, red) represent a confinement for cell migration (figure 21(A)). The MDA-MB-231 cell can overcome the constriction by degradation of collagen fibers (figure 21(B)). The MDA-MB-231 switches from a 3D migration towards a tube-like 1D migration, as it forms migration tubular migration channels (figure 21(B)). In line with this, the shape of cancer cells such as human melanoma cells migrating out of a spheroid is polarized and elongated, when the matrix fibers are aligned and changes towards a rounded and non-polarized phenotype when the matrix fibers are not so strongly aligned such as in the outermost regions of a cancer cell spheroid (Ahmadzadeh et al 2017). Moreover, it has been shown that the polarization of the cancer cells depends on the matrix stiffness, which is increased in regions with elevated matrix fiber alignment (Ahmadzadeh et al 2017).
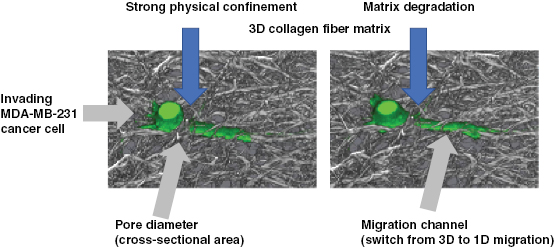
Figure 21. The migration and invasion of human MDA-MB-231 breast cancer cells through 3D collagen type I fiber networks depends on the confinement strength such as the pore-diameter or cross-sectional area. The cancer cells migrate through the matrix confinement in tubular channels that may represent a 1D rather than a 3D environment.
Download figure:
Standard image High-resolution image4.2.4. Embedded cells impact cancer cell migration and mechanics
Besides the mechanical properties of the extracellular matrix environment also embedded cells such as endothelial cells in this matrix can affect the migration and invasion of cancer cells. Thereby, these cells directly alter the extracellular matrix by secretion of molecules such as CXCL2 (synonymously termed Gro-beta) and CXCL8 (synonymously termed interleukin-8) that increase the motility and invasiveness of cancer cells such as breast cancer cells (Mierke et al 2008b). Moreover, the mechanical properties of human microvascular endothelial cells of the lung and skin are altered by cancer cells such as human breast cancer cells that in turn migrate at increased levels and deeper in 3D collagen fiber matrices (Mierke 2011). Hence it can also be suggested that endothelial cells themselves alter the mechanical properties of cancer cells, which needs to be further investigated in future studies. Besides endothelial cells, immune cells and fibroblasts have been reported to communicate with cancer cells in order to alter cancer cell invasion and metastasis (Malandrino et al 2018). Among immune cells are macrophages that can secrete TNF-alpha and TGF-beta in order to induce MMT1-MMP and MMP1 production and secretion in cancer cells, which causes enhanced migration speed and increased persistence of their migration and invasion through 3D collagen matrices (Li et al 2017). Cancer-associated fibroblasts regulate the mechanical properties of the tumor microenvironments in a number of ways. Firstly, they can utilize transmembrane receptors to exert pulling forces towards cancer cells that cause the dissemination of cancer cells out of the primary tumor into the local extracellular matrix microenvironment (Labernadie et al 2017). This mechanism seems to keep the invasion of primary tumors localized in the nearby microenvironment. In turn cancer-associated fibroblasts additionally release pro-inflammatory factors that lead to the recruitment of macrophages, increases the MMP activity and promotes the process of angiogenesis around the solid primary tumor (Erez et al 2010).
4.2.5. General requirements of experimental models for cell migration including microenvironmental cues
As the basic requirements of in vitro cell culture assays have an impact on in vivo cell migration during physiological processes such as development and tissue repair mechanisms after injury and malignant cancer progression. There exists a large variability of experimental migration models for the adaption of several parameters. Moreover, low costs and high throughput of these experimental migration models are both needed for efficient and reliable testing for migratory capacities of cells (Paul et al 2016). Engineered models for confined cell migration are indeed promising for the analysis of various determinants of cellular motility and distinct migration modes.
4.2.5.1. Artificial migration and 'tubular' invasion assays in 1D and 2D.
Even artificial migration and invasion models based on tubular structures such as channels represent mostly migration mechanisms that are utilized by cells in 1D or 2D environments and can reveal a novel migration mode such as the osmotic engine model that cells utilize in channel migration modes inside extracellular matrix connective tissue that represent physical tubular confinements (Stroka et al 2014). Moreover, the channel-based migration analysis can reveal easily the deformation of the nucleus under a distinct amount of spatial confinement that restricts the cell migration and reduces migration modes.
4.2.5.2. Biomimetic 3D extracellular matrix assays.
Dissimilar to real life tissues, in classical 2D cell cultures, cells are be kept under in non-physiological and hence artificial conditions, insofar that they adhere to flat and rigid surfaces and grow in rich and excessive nutrition, oxygenation and have sometimes the possibility to interact with highly modified extracellular matrix proteins (Alemany-Ribes and Semino 2014). These rigid surfaces, on which the cells are cultured, can evoke increased proliferation of the cells. However, the rigidity of these surfaces impairs cell differentiation, as the interactions with other cells among the same cell type or different cell types are restricted and the interaction of cells with the extracellular matrix scaffold is highly simplified (Cukierman et al 2002). Under natural conditions, cancer cells are one part of a highly complex microenvironment that allows them to interact through cell–cell adhesions within the same cancer cell type or distinct cancer cell populations or even with other cell types such as fibroblasts, macrophages or vascular endothelial cells. Moreover, cancer cells experience a crosstalk with the extracellular matrix of the tumor surrounding stroma such as collagen and connective tissue, chemical gradients such as metabolites, gas or pH and physical borders such as porosity or stiffness (Thoma et al 2014). Hence this tumorigenic and hence specific type of microenvironment enables cells to signal via transmembrane receptors that may react differently dependent on the dimensionality of their microenvironment. Indeed, altered activities of signal transduction pathways have been observed in 2D and 3D cell cultures. In more detail, HER receptors form heterodimers in 2D cultures and homodimers in 3D cultures. Subsequently, the kind of dimer promotes either a mitogen-activated protein kinase or a phosphoinositide 3-kinase pathway (Pickl and Ries 2009), which additional depends on the individual cell type.
Moreover, the 3D cell culture is associated with the enhanced secretion of cytokines such as interleukin 6, MMPs, transcription factors, proangiogenic factors that may serve as EMT markers. In turn, these markers indirectly impact the response to specific drug treatments, as they may cause drug resistance and affect the migration, invasion and subsequently the tumorigenesis of cancer cells (Leslie et al 2010, Alemany-Ribes and Semino 2014, Sung and Beebe 2014). In 3D cultures, it is possible to distinguish between malignant and non-malignant cancer cells based on their differences in morphology, whereas both cancer cell subtypes exhibit the same morphology in 2D monolayer cultures (Weigelt et al 2014). Another advantage of 3D cultures over 2D, is the pronounced effect of diffusion, which is present even in the absence of a microfluidic flow, and facilitates that secreted molecules are transported away by convection due to fluctuations of temperature, gas or solute concentration. Indeed, in a 3D cell culture system, diffusion has been observed to contribute to an increased imitation of natural tissues (Sung and Beebe 2014).
Another group of 3D culture systems is based on semisolid scaffolds such as collagen gels or on solid scaffolds such as titanium, ceramics or polystyrene scaffolds. Natural gels are usually based on alginate, collagen, hyaluronic acid, poly(lactic acid), poly(glycolic acid) or isolations of extracellular matrix such as MatrigelTM and Cultrex® BME. The latter two are isolations of extracellular matrices that are secreted by mouse sarcoma cells and they consist of collagen IV, laminin, proteoglycans and several growth factors. These natural biopolymers are biocompatible, biodegradable and rich in extracellular matrix binding motifs that all favor their usage in 3D cell culture assays. However, the lot-to-lot variability of natural polymers synthetic scaffolds may provide a major problem for the reproducibility and hence the usage of synthetic polymers such as poly(ε-caprolactone) and polyethylene glycol may be preferred under certain circumstances. These synthetic polymers are precisely defined by chemical composition and high reproducibility. In addition, they can be inert and display tunable degradability, but they may need promotors for cell adhesion, which additionally elaborates their production process. Hence, the synthetic matrices allow us to the fine tune the architecture of the matrices in terms of pore size, density, fiber architecture, adhesiveness and matrix stiffness to mimic distinct situations or properties of in vivo tissues (Thoma et al 2014, Knight and Przyborski 2015).
4.2.5.3. Parameters affecting the limits of cell migration indirectly.
The microenvironment is important for setting the limits of migration and invasion of cells. Hence a proper choice of the experimental migration model utilized for the investigations is required. In order to compare different approaches and studies the assessment and the comparison of the experimental migration models is also needed. For the usage of hydrogel-based motility assays, knowledge of the source of collagen type I such as bovine skin or rat tail collagen and its isolation procedure is highly recommenced, as the telopeptide status of the extracellular matrix and the polymerization temperature have both broad effects on the motility of cells (Wolf et al 2013).
In addition, physicochemical substrate properties such as the extracellular matrix telopeptide status and mechanical stiffness affect the migration rates of cells (Sabeh et al 2009, Miron-Mendoza et al 2010, Ehrbar et al 2011). In more detail, the difference between rat tail and bovine collagen fiber matrices may be attributed to differences in the telopetide content of the collagen types that may alter their polymerization dynamics and possibly also affect the entire polymerization process. Collagen extracted from the rat tails contains a high telopeptide content that leads to an increased polymerization of collagen fibrils and thereby increases the mechanical strength of the collagen scaffold, whereas the fibril diameter is reduced and the porosity of the entire network is decreased (Helseth and Veis 1981, Elbjeirami et al 2003, Sabeh et al 2009, Wolf et al 2009). All of which affects the migration of cells under non-proteolytic conditions. Inversely, when the temperature dependent fibrillogenesis is delayed, the pore sizes are larger and hence are in the range of the nuclear diameter of cells and hence are in the range of the nuclear deformability, which is important, when a non-proteolytic (MMP-independent) migration of the cells is preferred. Independently of the telopeptide content of the collagen preparation that is utilized for the polymerization of the collagen fiber matrices, a small pore-sized matrix requires excessive nuclear deformability to induce the migration of cells, whereas no large deformability of the cell nucleus prevents MMP-independent migration. In addition, the variation of the extracellular matrix stiffness under strong spatial confinement that abolishes cellular migration cannot reverse the immobilization of the cells and induce cellular migration. Hence, an effect of the matrix stiffness on cellular motility can under these conditions not be determined. Using reconstituted collagen fiber networks, the stiffness range is between 20 and 700 Pa, whereas the stiffness of polycarbonate membranes is above 106 Pa. Hence, the elastic moduli of in vivo tissues such as the mammary interstitium (in the range of 200–1000 Pa) and adipocytes and myofibers (in the range of 1000–10 000 Pa) can be addressed in these in vitro migration assays (Butcher et al 2009, Levental et al 2009, Stein et al 2008, Buxboim et al 2010, Fischer et al 2017). Subsequently, when the migration capacity of the cells is normalized to the matrix pore-size of soft and rigid matrices, nearly identical mean and absolute migration limits are obtained independently of the stiffness of the matrix, when it exceeds a value of 20 Pa (Sabeh et al 2009). However, if this result can be found for different cell types needs to be determined.
4.2.5.4. Biocompatibility issues impact microenvironmental mechanics.
The biocompatibility of materials used for cell culture assays plays an important role. In electrode-based physiological assays, the material of electrode and its insulation such as polyimide or PDMS needs to be suitable for cells and tissues and hence should not alter cell or tissue reaction in the biophysical assay (Hassler et al 2011). There is always a requirement to explore that a certain assay architecture or composition does not lead to changes in cell morphology or adhesion such as between a scaffold and the electrode or the insulation material. In certain cases, coating with extracellular matrix proteins such as fibronectin or RGD peptides may be applicable to increased cell adhesion. A disagreement is still on the biocompatibility of nanomaterials such as carbon nanotubes or graphene such as the occurrence of sharp edges of material sheets (Pinto et al 2013, Ping et al 2015). Although the uptake of biomaterial can challenge the outcome of an experiments and needs to be determined. Besides these materials diamond is known to be highly biocompatible and hence has been used as coating material for implants (Kromka et al 2010).
4.2.5.5. Models for the motility of cancer cells out of the solid tumor: Aggregate cultures and the formation of spheroids.
Multicellular aggregates can build rather rounded complexes, which are termed spheroids. These spheroid cell cultures are scaffold-free systems that produce their own extracellular matrix components and create possible their own internal structures. The spheroids can be generated by cells that are grown simply in suspension or embedded in 3D matrices, when 3D culture methods are applied (Mueller-Klieser 1987, Gottfried et al 2006, Hirschhaeuser et al 2010, LaBarbera et al 2012, Fennema et al 2013). Although the 3D spheroid models are more expensive and time consuming compared to simple 2D cell culture assays, they are frequently used as model system in cancer research and physical oncology. However, cancer cell spheroids, which are synonymously termed multicellular tumor spheroids (MCTS) are a model for a vascularized primary tumor nodules and/or micro-metastases (Friedrich et al 2009, Katt et al 2016) and have been investigated for at least four main applications: the first application is the analysis of cellular functions such as proliferation, migration and invasion in an avascular tumor microenvironment, the second application is the invention and development of novel cancer therapies and drug screening assays, the third is the analysis of the angiogenesis of tumors by adding endothelial cells to tumor spheroids and the fourth is the investigation of primary tumor and immune cell interactions (Mueller-Klieser 1987, Gottfried et al 2006, Lin and Chang 2008, Hirschhaeuser et al 2010, Fennema et al 2013, Katt et al 2016). The formation of spheroids can be performed by using a method based on a hanging drop culture (Keller 1995), in which the cells are indeed cultured in drops of cell culture medium that hangs upside down on the lid of dishes. Another method of spheroid generation is the culture of cell aggregates on a low adhesive substrate, on which the cells cannot adhere and hence assemble to cell clusters in the medium suspension. The hanging drop generated 3D spheroids can maintain a stem cell phenotype of bone marrow stroma cells over a prolonged time (Banerjee and Bhonde 2006) and provide a more homogeneous differentiation within the entire spheroid (De Smedt et al 2008) compared to ordinary cell 2D culture procedures. In addition, the hanging drop method for 3D spheroid cultures keeps a high local concentration of endogenous factors such as hedgehog proteins and provides a more reliable model for tissue functions compared to 2D monolayer cell cultures (Szczepny et al 2009).
Cell-cell and cell-matrix adhesion based interactions such as between cancer cells and their microenvironment can be analyzed in 3D spheroids (Hirschhaeuser et al 2010, Mehta et al 2012, Fennema et al 2013). Additionally, transport properties within the 3D spheroid can be investigated (Mehta et al 2012). In MCTS, it is possible to create 3D spheroids with variant sizes, which allows us to mimic the effect of tumor sizes in vivo on mechanical properties and functions of cancer cells. Since the tumor size is highly correlated with cellular functions such as drug penetration and transport, this feature can be effectively addressed in MCTS. 3D spheroids exhibit usually a size between 200 and 500 µm in diameter and hence they are sufficiently large to possess gradients of oxygen, nutrients and catabolites (Hirschhaeuser et al 2010). However, when a handing drop method is used, these large 3D spheroids have difficulties in the exchange of media. In contrast, hypoxic conditions within the 3D spheroid can be an advantage in remodeling the developmental growth and progression of primary solid tumors.
Above a certain diameter-size of 400–600 µm, the 3D spheroids are subject to the phenomenon of secondary necrosis in its core, where cells undergo apoptosis or necrosis (Gottfried et al 2006, Friedrich et al 2009, Hirschhaeuser et al 2010, Yoshii et al 2011). Around the necrotic core within these larger 3D spheroids, a viable cell rim of 100–300 µm thickness mimics the in vivo situation in a primary tumor (Gottfried et al 2006, Hirschhaeuser et al 2010). Small Spheroids of 200 µm size are used for drug testing and for investigating cell–cell and cell–matrix adhesive interactions, whereas they are suitable to generate sufficient oxygen gradients containing hypoxic regions or proliferation gradients in order to mimic primary tumors (Friedrich et al 2009).
3D spheroids contain heterogeneous populations of cells due to their location within the spheroid, proliferating cells are located in the outermost regions near the surface of the spheroid, whereas quiescent cells are located in the spheroid core under conditions with limited oxygen and nutrient transport (Mehta et al 2012). As a result of these structural features, 3D spheroids possess similar protein and gene expression profiles as in vivo primary tumors (Friedrich et al 2009, LaBarbera et al 2012).
Besides cancer research, multicellular aggregates are used in cell biology for the in vitro differentiation of stem cells. The aggregates are termed embryoid bodies and are created by using the hanging-drop method and even other techniques (Kurosawa 2007, Antonchuk 2013). The generation of uniform-sized embryoid bodies is necessary, as they impact the differentiation of the cells (Messana et al 2008, Bratt-Leal et al 2009). These embryonic stem cell-based aggregates assemble simple embryoid bodies that contain morula-like structures or even cystic embryoid bodies and additionally a cavity in its core similarly to the blastula stage (Abe et al 1996). Limitations of embryoid bodies are the relatively low complexity of the ordered structures and their short overall maintenance. Similar to normal stem cell cultures, multicellular spheroids culture methods have been utilized to culture CSCs by maintaining their specific properties such as cell surface receptor expression or invasiveness, as they are seen as critical determinants for the relapse of cancer after drug treatment (O'Connor et al 2014).
There exist four main methods for 3D spheroid cultures. The first method is a suspension culture that causes the aggregation of cells through agitation or addition of carboxymethyl cellulose that increases the viscosity to induce spontaneous aggregation (Lin and Chang 2008, Metzger et al 2011). An advantage of suspension cultures is the high throughput, whereas a disadvantage is that the size and uniformity cannot be controlled (Lin and Chang 2008, Mehta et al 2012). The second method employs cell cultures on non-adherent surfaces that impair cell adhesion to the substrate and enable the initiation of spheroid formation. The technique is based on a liquid overlay, where suspended cells are seeded and grown on non-adherent surfaces such as agar, 1%–1.5% agarose or poly-HEMA (Yuhas et al 1977, Ivascu and Kubbies 2006, Metzger et al 2011). When non-adherent surfaces are utilized for spheroid cultures, it presents a straightforward and reproducible technique to generate spheroids. However, the weakness of this method is the lack of control regarding the size of the spheroid and uniform structure. Using microarrays, the growth of spheroids can be increased by enhancing the throughput under controlled spheroid size growth (Hsiao et al 2012, Mehta et al 2012, Fennema et al 2013). In particular, the growth of spheroids is covered and directed by using round-bottom non-adherent 96-well plates or even stamped agarose that serves as microwells for spheroid culture (Fennema et al 2013). The third method is based on the hanging drop cultures that allow us to regulate precisely the size and the composition of spheroids (Mehta et al 2012). The hanging drop-based spheroid generation uses a droplet with suspended cells that are positioned on the lower side of an adherent tissue culture dish lid. Due to gravity, the aggregation of cells results into a cell cluster that appears directly at the lowest point of the hanging drop and assembles to a full spheroid after cell proliferation within a few culture days (Kelm et al 2003). Whereas a uniform size of the spheroid can be archived, the throughput is rather low depending on the frequent manual media replacements. The fourth technique is based on microfluidic devices which help to precisely control the formation spheroids (Wu et al 2008, Mehta et al 2012, Fu et al 2014). During the spheroid formation, a continuous perfusion under physiological conditions provides a faster spheroid assembly, leads to larger and more uniform spheroids (Mehta et al 2012). Several cellular functions can be easily and more closely related to the in vivo situation of primary tumors that has been tested by using spheroid cultures. These spheroids mimic the situation in a solid primary tumor and hence growth kinetics, composition, differentiation, apoptosis, protein and gene expression profiles, and resistance to drug therapy can be determined (Sutherland et al 1971, Freyer and Sutherland 1986, Durand 1990, Friedrich et al 2009, Hirschhaeuser et al 2010).
As the capacity of cells to migrate and invade is a hallmark of cancer and requires the transition of epithelial cells to mesenchymal cells (Gagliano et al 2005), cancer cell migration assays are employed to determine the effect of therapeutics and their potential in the impairment of cancer cell migration as well as their abolished ability to switch to an invasive and metastatic phenotype (Rao et al 2005, Vinci et al 2013). Hence, cancer cell invasion assays are performed by using a spheroid of cancer cells that is placed on coated surfaces such as vitronectin-coated surfaces or embedded in collagen type I matrices to measure the migratory capacity or invasiveness under proteolytic matrix degradation such as such as cathepsin-B and MMPs (Tamaki et al 1997, Lakka et al 2004, Wolf et al 2007, Ilina et al 2011).
How can the process of angiogenesis be investigated in primary tumors using cell culture model systems? Indeed, the capacity of a primary tumor to build a vascularization (termed neovascularization) can be mostly probed by allowing endothelial cells to migrate into tumor spheroids or investigate the assembly of vascular networks in spheroids (Timmins et al 2004). Among these assays are cultures of MCTS on closed endothelial cell monolayers, cocultures of MCTS spheroids and endothelial cell spheroids, as well as spheroids produced by a mixture of cancer and endothelial cells (Jadhav et al 2004, Timmins et al 2004, Ghosh et al 2007, Upreti et al 2011). The tumor-driven neoangiogenesis elevates the oxygen consumption and upregulates the expression of hypoxia-related and hence proangiogenic genes (Wartenberg et al 2001). However, there exist factors such as MMP-9 that decrease or impair angiogenesis, which is crucial for the organization of endothelial vascular networks (Jadhav et al 2004). In conclusion, the 3D spheroid coculture models are preferred for angiogenesis studies in physical oncology and tissue engineering (Korff and Augustin 1998, Korff et al 2001, Wenger et al 2004, Wenger et al 2005).
4.2.5.6. Biomimetic scaffold matrix models for 3D cancer cell invasion.
The usage of scaffold-based 3D culture models can even widen the range of options for investigations (Knight and Przyborski 2015). The scaffold components can be natural and synthetic materials. The natural biomaterials are mostly various components of the extracellular matrix of connective tissues such as collagen type I (Baharvand et al 2006), fibrin (Willerth et al 2006, Robinson et al 2017) and hyaluronic acid (Gerecht et al 2007), whereas also other natural materials such as silk (Mauney et al 2007), gelatin (Awad et al 2004) and alginate (Li et al 2010) can be included into extracellular matrix scaffolds and thereby become a major structural component of individual matrices. All these materials are biocompatible, possess many cellular adhesion sites and they have an advantage over synthetic materials, as they can be naturally degraded by cells. However, in cell culture usage, the biodegradation of the matrix scaffold may also be a disadvantage, as it may be difficult to control and therefore may impact the outcome of the experimental approach. In addition, lot-to-lot variations due to the isolation or source of the material are adding other variable to the in vitro system. Moreover, the limited range of mechanical properties has also to be taken into account. Scaffolds of synthetic materials have the advantage of a defined chemical composition and possess a wide range of tunable mechanical properties that affect differentiation (Engler et al 2006) and adhesion of cells (Hayward et al 2013). Among the synthetic materials of artificial 3D scaffolds are biopolymers (Gunatillake and Adhikari 2003), titanium (van den Dolder et al 2003), bioactive ceramic-based glass materials (Lu et al 2003) and self-assembled peptide-based materials (Garreta et al 2006). A synthetic scaffold usually can be produced highly reproducible. It can either be inert and non-degradable or arranged as a material with tunable degradability. A weakness of synthetic materials seems to be the lack of cellular adhesion sites, which can be obtained by coating synthetic materials with extracellular matrix proteins such as fibronectin or collagen in order to archive a natural adhesion site. In general, there exist distinct methods for scaffold-based 3D cell culture systems that can be grouped widely into two main approaches for biomimetic scaffolds: the hydrogels and solid scaffolds.
4.2.5.6.1. Hydrogels as biomimetic environmental migration models.
A highly used variant of the 3D cell culture is the procedure with the embedding of cells in a non-polymerized hydrogel that then polymerizes to a loose scaffold network that is characterized by a crosslinked natural basic material such as agarose, fibrin, collagen or hyaluronic acid, which has a high content of water (Tibbitt and Anseth 2009). In particular hydrogels can be adapted to provide the selected growth of distinct cell types and enable them to function, when these cells are either trapped within these artificial extracellular matrix scaffolds (Heywood et al 2004, Jongpaiboonkit et al 2008). Moreover, cell populations of different invasive potential can be selected, when cells start their migration through the matrix scaffold's interior of the hydrogel, when they have been seeded on top of the gel's surface (Mierke et al 2011a, Topman et al 2013). Modifications of the hydrogels are possible by either adding distinct biologically active molecules or crosslinking proteins. The cells can be embedded into the hydrogels by adding them before the gels are self-assembled or injecting after ionic crosslinking or radical polymerizations by UV exposure of the hydrogels (figure 22). A disadvantage of hydrogels is the effect of UV light, when the cells are embedded in the hydrogels, as the exposure of cells to UV light may cause severe effects on cellular functions and induce an apoptotic cell death (Nicodemus and Bryant 2008). Another disadvantage of hydrogels is the short culture period for certain cells due to the slow or impaired diffusion of nutrients to the embedded cells (Jongpaiboonkit et al 2008). However, the impact of hydrogel matrix degradation may also affect the outcome of the experiment and hence needs to be impaired by electron beam irradiation or chemical crosslinking such as glutaraldehyde.
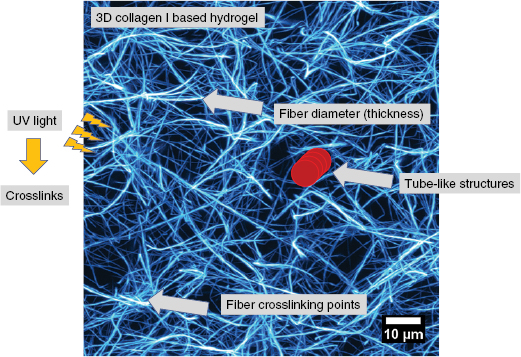
Figure 22. 3D microenvironmental confinements such as 3D collagen type I hydrogels can be altered by UV light, which additionally crosslinks the collagen fiber bundles. The migration of cells depends on parameters such as the pore-size diameter (often referred to as cross-sectional area), fiber thickness, stiffness of the matrix network and matrix components.
Download figure:
Standard image High-resolution imageHowever, hydrogels are commonly used to model the branching morphogenesis of cells that is regulated by chemical and physical properties of the extracellular matrix. As in vivo models are highly complex, the molecular and cellular mechanisms underlying the tube formation of cells can hardly be revealed and simple 2D in vitro cell culture models cannot be utilized to investigate cellular branching without an interaction with the extracellular matrix, 3D hydrogels such as 3D collagen fiber matrices (Wells et al 2013) or reconstituted basement membrane extracts such as Matrigel (Debnath et al 2003) seem to be promising to detect the mechanism (Zegers 2014). Both 3D tube formation hydrogels require different extracellular matrix compositions for cell polarization and the formation of the tube lumen and therefore are not equally well-suited for each cell type (Santos and Nigam 1993, Campbell and Watson 2009). However, hydrogels are mostly used to investigate angiogenesis in vitro such as the vascular morphogenesis and are also suitable for analyzing the effect of certain drugs on angiogenesis (Zeitlin et al 2012). Moreover, these hydrogels allow us to encapsulate bioactive drugs and create a time-delayed release. Regulatory factors for the enhanced angiogenesis can also be immobilized within these hydrogels such as it has been shown for the vascular differentiation of human embryonic stem cells (Ferreira et al 2007). The tube formation assays have also been used to investigate the neoangiogenesis within primary tumors, as an endothelial and cancer cell co-culture system has been established (Chwalek et al 2014).
4.2.5.6.2. Solid scaffold-based environments serve as biomimetic environmental migration models.
When cells are cultured in a solid and rather rigid scaffold, the 3D space of the scaffolds may initiate and enhance the formation of tube-like structures similar to natural 3D tissue structures. A major advantage of solid scaffold-based culture matrices is that they create an environment for cells that supports the 3D culture and a 3D architectural arrangement of the cells exhibiting a highly ordered structure in vitro. This arrangement of cells is controllable and reproducible and hence these solid scaffolds are more appropriate for routine applications. These scaffolds are purely synthetic, free of animal-based components, can be sterilized and easily used, and are manufactured by precisely controlled procedures in order to minimize batch to batch variations and support reproducibility. A future application of these scaffolds seems to be most likely layered structures of dermal and epidermal components for artificial skin constructs.
4.2.6. Organotypic matrix model for cell invasion.
Organotypic co-cultures may fill the gap between the simplest standard 2D culture and the highly elaborative in vivo mouse models. Organotypic co-culture means the recombination of two different cell types that are each previously disaggregated and maintained in culture as separate cell lines. These organotypic assays may increase the predictability of pre-clinical studies for the testing in clinical studies in the patient. In order to provide a faster lead compound development for pharmaceutical drugs, these organotypic matrix models are more reliable and suited for drug screening that are still commonly performed by using less faithful 2D cell culture models. In conclusion, organotypic co-cultures are an easy, affordable and scalable cell culture in vitro model for analysis of drug responses in a physiologically relevant 3D platform with a high throughput compared to in vivo animal models (Conway et al 2017).
5. Classical molecular and cell biological models can induce migration mode switches based on environmental cues
Mostly 2D migration models have been developed, as many experimental results have been obtained from simple 2D migration models. These models are the basis for many theoretical physics-based approaches to predict, simulate and describe the transition of migration modes that are based on a morphological transition such as EMT and MAT. In the following the focus is on the switch between different migration modes based on morphological features.
In multiple studies, the classical migration modes such as the mesenchymal and amoeboid migration modes have been precisely defined (Friedl and Wolf 2003, Wolf et al 2003, Taddei et al 2014). Despite the fact that these migration modes have been described by with mechanistically concepts, they are based on the morphological description of the migration modes and hence still lack a well-defined and exact definition based on other measurable criteria such as cellular stiffness, which can be used to distinguish those migration modes (Lämmermann and Sixt 2009). Hence, it still needs to be figured out how each of these modes are regulated in terms of the mechanical properties of the cells and the environmental constraints. In more detail, it needs to be characterized whether a cancer cell prefers a certain mode of migration that displays a distinct mechanical phenotype and hence defines its mode of migration exactly that it can be distinguished from the other migration modes. Moreover, the term amoeboid is not precisely defined by function, instead it is rather defined by morphology, as migrating cells with roundish cell shape are categorized as amoeboid migrating cells. The mechanistic aspects such as the cytoskeletal remodeling dynamics and mechanical forces are not yet integrated in the definition of the amoeboid migration mode. Thus, a definition of the amoeboid migration has been purely based on the morphological aspects of the cell, its adhesive behavior and the capacity to remodel the extracellular matrix environment by proteolytic enzymes, as it has been employed by leukocytes during the interstitial migration (Wolf et al 2003, Sabeh et al 2009). The proteolytic degradation of the extracellular matrix in connected to the amoeboid migration mode, which also is associated with cellular deformability (Sabeh et al 2004, Wolf et al 2007, Rowe and Weiss 2009). In addition, the proteolytic degradation enables the interstitial invasion of mesenchymal cells into dense collagen-rich 3D extracellular matrices. Moreover, these mesenchymal cells migrate in and invade through the extracellular matrix confinement by using both proteolytic matrix degradation and non-proteolytic cell squeezing, when passing through narrow spatial constrictions. There are still important questions not yet clearly answered: What role play cellular forces such as contractile or protrusive forces in regulating the switch between the different migration modes? How can protrusive or blebbing-based or lobopodial migration modes fit into this scenario? What are the criteria for a switch between the migration modes? On what timescale is the switch between the migration modes detectable?
5.1. Epithelial-mesenchymal transition (EMT)
The EMT has been introduced for the migration of single cancer cells out of the primary tumor mass at the onset of cancer dissemination. In this process, the complex regulation of EMT is provided by environmental cues such as increased compression of cancer cells. There exists accumulated evidence that E-cadherin fulfills a crucial role together with beta-catenin in facilitating the dissociation and spreading of cells such as epithelial cancer cells. The hallmarks of cancer in tumor invasion and metastases formation are associated with a lack of E-cadherin in cell-cell adherence junctions of epithelial cells that is based on downregulation of E-cadherin (figure 8) (Cavallaro and Christofori 2004). Moreover, several approaches revealed that the reestablishment of a functional E-cadherin adhesion complex can repress the invasive phenotype of several cancer cell types (Vleminckx et al 1991, Luo et al 1999, Hsu et al 2000) and hence E-cadherin acts as a tumor suppressor based on its crucial role in the maintenance of functional intercellular junctions. Moreover, the increased expression of E-cadherin impairs the transitory process of EMT in the malignant progression of epithelial tumors. The EMT-inducing signals are partly induced by growth factor stimulation such as hepatocyte growth factor, epidermal growth factor and TGF-β. In more detail, the growth factor dependent stimulation, causes the downstream activation of various EMT facilitating transcription factors such as Snail, Slug, Twist and zinc finger E-box binding homeobox 1 and 2 (Cano et al 2000, Thiery 2002, Peinado et al 2007, Medici et al 2008). The EMT creates a decrease in the expression of epithelial cell junction proteins such as E-cadherin, α-catenin, several claudins, occludin and ZO-1, whereas several mesenchymal markers such as N-cadherin, vimentin and fibronectin are increased expressed on molecular and protein levels. In addition, the EMT is associated with the reorganization of the cytoskeleton that is indicated by the abolishment of the apical-basal cell polarity and the adaption of spindle-shaped morphology and thereby large cellular polarity is based on protrusions at the leading and trailing edge (Huber et al 2005, Trimboli et al 2008). During EMT, cells acquire the production of extracellular matrix proteolytic enzymes such as MMPs. The total shift in the course of EMT ends in the characteristic mesenchymal migration mode phenotype (Thiery 2002, 2003, Kalluri and Wenberg 2009, Giannoni et al 2012). Besides the classical EMT model that is based on a full transition of cancer cells, there has been a concept revisited that describes a state of a hybrid epithelial/mesenchymal (E/M) state in cancer disease (figure 8) (Lecharpentier et al 2011, Strauss et al 2011, Huang et al 2013b, Naber et al 2013, Bronsert et al 2014, Sampson et al 2014, Schliekelman et al 2015, McCart Reed et al 2016, Grosse-Wilde et al 2015, Bocci et al 2017, Jia et al 2017, Lambert et al 2017, Seager et al 2017), which has been reported in in vitro cell culture assays over a long culture time period, including multiple cell passages, to be stable (Jolly et al 2016b, 2017).
5.2. Mesenchymal-amoeboid transition (MAT)
Besides the EMT transition, a second type of transition such as the mesenchymal to amoeboid transition (MAT) has been demonstrated to be crucial for the malignant progression of cancer (Friedl and Wolf 2010). MAT is chemically induced in cancer cells by the inhibition of integrin function by adding pharmacological inhibitors or by induction of MMP activity evoked by p53 or p27 deficiency (Gadea et al 2007, Berton et al 2009), and through activation (or reexpression) of EphA2 (Wolf et al 2003, Friedl 2004, Parri et al 2009, Taddei et al 2009). Although MMP inhibitors have been extensively tested in clinical approaches, but they have failed to reduce the malignant progression of tumors such as metastasis (Lah et al 2006). The disapprovement of MMP inhibitors as a therapeutic treatment may be based on the problem of the large plasticity of cancer cells and thereby their ability to adapt to proteolytic enzyme inhibition by switching an amoeboid based migration mode in the absence of protease activity (Wolf et al 2003, Taddei et al 2014). However, there remain still some questions unanswered: Can all cell reverse their mesenchymal to amoeboid transition? How fast is the reversion? Is reversion periodically? How is cell division integrated in the migration mode reversion or switch?
6. Physical model for the initiation of cell migration: Jamming to unjamming transition
Dissimilar to single cell migration, for the collective migration of cancer cells exist at least two major modes of migration. One mode of cells can be described as a phase, in which the cells rearrange in a cell collection by exchanging their positions within the cell collection and hence they flow through the stromal connective tissue. This phase of the cells can be determined by a hypermobile (highly invasive) and fluid-like behavior of the constituent cells. In particular, the cells are no longer tightly bound to their neighbors via the formation of strong cell-cell adherence junctions and therefore, they are identified as cells that exhibit an unjammed phase. Another mode of cells is a phase, in which cells are impaired in their movement and exhibit a frozen or fixed state with a collection of cells and hence cells are stably associated with distinct individual positions such as the atoms in a crystal structure that are tightly stabilized and integrated into a fixed matrix, which restricts cellular movement. This second phase is characterized by a quiescent and solid-like state. Moreover, the cells are pronouncedly confined in their motion and hence are caught in a distinct space. Thus, these cells are in a so-called jammed phase (Park et al 2015, Park and Fredberg 2016).
Movement of cells might be only possible when the entire cell-cluster moves through a tissue or single cells undergo an individual transformational step. In particular, the cells typically transit from a maturating cell layer into a quiescent, solid-like and jammed phase and in turn convert then to an immature cell layer into a hypermobile, fluid-like and unjammed phase, in which the cells are solely weakly associated and loosely bound to neighboring cells via intercellular adhesions. However, the cells migrate still as a collective such as a distinct cluster of cells. The whole concept of jamming is still not yet fully understood in great detail and hence requires further investigation. There remain still open questions such as whether the concept of cell jamming can be applied to the migration and invasion of single cells or to the transmigration of cells through a cell layer of different types of cells in the malignant progression of cancer such as cancer metastasis. In the collective cell migration mode, the unjammed phase represents a migratory phenotype that may be induced by the compressive stresses evoked by the surrounding connective tissue of primary tumors with are usually stiff. A wide variety of densely-packed collective systems ranging from inert materials to living soft matter such as cells have the capacity to exhibit jamming behavior. Under a certain condition, the collective of particles or cells can flow like a liquid and exhibit a unjamming phase and under other circumstances, particles or cells appear to be jammed and hence rigidify as a solid material. How are these phases characterized by structural properties of physical signaling processes or transduction pathways? How the jamming to unjamming transition of cells related to the classical EMT? Can the process of jamming describe partly EMT behavior? Do intermediate state or other intermediate phases in the jamming to unjamming behavior exists?
The jamming transition seem to be linked to the cell's shape and morphology that represents solely a structural criterion for cell jamming. Thus, there exists a counter-intuitive relationship between the jamming behavior of cells, their cell shape and their cell-cell adhesive stresses. Can cell-matrix adhesive stresses similarly to cell-cell adhesive stresses facilitate the transition between the jamming and unjamming phase? However, tissue-remodeling events such as morphogenesis, tissue injury based wound healing processes, chronically inflamed tissue as well as the migration and invasion of cancer cells have been linked to collective cellular migration involving the classical EMT (Thiery 2002, 2003, Kalluri and Weinberg 2009, Giannoni et al 2012). Contrarily, cancer cell motility has been shown to be based on a partial EMT (Jolly et al 2016b, 2017). In conclusion, no clear physical picture can be drawn to describe collective biological processes and their interconnections. The statistical analysis of cellular motions in a confinement that is created by their own direct cellular neighbors has confirmed the solid-like layer and the jammed behavior (Jolly et al 2016b, 2017).
7. Effect of the microenvironment on cancer cell migration persistence
In cell migration and invasion, the matrix mechanics such as stiffness has been revealed as a key player for regulating cell survival and motility. Therefore, multiple synthetic and natural materials have been employed to investigate the functional role of matrix stiffness, crosslinkers, proteolytic enzymes, fibrillary diameters, structural architecture including pore-sizes and the interaction with stromal cells such as fibroblasts or immune cells for the migration of cells. However, for natural materials, a major effort is provided on deciphering the matrix mechanical properties from structural effects. In addition, synthetic hydrogels have been designed to regulate the physical parameters of the matrix independent of structural parameters. Experiments employing these novel synthetic materials will help the decipher the dependence between cellular function and material stiffness.
7.1. Anisotropic persistent random walk in 3D microenvironments
Most knowledge on the mechanistic molecular level regulating eukaryotic cell migration has been obtained from 2D migration studies on flat surfaces. In the absence of external cues, cell trajectories can usually be described as random paths. On the mesoscopic scale (cell level), randomness is indeed inevitable and arises from both the internal cell dynamics, which is regulated by stochastic processes, and the microenvironment. Different types of random processes ranging from simple Brownian motion to persistent random walks (Selmeczi et al 2008), Levy walks (Harris et al 2012) or mixed processes, such as intermittent random walks (Benichou et al 2011) can alter the trajectories of a migrating cell. These trajectories are characterized by both the instantaneous cell velocity and the cell persistence time, which quantifies the cell's ability to sustain its direction of migration. Although the propulsion force imposes the cell velocity and the maintenance of polarity regulates cell persistence, both parameters rely crucially on actin flows (Callan-Jones and Voituriez 2016).
Therefore, large scale migratory patterns are severely restricted, since cell velocity cannot be adjusted independently of cell persistence. When a cell migrates faster, it requires to increase its propulsion force and thereby increase the actin retrograde flow, which leads to reinforced cell polarity and a larger persistence of motion (Tjhung et al 2015). This phenomenon is termed universal coupling between cell velocity and persistence (Maiuri et al 2012, 2015, Gorelik and Gautreau 2015). In fact, it is detected in different cell types, migration modes (mesenchymal or amoeboid), and confined and non-confined microenvironment.
The universal coupling between cell velocity and persistence not only takes into account for the observed relationship between speed and persistence in cell trajectories. Moreover, its theoretical analysis showed that the adaption of their strength, which is primarily regulated by the affinity of polarity cues to actin, is sufficient to generate most of the classical migration patterns, ranging from diffusive Brownian trajectories, via persistent random walks to intermittent random walks. Subsequently, these results demonstrate that the dynamics of cellular components, such as actin and myosin, on the molecular scale, influence the behavior of cells in a predictable manner on the mesoscopic scale.
Cell motility of eukaryotic cells in the absence of gradients has been reported to be a persistent random walk (PRW) similar as the movement of individual particles such as atoms or molecules (Berg 1993) (figure 23). These random walks can be found ubiquitous in biological systems. In absence of symmetry-breaking gradients, prokaryotic cells such as bacteria and eukaryotic cells such as cancer cells show a movement behavior that can be described by using random walk statistics (Wu et al 2014b). Cellular motility in in vivo 3D microenvironments requires the remodeling of the extracellular matrix and exertion of cellular pulling forces, when the extracellular matrices represent a restriction for cellular movement (Zaman et al 2006, Fraley et al 2010, 2012, Khatau et al 2012, Yu and Machesky 2012, Giri et al 2013, Tang et al 2013). In contrast, the migration on 2D surface induces an actomyosin contractility facilitated basal-apical polarization of the cells, when they adhere to the matrices. The contractility is provided by F-actin stress fibers and large focal adhesions and leads to the formation of a lamellipodium with filopodial structures on its external borders towards the direction of migration (Ridley et al 2003, Kim and Wirtz 2013). In 3D matrices, the situation is different, as cells cannot build a large lamellipodium or filopodia, instead they display dendritic and pseudopodial or lobopodial protrusions that are regulated by actomyosin contractility and the dynamic assembly and disassembly of microtubules (Friedl et al 2012, Giri et al 2013). The migration of cells through the 3D extracellular matrix can also involve the expression of MMPs, which are unnecessary for the migration in 2D, but facilitate the switch of the cells from a protrusive migration mode towards an amoeboid migration mode, in which the cells alter the mechanical properties in dense 3D extracellular matrices such as pore-sizes (Bloom et al 2008, Wirtz et al 2011, Wolf et al 2013). In addition, the signaling via zyxin, alpha-actin and p130 CAS is altered and regulates the spatial and temporal different movements sections through the matrix (Fraley et al 2012).
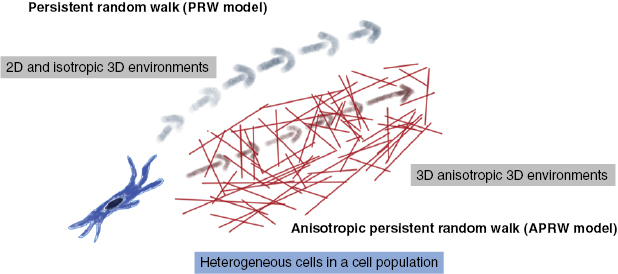
Figure 23. Persistent random walk (PRW) model is suitable for 2D migration, but not generally for the migration in anisotropic 3D microenvironments of a heterogeneous cell population. In the latter case the anisotropic persistent random walk model needs to be employed.
Download figure:
Standard image High-resolution imageThe traditional PRW model cannot be used for describing the migration of fibrosarcoma cells such as HT1080 in 3D collagen I matrices as it has been used to analyze the parameters such as cell speed and persistence time (Wolf et al 2013). A weakness of the persistent random walk model is that it does not account for cellular heterogeneity, which has been detected in single cancer types and is even increased during the malignant progression of cancer. In addition to 2D environments, cells migrating through a 3D matrix scaffold that is on larger length scales isotropic, but on smaller length scales highly anisotropic. Hence the anisotropy needs be integrated into the model as well as the heterogeneity within a cell population. Indeed, a new anisotropic persistence random walk (APRW) model has been proposed that can fit the migration of cells in 3D microenvironments (Wu et al 2014a). The findings indicate that the strategies for cell movement are matrix density dependent.
7.2. Adhesive structures such as adhesive tracks guide cells out of the primary tumor
Fibronectin is a major component of the extracellular matrix of tissues and is found to be increased around primary tumors (Astrof et al 2004, Levental et al 2009). A fibronectin gradient guides the cancer cells to move out of the primary tumor in a directed manner (Oudin et al 2016). The haptotaxis-driven migration on the fibronectin tracks requires the direct interaction between the alpha5 beta1 integrins and Mena, which belongs to the ENA/VASP family, undergoes alternative splicing (during cancer progression) and is an actin regulator by supporting the elongation of actin filaments (Gertler and Condeelis 2011). Pro-metastatic isoforms of Mena, termed MenaInv, induced through the interaction with alpha5 beta1 integrins during the haptotaxic migration of fibronectin. Moreover, high levels of MenaInv are correlating with increased metastatic lesions and decreased patient survival. In particular, MENA binds to the cytoplasmic C-terminal tail of the integrin α5 through an LERER repeat domain, which is not present in other ENA/VASP proteins (Gupton et al 2012). In fibroblasts, it has been demonstrated that MENA facilitates both outside-in and inside-out signaling at focal adhesions through the interaction with the alpha5 integrin subunit (Gupton et al 2012). MenaInv, which is an alternatively spliced isoform with an additional 19 amino acid inclusion ('INV' exon), is highly present in aggressive and invasive cancer cell subpopulations (Goswami et al 2009). Hence, MenaInv expression fosters metastasis through enhanced sensitivity towards EGF (Philippar et al 2008) and increases the efficiency of extracellular matrix degradation, cancer cell invasion and intravasation in blood or lymph vessels (Roussos et al 2011a). Based on these findings, it has been proposed that the extracellular matrix plays an additional crucial role in MENA/ MenaInv facilitated metastasis. Based on clinical models and the analysis of samples from patients, it has been proposed an unappreciated mechanism of metastasis, where increased levels of MENA and its invasive isoform endows cancer cells to migrate up fibronectin gradients and create their own pathway to reach the bloodstream.
Cancer cells react to a variety of cues to facilitate the invasion and metastasis in targeted organs and tissues. A growth factor driven chemotactic response is found to be crucial for local invasion and metastasis (Roussos et al 2011b). However, less is known about the process of haptotaxis that is utilized for cell migration and guided by gradients of surface-bound molecules such as extracellular matrix proteins. This process is highly relevant in cancer, where the levels of fibronectin in primary tumors can vary greatly. There are typically high concentrations of fibronectin near blood vessels, at the periphery of primary tumors and in targeted sites for metastases (Astrof et al 2004, Levental et al 2009). Besides the function of fibronectin in the activation of intracellular signaling pathways through the binding to integrins (Huttenlocher and Horwitz 2011), the cells are able to remodel the extracellular matrix by facilitating the integrin-mediated assembly of soluble fibronectin into fibrils (Schwarzbauer and DeSimone 2011). Moreover, fibronectin plays a prominent role in fibrillogenesis of collagen (Kadler et al 2008). Finally, the bidirectional communication between cells and extracellular matrix cues controls cellular behavior and compromises the composition and structure of the tumor surrounding stroma.
Besides adhesive tracks that guide cells out of a primary tumor, the presence of specific extracellular matrix proteins such as fibronectin, laminin or various collagen types such as collagen type IV and V can vary the migratory behavior inside these 3D extracellular matrices in terms of migration efficiency such as percentage of invasive cells, invasion speed, invasion path (random or directed persistent motion) and their invasion depths (Mierke et al 2011a). The presence of fibronectin inside 3D extracellular matrices increases the migration efficiency of human breast cancer cells significantly, when these cells express the fibronectin receptor, the integrin alpha5 beta1, on their cell surface, whereas otherwise the cells do not show an altered migration response (Mierke et al 2011a).
7.3. Effect of chronic fibrosis and stroma stiffening on cancer metastasis
During limited tissue repair is the direction of cellular movement impaired (Qu et al 2017). In dense connective extracellular matrix tissue environments are repair and wound healing processes often restricted due to the lack of immune cells and stroma cells at the tissue injury site. Decreasing the density of the local extracellular matrix has been hypothesized together with the release chemoattractive signals to cause an enhanced cellularity of the injured tissue and increase the formation of new tissue after injury (Zhang and Xu 2017). Indeed, in a knee meniscus model system, it has been found that interstitial cell migration under confinement such as a migratory barrier, which is represented by a novel tissue Boyden chamber, depends on a gradient of platelet-derived growth factor-AB (PDGF-AB), which increases the migration speed and efficiency through native tissue (Zhang and Xu 2017). Hence it may be hypothesized that cancer cells experience a similar confinement and hence may behave similarly. To analyze this effect in situ, nanofibrous scaffolds are utilized consisting of a distinct fiber fraction, which sequentially release active collagenase to enhance the extracellular matrix porosity and PDGF-AB dependent recruiting of endogenous cells in a localized and highly coordinated manner. When these scaffolds are placed into a meniscal defect, the controlled release of collagenase and PDGF-AB increases the number of cells at the interface as well as within the entire scaffold and subsequently increases their integration into the surrounding tissue. An increasing number of old and recent studies show that the chronic inflammation can facilitate the formation of a tumor (figure 24). Indeed, it has been shown that the cystic fibrosis elevates the risk of cancer (Neglia et al 1995, Oudin and Weaver 2016). The complex interactions between inflammatory cells, stroma and tumor parenchymal cells are closely related to the in vivo situation when a primary tumor is formed. A chronic inflammatory microenvironment causes long-term interaction of inflammatory cells, stromal cells and parenchymal cells that alters the signaling in parenchyma cells from an ordered to a disordered state. In line with this, multiple gene level editor modifications, epigenetic alterations and the deregulated transcription and translation of distinct genes are supposed to be evoked by a disorder of signaling pathways due to the chronic inflammation. These alterations subsequently cause cell mutations and a phenotypic transformation.
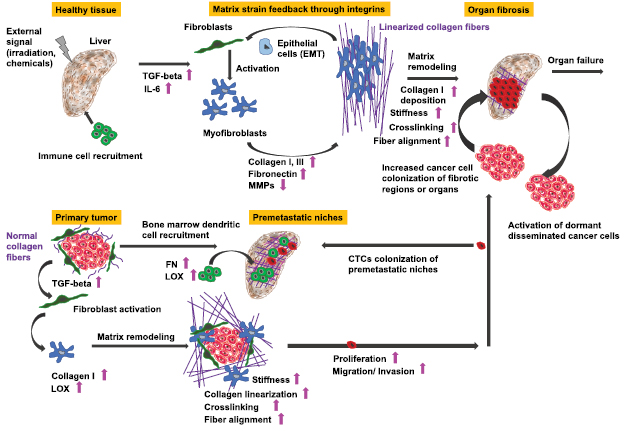
{kind=link}
{kind=link}
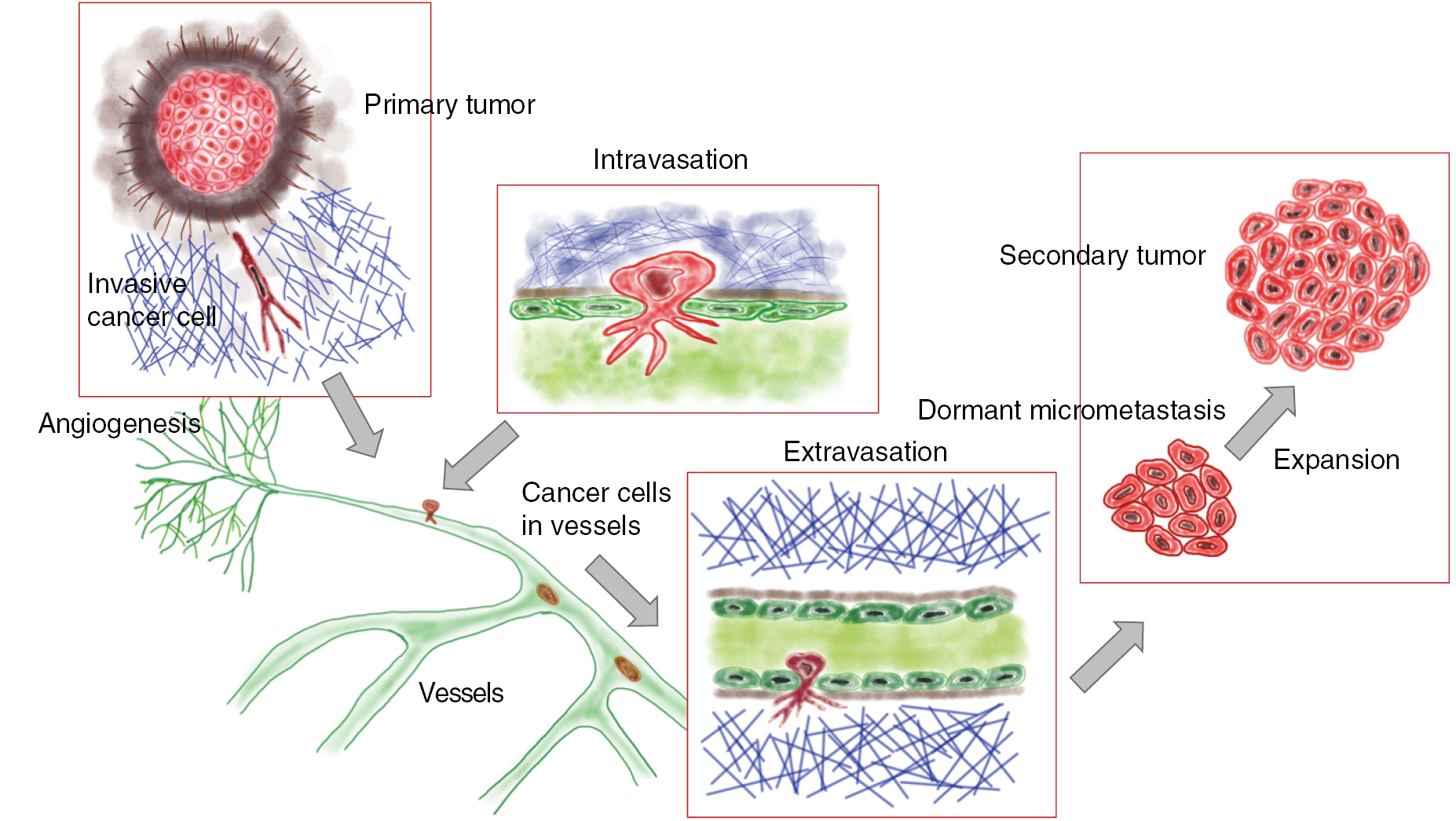{kind=link}
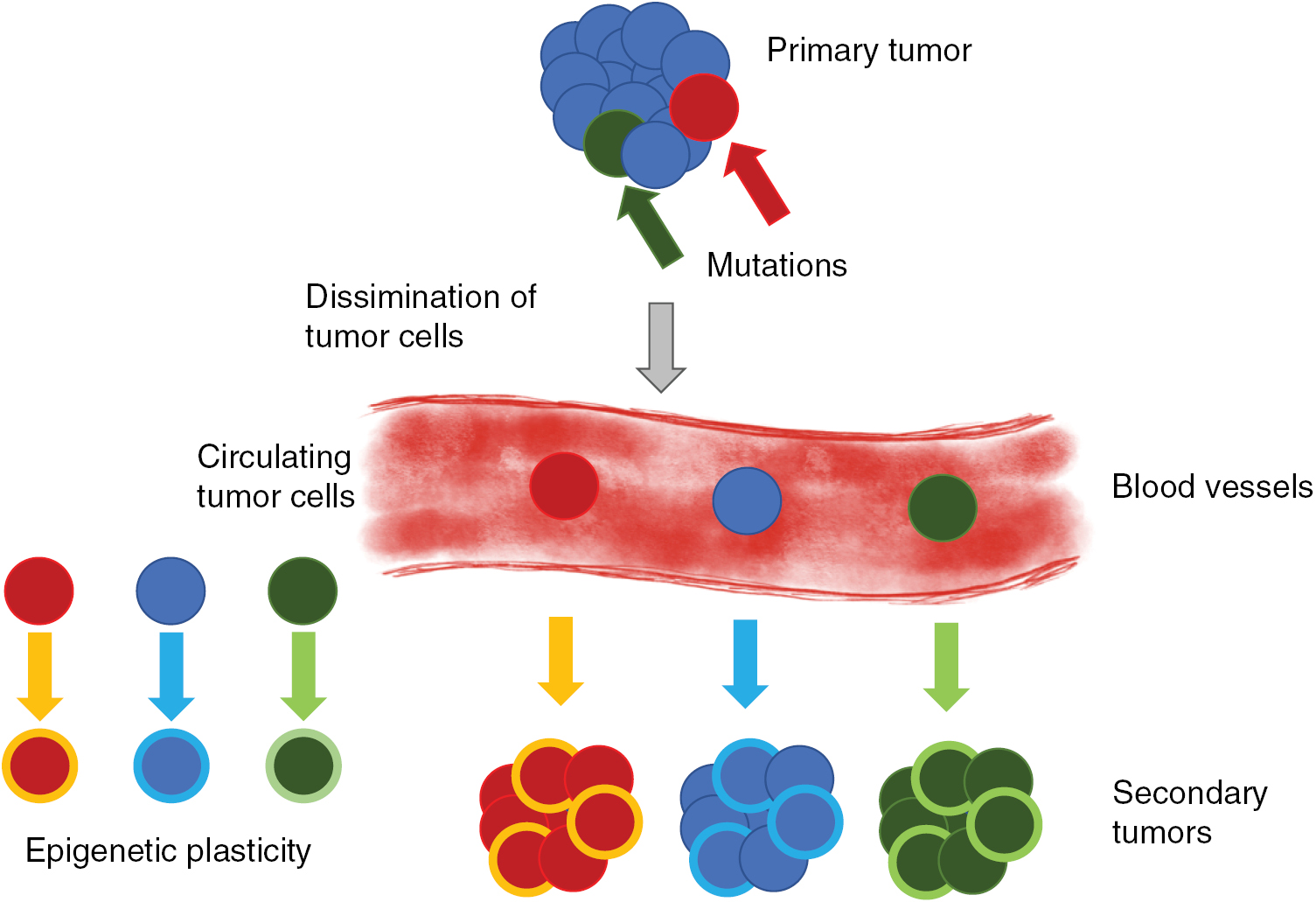{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
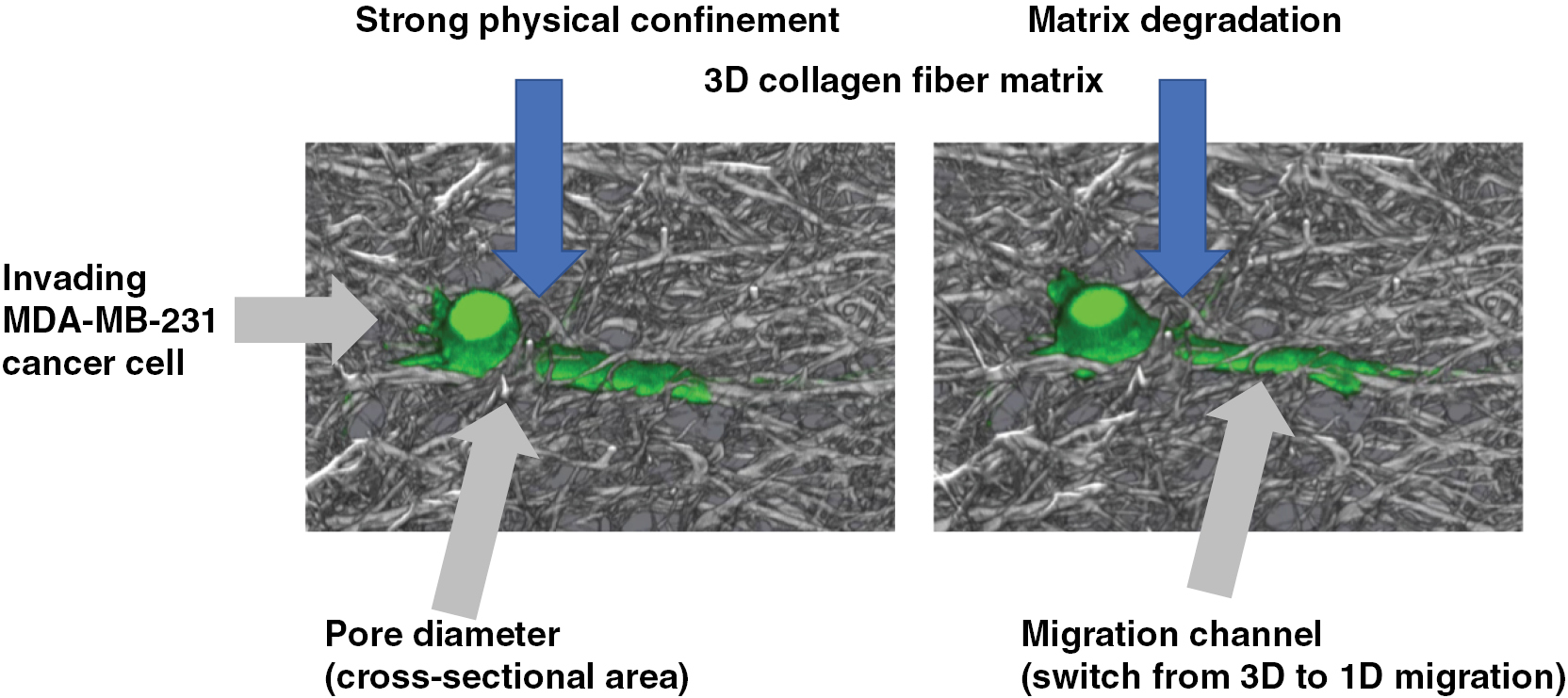{kind=link}
{kind=link}
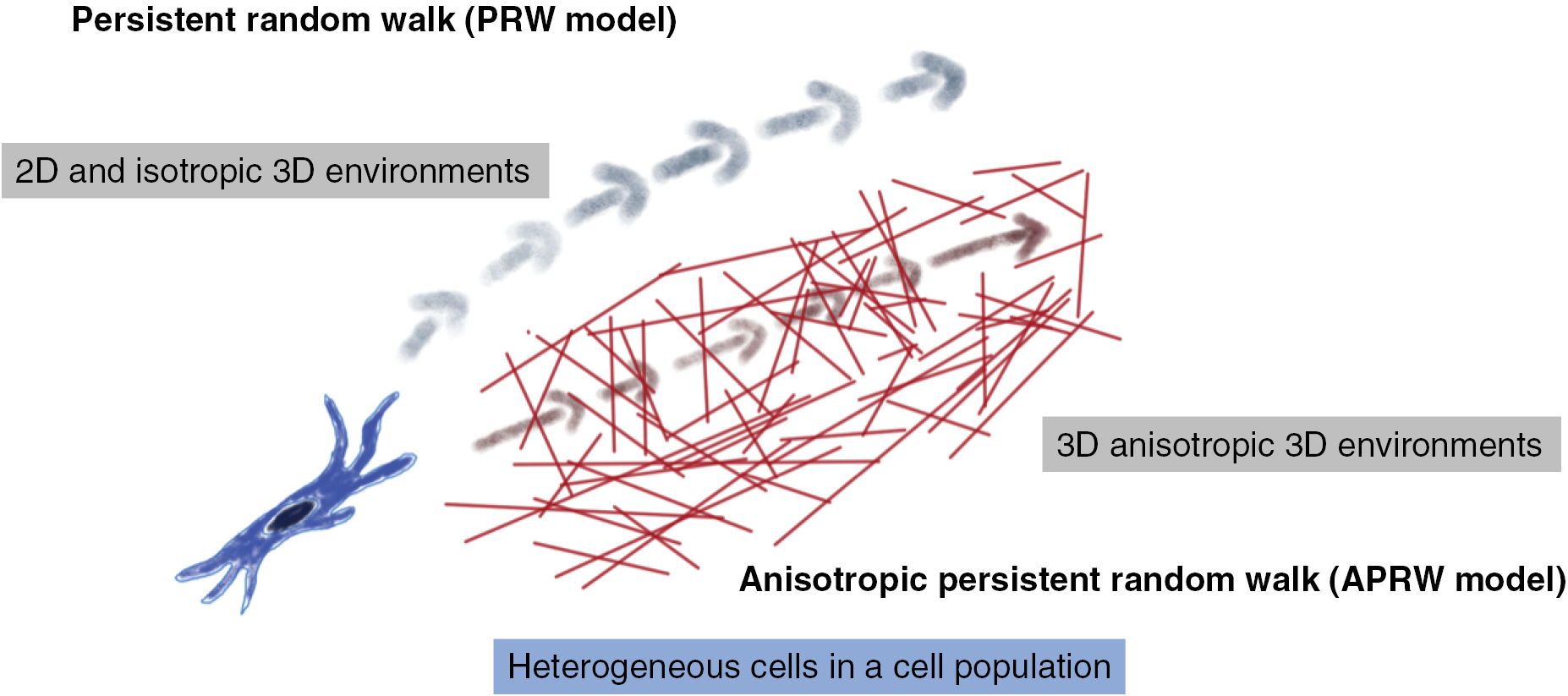{kind=link}
Figure 24. Tumor-stroma interactions. Healthy tissue can activate repair mechanisms upon tissue damage. The onset of fibrosis is reached after repetitive or chronic inflammation through the persistent activation of myofibroblasts that alter the mechanical properties such as increases stiffness of tissues by collagen fiber alignment (linearization) or extracellular matrix deposition. The affected fibrotic tissue is a niche for dormant cancer cells to reside and generate a prometastatic microenvironment, which in turn recruits more CTCs. Both tumor microenvironment and fibrotic tissue lesions favor the metastatic cascade of the malignant progression of cancer and causes subsequently organ failure and the formation of secondary tumors in targeted tissues or organs.
Download figure:
Standard image High-resolution image{kind=link}
7.4. Effect of matrix degradation or remodeling on cancer cell migration and persistence
As the mechanical properties of the extracellular matrix environment such as connective tissue can alter the mechanical properties of cells and hence determine cellular functions such as motility, the extracellular matrix environment can be altered by the cells that migrate in and invade through it (Wolf et al 2003, Mierke et al 2018). Using particle tracking of intracellular beads, it has been found that the intracellular remodeling dynamics and cellular cytoplasmic stiffening is coupled to cell motility and can perturb the mechanotransduction process in breast cancer cells. These results support the hypothesis that the intracellular contractility and stiffness are altered due to extracellular matrix stiffness changes (Baker et al 2010). In addition, matrix-embedded beads can be tracked and combined with proteolytic tracks analyses in extracellular matrices (Bloom et al 2008). In more detail, beads can be tethered to collagen I fibers in close proximity to migrating fibrosarcoma cells that are cultured in the absence and presence of proteolytic inhibitors and acto-myosin dependent contractile force inhibitors. However, the bead internalization and thereby the migratory impairment needs to be determined (Mierke 2013). When the axis of cell migration is used as a reference during forward cell motion, the release of extracellular matrix due to proteolytic activity near the cell's trailing edge has been detected. As the extracellular matrix degradation has been found to be asymmetric to the axis of cell migration and evoked inelastic deformation and thereby may support increased persistence of motion, symmetric to the cell axis it has been shown that extracellular matrix deformations near the cell's leading edge are elastically recoverable deformations.
8. Conclusions and outlook
The mechanical properties of the matrix microenvironment of primary solid tumors and the cellular mechanical properties regulate the invasive phenotype of cancer cells. However, there remain still many questions that are not yet fully answered in the vastly expanding field of mechanobiology, physical oncology and cell migration. Cancer cells are known to be extremely plastic in terms of their mechanical properties and may be able to adapt to commonly applied cancer treatments and microenvironmental changes rapidly (Gong et al 2012, Huan et al 2013, Liang et al 2013, Tang et al 2014, Tu et al 2015, Frankel et al 2017, Gouirand et al 2018). This behavior represents one of the factors that is responsible for the poor prognosis of malignant cancer progression. In addition, the tumor microenvironment is adapted to the ongoing progression of cancer by altering the mechanical properties of the extracellular matrix that creates an even more advanced tumor microenvironment. Besides the cellular mechanical properties of cancer also the cancer signal transduction processes are altered by the matrix mechanical properties.
Multiple biophysical reports have previously been concentrated on single proteins, which act as mechanosensory proteins, when mechanically stretched, that allows them to bind other interacting proteins. However, it will be crucial to reveal whether the stretching of tumor associated and promoting proteins can induce their enzymatic activity by unmasking of protein-binding sites that have been sterically impaired. Stretching is not limited to protein structures, it can also cause formational alterations of a membrane by the exertion of pulling forces, induction of curved membranes (increased tension) or exertion of protrusions that are known to be critical for a distinct migration type during cancer cell migration or normal cell migration. Hence, although there exists a large heterogeneity in cancer types, it seems to be likely that hallmarks of cancer cell migration are unique for various cancer types. A key question still needs to be answered is: Are mechanobiological signals and cues are drugable? Can they be used for cancer treatment? During fibrosis or tumor growth and invasion, medication of the mechanobiological cellular properties and the surrounding stroma appears to be a valid treatment option. Besides a single treatment approach of cancer, it is proposed that various mechanisms of cancer progression need to be attacked at the same time in order to achieve that best outcome of cancer treatment and also for the prevention of cancer. Hence, the identification of predictive biomarkers such as mechanical or biochemical properties of cancer cells that address the complex scenario of cancer, will further promote the development of novel therapies that are adapted to individual patients and tailored to their special needs.
As the extracellular matrix microenvironment fulfills such an important task in the malignant progression, the alteration of the extracellular matrix mechanical phenotype may possess a great potential for the development of novel approaches for cancer treatment. Indeed, novel methods are being actively trailed to reduce the spreading of cancer cells from the primary solid tumor by the use of drugs altering the mechanical properties of the tumor microenvironment that subsequently impairs the invasive phenotype of cancer cells (Oudin and Weaver 2016). Another approach has been the investigation of the mechanisms that cause matrix remodeling in order to develop inhibitory drugs impairing this specific process. However, it is needed to fully characterize the matrix mechanical phenotype together with the mechanical phenotype of cancer cells and their interplay to understand the underlying mechanisms and how those can be modified in order to achieve therapeutic benefit. The tumor microenvironment has become broadly accepted as a target for cancer therapy, whereas synergistic approaches targeting also the mechanical properties of cancer cells or the identification of multiple target markers seem to be promising in future cancer therapy to circumvent tumor plasticity and the impair the progression of cancer in the long term.
Acknowledgments
This work was supported by the DFG (MI1211/18-1 and INST268/357-1 FUGG), the ESF-SAB founded young scientist research group (No. 100147954) and EFRE-SAB infrastructure (No. 100299919).
Biographies

Claudia Tanja Mierke studied biology at TU Braunschweig in Germany and received her doctoral degree from the Medical School of Hannover 2001. Her postdoctoral research was performed in different institutes of the University of Erlangen-Nuremberg. In 2012 she habilitated in biophysics. Since 2010 she has worked as a professor at the University of Leipzig and is now head of the Biological Physics Division. She has published recently two books "Physics of Cancer", many review articles and book chapters on the subject of physical-driven cancer research.