Article Text

Abstract
Background Families with a child who died of severe, maternally inherited mitochondrial DNA (mtDNA) disease need information on recurrence risk. Estimating this risk is difficult because of (a) heteroplasmy—the coexistence of mutant and normal mtDNA in the same person—and (b) the so-called mitochondrial bottleneck, whereby the small number of mtDNAs that become the founders for the offspring cause variation in dose of mutant mtDNA. The timing of the bottleneck and of segregation of mtDNA during foetal life determines the management options. Therefore, mtDNA heteroplasmy was studied in oocytes and placenta of women in affected families.
Results One mother of a child dying from Leigh syndrome due to the 9176T→C mtDNA mutation transmitted various loads of mutant mtDNA to ≤3 of 20 oocytes. This was used to estimate recurrence as ≤5%. She subsequently conceived a healthy son naturally. Analysis of the placenta showed that some segregation also occurred during placental development, with the mutant mtDNA load varying by >10% in a placenta carrying 65% 3243A→G mutant mtDNA.
Discussion This is the first report of (a) an oocyte analysis for preconception counselling, specifically, refining recurrence risks of rare mutations and (b) a widely different load of a pathogenic mtDNA mutation in multiple oocytes, apparently confined to the germline, in an asymptomatic carrier of an mtDNA disease. This suggests that a major component of the bottleneck occurs during oogenesis, probably early in the foetal life of the mother. The variable mutant load in placenta implies that estimates based on a single sample in prenatal diagnosis of mtDNA disorders have limited accuracy.
- Metabolic disorders
- Molecular genetics
- Neuromuscular disease
- Reproductive medicine
Statistics from Altmetric.com
Maternally inherited mitochondrial DNA (mtDNA) diseases are common genetic disorders, with the population prevalence of deafness associated with the 3243A→G mtDNA mutation being 1 in 400.1 The more severe types often present in the neonatal period, with a range of devastating conditions, including cardiomyopathy, loss of vision and fatal liver disease. The severity of their expression varies because of heteroplasmy—the coexistence of mutant and normal mtDNA in the same person. A threshold effect (ie, tissues function normally unless the load of mutant mtDNA rises above a particular level) also exists in most mtDNA diseases, the progression of which is explained by preferential accumulation of mutant mtDNAs in affected tissues.
Couples who have had a child severely affected by a maternally inherited mtDNA disorder frequently ask about the much publicised nuclear transfer as a way of reducing transmission of mtDNA disease; however, this is not a realistic option at present.2 They then request prenatal diagnosis in a subsequent pregnancy, which has not been widely applied to mtDNA disease.3 For instance, chorionic villus sampling (CVS) may be unreliable.4 High levels of mutant mtDNA in placental tissue indicate that the foetus is probably affected; zero levels indicate that it is probably unaffected, but intermediate levels5 are unhelpful. The correlation between mutant dose and disease severity is often poor.6–8
Providing accurate information about risk recurrence is similarly difficult, and so many families reluctantly decide not to have more children. Preconception counselling is problematic because of the mitochondrial “bottleneck”.9–11 The most extreme example occurs after complete switching of mtDNA type in a single transmission, probably as a result of a mutation in a single mtDNA molecule, so that all a mother's mtDNA copies differ from those in her offspring by a single nucleotide. Significant mtDNA segregation occurs during oogenesis in both woman and mouse.5 10 12–15 Furthermore, this may enable selection against detrimental mtDNA mutants in the mouse germline.16 17
Investigation of the bottleneck has had three foci. First, its exact timing and molecular basis remain controversial: the major component is held to occur at E7.5 in the mouse,13 early in oogenesis, while the mother is still herself a developing foetus. Second, the mutant load in the foetus may be determined by segregation between the blastocyst stage and fertilisation. This is because the majority of cells in the blastocyst will develop into extra-embryonic structures, with perhaps as few as three cells of the inner cell mass developing into the embryo. Third, segregation of mutant mtDNA in extra-embryonic tissues would affect sampling of mtDNA at CVS. All of these are difficult to investigate in humans, but are clearly critical in advising families about their options.5
The current investigation of the mtDNA bottleneck is highly relevant because it significantly amplifies the existing dataset in humans. First, we have studied the segregation of both pathogenic and non-functional polymorphic mtDNA variants in the germline. Two women who sought preconception counselling enabled us to study two pathogenic mutants in their oocytes. Hence, we have documented a unique case of germline mosaicism for an mtDNA mutant. Second, we have investigated segregation after conception in a placenta from a woman who has mitochondrially inherited diabetes and deafness due to the pathogenic mtDNA 3243A→G mutant. We documented the segregation of mtDNA subpopulations, with estimates of heteroplasmic load varying by as much as 10% in different samples.
We conclude that a major component of the mitochondrial bottleneck has occurred by the time oocytes are mature, and that segregation, and potentially selection, may continue post conception. Oocyte sampling is useful for genetic counselling. Our data support the use of prenatal diagnosis with certain provisos.
Materials and methods
Informed consent was obtained from subjects (or their guardians), and ethics committee approval was from the Oxford Research Ethics Committee.
Patients and oocyte sampling
Case 1
The woman's first child, who died aged 4 years, had developmental regression, lactic acidosis, cardiomyopathy and MRI changes consistent with maternally inherited Leigh disease. An mtDNA mutation in the child was identified at 9176T→C. In this disorder, the published data suggest that individuals carrying less than 80–90% mutant mtDNA are asymptomatic.18 However, 9176T→C mutant mtDNA was undetectable in the mother's blood, buccal smear and urinary epithelial cells. She opted for oocyte sampling using conventional in vitro fertilisation methods because of the uncertainty surrounding the recurrence risk. Twenty oocytes were retrieved under ultrasound control following gonadotrophin stimulation, lysed and amplified by PCR as before.9
Case 2
The woman's first child died aged 21 months with liver disease, lactic acidosis and anaemia, which are characteristics of Pearson syndrome. She had been given ciprofloxacin around the time of conception, a drug known to cause mtDNA depletion in cell lines but never previously implicated in the pathogenesis of mtDNA rearrangements. Pearson syndrome is usually sporadic; however, maternal transmission was considered possible in this case because the affected child had duplications as well as deletions (rearrangements) of mtDNA, and these may be maternally inherited.4 19 Furthermore, the mother had a cardiac conduction defect, and these can be caused by mtDNA rearrangements. No rearranged mtDNA was detectable in the mother's blood; however, maternal muscle was not available for analysis. The parents requested oocyte sampling even though the recurrence risk was believed to be low (≤5%4 20) because they could not contemplate the possibility of another affected child. Gonadotrophin stimulation and oocyte collection were carried out as above. Seventeen oocytes were collected (see supplementary information for the detailed analytical method).
Placental donor: case 3
A known patient with diabetes and deafness due to the mtDNA 3243A→G mutation, who has one apparently healthy child, became pregnant for the seventh time at age 40 years. Weight gain was poor and insulin requirement barely increased. Following pre-eclampsia, which was a feature of all her pregnancies and an emerging complication of this mutation,21 she gave birth to a severely growth-restricted stillborn foetus weighing only 350 g. About 10% of the foetal component of her 30 g placenta was frozen for analysis.
Isolation of DNA from single oocytes
Detailed methods for isolating oocytes and extracting DNA are in supplementary information B.
PCR analysis of oocytes and placenta (see supplementary information B)
The 9176T→C mutation was quantitated using standard methods.22 23 Last-cycle labelling with 32P-dCTP and Phosphorimager analysis were used. The sensitivity of the method was generally high, at least 2% 9176T→C mtDNA being detectable on a wild-type background (supplementary information D figure 2). The 3243A→G mutation was quantitated using last-cycle fluorescent labelling followed by capillary electrophoresis on ABI3100,22 23 the sensitivity being <2%.
The method for estimating risk used a mathematical model of the bottleneck with a range of bottleneck sizes9 (Supplementary information A).
To detect mtDNA carrying the rearrangement, primers that straddled the deletion junction were used as previously shown.24 The sensitivity of the method was again high (1% could be detected on a wild-type background; data not shown).
To distinguish between maternal and foetal DNA in the mtDNA 3243A→G placenta, we carried out microsatellite analysis using the AmpflSTR Profiler Plus kit (Applied Biosystems, Foster City, California, USA).
Statistical analysis
Analysis of the bottleneck in human oocytes used a binomial sampling distribution and is outlined in Supplementary information A. The mtDNA mutant in individual oocytes was compared with the Kimura distribution as previously shown 25 (Supplementary information). Differences in mtDNA 3243A→G mutant load in placenta were assessed by using t test.
Results: oocyte study
We aimed to study the mtDNA bottleneck by determining the levels of mutant mtDNA in oocytes from two mothers of affected children, to refine the recurrence risks for subsequent pregnancies.
Case 1: maternally inherited Leigh disease due to 9176T→C
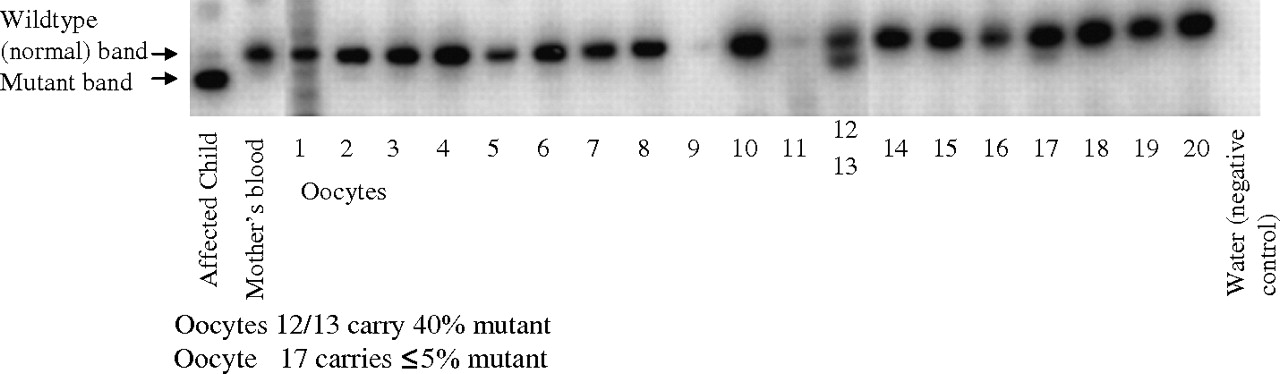
Figure 1 shows that levels of ≤80% 9176T→C mtDNA were detected in ≤3 of the 20 oocytes obtained. Calculation of recurrence risk is complex, with two sources of uncertainty. One is of the actual load of mutant mtDNA molecules in the germline, given the observations and the accuracy of the quantitation. The other is the probability that any particular oocyte will develop into an individual with a mutant load above the disease threshold (taken as 80% for 9176T→C).18 22 26–31 We estimated the recurrence risk to be ≤5%; the subsequent more detailed analysis suggests that it is ≤3% (Supplementary information). The distribution of the mutant load of mtDNA was compared to the Kimura distribution. It was consistent with the theoretical distribution (Supplementary information) with an unusually high variance, twice as high as the heteroplasmy variance in oocytes that was previously reported.32 The woman subsequently decided to conceive naturally, and happily3 delivered a healthy baby at term. CVS was carried out as in a previous family18 and mutant mtDNA was undetectable at 10 weeks of gestation, in 16 different samples of term placenta and in cord blood.

Detection of the 9176T→C mutation in 20 oocytes after superovulation, in the mother's blood and in muscle from the affected child who died. The 9176T→C mutant mitochondrial DNA (mtDNA) generates a new restriction site, so that, in the affected child, the upper wild-type (normal) band is replaced by a lower (mutant) fragment. The child carried ∼99% 9176T→C mutant mtDNA in all tissues examined (marked “affected”), but none was detectable in the mother's blood, CVS, placenta or cord blood from her foetus. Oocyte 17 (mutant band judged just visible) carried ≤5% 9176T→C mutant mtDNA. Oocytes 12 and 13 could not be dissected apart and carried 40% mutant mtDNA, consistent with 40–80% in one and 0–40% mutant in the other. The PCR efficiency was too low to be used in two oocytes (No 9 and 11) and these were excluded from the analysis. Assuming a mutant mtDNA load of 80% in oocyte 12, 0% in oocyte 13 and 5% in oocyte 17, the average load in the 18 useable oocytes was 5%. Assuming a threshold mutant load of 80% for symptomatic disease and taking into account a range of bottlenecks, the estimated recurrence risk was ≤5% (supplementary information).
Case 2 Pearson syndrome due to mtDNA rearrangement
No rearranged mtDNA was detected in any of the 10/17 oocytes from which PCR product was obtained. The recurrence risk was estimated as <1% (Supplementary information A). She has not yet conceived.
Results: placental study
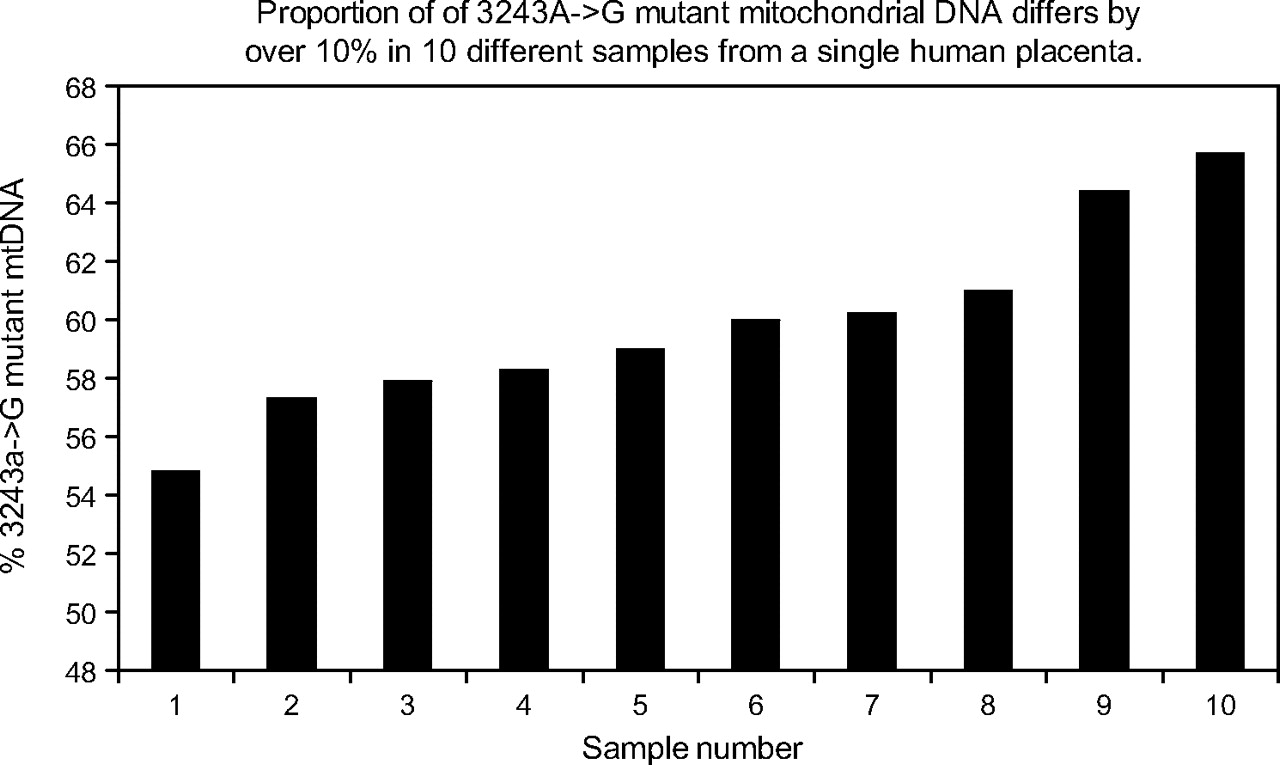
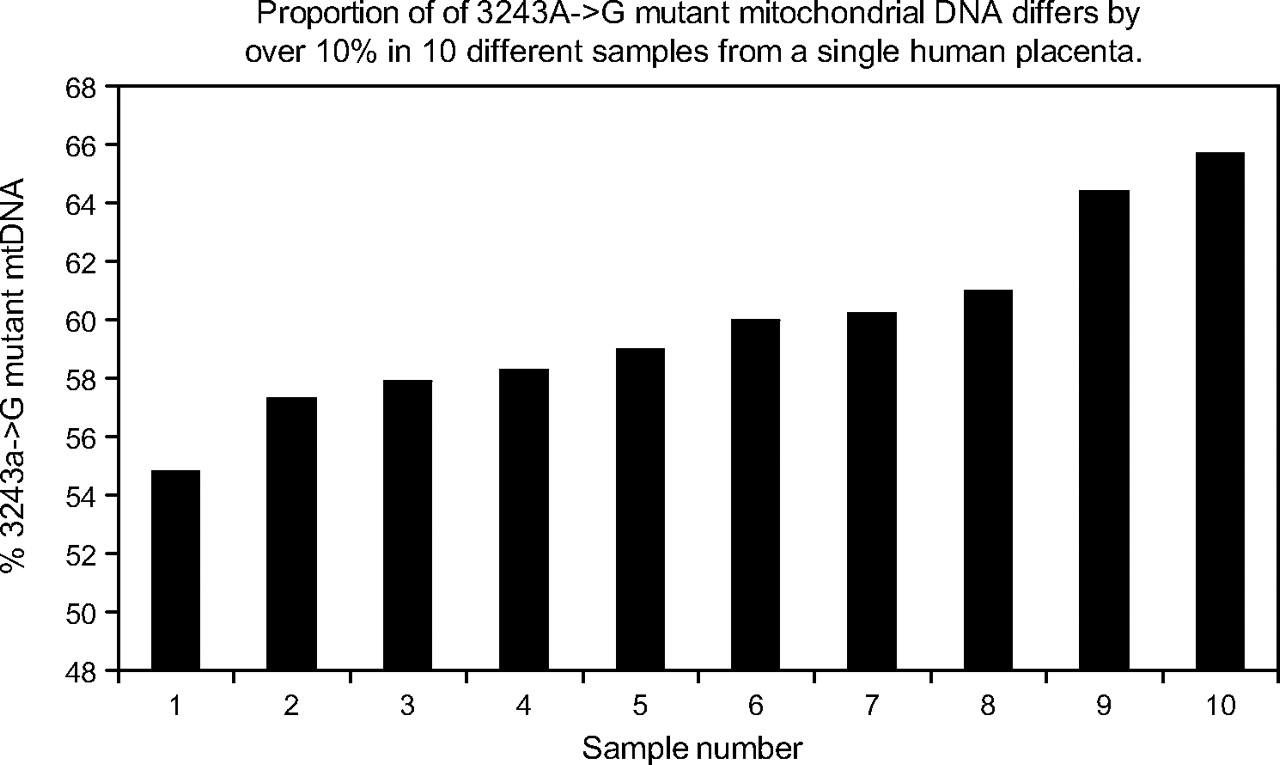
DNA from 10 samples of the mtDNA 3243A→G placenta, each approximately 10 mg, were analysed for both the mutant load and mtDNA copy number as shown in figure 2. The mother's mtDNA 3243A→G mutant load was 24% in blood DNA. The percentage mutant ranged from 55% to 64% and the mtDNA copy number varied almost fivefold (supplementary figure). To assess the reproducibility of these differences, the samples containing the highest and lowest mutant loads were rerun (including PCR and restriction digestion) in quadruplicate (each one being loaded in duplicate) on two further occasions. The average mutant loads differed by 11.95% (being 49.41% (0.42%) and 61.42% (0.42%), p<0.0025, t test), confirming that this reflects a real difference in mutant load in different regions of the same placenta. Microsatellite analysis of all placental samples (using the AmpflSTR Profiler Plus kit, Applied Biosystems) showed that all samples were entirely composed of foetal and not maternal DNA.
{kind=link}
{kind=link}
Proportion of 3243A→G mutant mitochondrial DNA (mtDNA) differs by over 10% in 10 different samples of human placenta.
Discussion
These data significantly increase the data demonstrating segregation of pathogenic mutants in the human germline and in embryonic development. We have successfully used oocyte sampling to predict patient-specific recurrence risk for preconception counselling in heteroplasmic mtDNA disorders. Our demonstration of a widely different load of a pathogenic mtDNA mutation in multiple oocytes, apparently confined to the germline in an asymptomatic carrier of an mtDNA disease, demonstrates that segregation occurs during oogenesis. We have shown variation in the load of pathogenic mtDNA between small placental samples. In agreement with previous studies,9 13 15 17 our findings suggest that much of the segregation for these mtDNA variants, known as the mitochondrial bottleneck, occurs during oocyte development, with additional mtDNA segregation during differentiation of embryo and extra-embryonic structures.
As far as we know, this is the first report of the use of oocyte sampling to predict patient-specific recurrence risk for these disorders. Recurrence estimates given to the mothers were ≤5% and <1%. The new risk estimations enabled both women, who had decided against conceiving again, to change their minds. This is also the first demonstration of an mtDNA mutation in multiple oocytes, apparently confined to the germline, in an asymptomatic carrier of an mtDNA disease. As the 9176T→C mtDNA mutation was confined to between one and three oocytes, it presumably arose within the germline during the foetal life of the mother. This would be consistent with the high heteroplasmy variance (thus, small number of mtDNA founders) identified in the statistical analysis (supplementary information). The mutation could have been initially present but selected out of other tissues such as blood, or at a level below the detection limit. In case 2, there was no evidence of transmission of rearranged mtDNA to the oocytes, and a work that was unpublished at the time33 supports our advice that she had a low recurrence.
A limitation of using oocyte sampling for estimating recurrence risks is the scanty literature correlating the load of mutant mtDNA and disease severity. Individuals carrying a mutant load of <80% 9176T→C mutant mtDNA in blood mtDNA have minimal problems18 but many mtDNA disorders do not have clearly defined thresholds, which limits the accuracy of recurrence risk estimations. In particular, prenatal diagnosis has limited application in most families with Leber hereditary optic neuropathy, because these tend to be homoplasmic with a low penetrance of the disease phenotype: where they are heteroplasmic, there is a substantial risk of developing visual impairment over a wide range of mutant loads.34 Clinical geneticists have performed preimplantation genetic diagnosis (PGD) in these families, selecting embryos for female sex as women have a substantially lower risk of developing symptoms than men.35 We cannot be sure that the mutant load in oocytes precisely reflects that in live born children. However, the load in oocytes is likely to be more relevant than the load in maternal muscle and has some support from animal studies.36
Segregation of the 3243A→G mtDNA in the placenta showed that mtDNA mutant load differed across the placenta by as much as 10%, and this was not due to maternal contamination. In addition, mtDNA copy number varied approximately fivefold (see online figure 3). Similarly, we saw a statistically significant variation of similar magnitude in the distribution of one of two polymorphic variants, consistent with previous data.37 We previously demonstrated that the average level of polymorphic heteroplasmic mtDNA variants is very similar in mother, cord blood and placenta. However, where placental samples were very small (<10 mg), there was some evidence of variation in the distribution of mtDNA polymorphic variants. Differences in mtDNA content of the different cell types present in the 10 samples of 3243A→G placenta might underlie the fivefold difference in mtDNA copy number that we identified. Indeed, we have documented a similar degree of variation in mtDNA content in control mouse placenta (Sajida Malik and Jo Poulton, unpublished data 2008). However, variation of both mutant load and mtDNA copy number is a well-known characteristic of the 3243A→G mtDNA mutant, resulting in “ragged red” fibres in skeletal muscle probably due to a combination of random drift38 and compensation for the mitochondrial dysfunction.39 The magnitude of the amplification of mtDNA copy number is more comparable to that seen in ragged red fibres in skeletal muscle40 than tissue culture (see online figure 3). As this mother was prone to pre-eclampsia, the placenta may have been under considerable oxidative stress quite apart from the 3243A→G mtDNA mutant. We conclude that we documented variation in mtDNA content in different regions of a single placenta, and that oxidative stress and/or variable mitochondrial function may contribute to this.
The current study supports our previous suggestion that a single sample at CVS, or indeed PGD, may be insufficient to assess the mutant load. In preliminary data from human blastomeres41 42 (supplementary information 2), we provide some support for earlier findings from mouse43 and human44 that heteroplasmy in blastomeres varies more between embryos than within embryos. Nevertheless, in the absence of extensive data, it may be necessary to take two samples at CVS37 because of the considerable variation in the level of mtDNA mutant that we documented in placental patches. This also argues for repeated sampling at PGD, say two separate blastomeres. However, taking two samples would likely reduce the viability of the embryo. Another less invasive possibility would be to sample a blastomere and the cytoplasm surrounding a polar body, as preliminary data from mouse43 suggest that the mutant load is identical. As an alternative strategy,45 initially a single blastomere would be sampled from each embryo to identify those with low levels of mutant mtDNA. All blastomeres from the unsuitable embryos would then be analysed to ensure that the distribution of mutants is uniform within embryos. Only if this was reassuring would an embryo with a low level of mutant mtDNA be implanted.
The management options in clinical practice are extremely complicated.3 It is therefore very taxing and ethically challenging to advise women what to do. It is especially difficult for geneticists to estimate recurrence risk in heteroplasmic mtDNA diseases. In these disorders, prenatal diagnosis based on respiratory chain enzyme activities is unreliable,46 and that based on mtDNA is complex (because of the problem of heteroplasmy). PGD, which involves sampling blastomeres from early embryos and only transferring low-risk ones, could potentially avoid this dilemma. This procedure is hard to apply to women whose oocytes all have an unacceptably high mutant load. While there are scanty published reports of this technique for mtDNA disease,5 41 42 47 recent mouse studies suggest that strong selection in the germline against certain types of mutation16 17 may increase the chance of identifying embryos with an acceptably low mutant load. More data are needed to determine whether there is selection against the 3243A→G mutation in the human germline. Other authors discuss more radical measures such as replacing the zygote's mutant mitochondria with wild type.18 44 48 49
In conclusion, despite the uncertainties, two women willingly accepted oocyte sampling to estimate their risk, and one has since had a healthy child who is now aged 5 years old. We feel that undergoing two procedures is justified by the problems attributable to heteroplasmy. We advocate that clinicians consider whether taking two samples at CVS may be appropriate in specific cases. Furthermore, interpretation of CVS may be much more feasible where PGD has been used to select suitable embryos. The positive outcome of oocyte sampling has important practical and theoretical implications for genetic counselling of families affected by devastating mtDNA disorders. Even though we have demonstrated a major component of the bottleneck occurring during oogenesis, an essential precondition for PGD, our analysis also suggests that a considerable variation may develop during the later stages of placental development. It may therefore be advisable to take two samples at CVS in selected cases. These studies are generally encouraging for prenatal diagnosis in selected mtDNA diseases.
Acknowledgments
We thank the patients, their families and the physicians for their help, particularly Drs A Dornhurst and J Wyatt-Ashmead. We are grateful to Professor DH Barlow for initiating this project and to Dr Anneke Seller for provision of laboratory facilities, Julie Evans and Passorn Wonnapinij for technical support, Mrs Annie Shrier and Dr J Marchington for critical appraisal of the manuscript and Dr J Birks for statistical advice. This work was supported by Action Medical Research, the Wellcome Trust, the Medical Research Council, the Royal Society and the Oxford Partnership Comprehensive Biomedical Research Centre with funding from the Department of Health's National Institute for Health Research Biomedical Research Centres funding scheme. The views expressed in this publication are those of the authors and not necessarily those of the Department of Health.
References
Supplementary materials
Web Only Data jmg.2009.072900
Files in this Data Supplement:
Footnotes
Funding Action Medical Research. Other funders: Wellcome Trust; Medical Research Council, Royal Society and Department of Health's National Institute for Health Research.
Competing interests None.
Patient consent Obtained.
Ethics approval This study was conducted with the approval of the Oxford Research Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.