Article Text

Abstract
Objective This double-blind, placebo-controlled, dose-finding phase IIb study evaluated the efficacy and safety of ponesimod, an oral selective S1P1 receptor modulator, for the treatment of patients with relapsing–remitting multiple sclerosis (RRMS).
Methods 464 patients were randomised to receive once-daily oral ponesimod 10, 20 or 40 mg, or placebo for 24 weeks. The primary endpoint was the cumulative number of new T1 gadolinium-enhanced (T1 Gd+) lesions per patient recorded every 4 weeks from weeks 12 to 24 after study drug initiation. Secondary endpoints were the annualised confirmed relapse rate (ARR) and time to first confirmed relapse. Safety and tolerability were also evaluated.
Results The mean cumulative number of new T1 Gd+ lesions at weeks 12–24 was significantly lower in the ponesimod 10 mg (3.5; rate ratio (RR) 0.57; p=0.0318), 20 mg (1.1; RR 0.17; p<0.0001) and 40 mg (1.4; RR 0.23; p<0.0001) groups compared with placebo (6.2). The mean ARR was lower with 40 mg ponesimod versus placebo, with a maximum reduction of 52% (0.25 vs 0.53; p=0.0363). The time to first confirmed relapse was increased with ponesimod compared with placebo. The proportion of patients with ≥1 treatment-emergent adverse events (AEs) was similar across ponesimod groups and the placebo group. Frequently reported AEs with higher incidence in the three ponesimod groups compared with placebo were anxiety, dizziness, dyspnoea, increased alanine aminotransferase, influenza, insomnia and peripheral oedema.
Conclusions Once-daily treatment with ponesimod 10, 20 or 40 mg significantly reduced the number of new T1 Gd+ lesions and showed a beneficial effect on clinical endpoints. Ponesimod was generally well tolerated, and further investigation of ponesimod for the treatment of RRMS is under consideration.
Trial registration number NCT01006265.
- Multiple Sclerosis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of the central nervous system affecting an estimated 2.5 million people worldwide.1 ,2 Disease-modifying drugs for its treatment aim to reduce the relapse rate and slow the progression of disability. These include interferon β, glatiramer acetate, mitoxantrone, teriflunomide, dimethyl fumarate, alemtuzumab and natalizumab. However, due to the complex interplay of efficacy and safety, the more efficacious therapies may be reserved for disease states sufficiently aggressive to justify the associated toxicity.1 Furthermore, some available treatments, such as glatiramer acetate, require long-term self-injection by the patient, which may lead to a reduction in treatment adherence and, consequently, suboptimal efficacy.3 New therapies should have improved safety and efficacy and reduce the burden on the patient. Hence, there has been a trend towards the development of oral therapies. Recently approved therapies include fingolimod, which modulates sphingosine 1-phosphate (S1P) receptors, including S1P1, S1P3, S1P4 and S1P5; teriflunomide, an inhibitor of pyrimidine synthesis, and dimethyl fumarate, which were first approved in 2010, 2012 and 2013, respectively.4–8
Ponesimod [(Z,Z)-5-[3-chloro-4-((2R)-2,3-dihydroxy-propoxy)-benzylidene]-2-propylimino-3-o-tolyl-thiazolidin-4-one], an iminothiazolidinone derivative, is a reversible, orally active, selective S1P1 receptor modulator.9 ,10 Unlike fingolimod, which is a structural analogue of sphingosine, ponesimod is selective for S1P1; In vitro, ponesimod is at least 10-fold more potent on the S1P1 receptor than on any other S1P receptor subtype.10 Lymphocyte exit from lymph nodes and their migration into the blood and target tissues is mediated by S1P binding to S1P1 receptors. Binding of ponesimod to the S1P1 receptor results in rapid and efficient receptor internalisation, degradation and functional antagonism,10 ,11 thereby causing lymphocyte sequestration in the lymph nodes.
Phase I studies demonstrated that ponesimod pharmacokinetics are dose-proportional and characterised by low variability,12 and circulating lymphocyte counts are reduced in a dose-dependent manner.12 ,13 Ponesimod has a rapid onset of action, with maximal plasma concentrations observed 2.5–4 h after dosing,13 ,14 and maximum lymphocyte count reduction achieved 6 h after dosing.12 ,14 Ponesimod has a terminal half-life of approximately 32 h,13 ,14 and following treatment discontinuation, mean lymphocyte count returns to normal range within 7 days, with no rebound phenomena observed.12–14
We conducted a double-blind, placebo-controlled, dose-finding phase IIb study to evaluate the efficacy, safety and tolerability of three doses of once-daily ponesimod (10, 20, 40 mg) for the treatment of patients with relapsing–remitting multiple sclerosis (RRMS) (ClinicalTrials.gov identifier: NCT01006265).
Methods
Patients
Eligible patients were men and women aged 18–55 years with RRMS (as defined by the revised 2005 MacDonald Criteria15) and an Expanded Disability Status Scale (EDSS) score of 0–5.5, with at least one of the following characteristics: ≥1 documented relapse(s) within the 12 months before screening; ≥2 documented relapses within the 24 months before screening or at least one T1-weighted gadolinium-enhanced (Gd+) lesion detected on brain MRI at screening.
Study exclusion criteria were use of systemic corticosteroids within 30 days of randomisation; use of immunomodulators (interferon β, glatiramer acetate) and some immunosuppressants (cyclosporine, sirolimus and mycophenolic acid) within 3 months of randomisation; use of other immunosuppressants (azathioprine, methotrexate and natalizumab) and non-lymphocyte-depleting biologic agents (eg, daclizumab) within 6 months prior to randomisation. Patients treated at any time with certain immunosuppressive (cyclophosphamide, mitoxantrone and cladribine) or lymphocyte-depleting biological agents (alemtuzumab and rituximab) were excluded from the study.
Study design and procedures
This was a prospective, multicentre and multinational (94 centres in 23 countries, including Europe, Australia, Canada and USA), randomised, double-blind, placebo-controlled, four-arm, parallel-group, dose-finding phase IIb study, conducted in accordance with the Declaration of Helsinki16 and adhering to the International Conference on Harmonisation Guidelines for Good Clinical Practice.17 Within each investigation site, patients were randomised in a 1:1:1:1 ratio to once-daily treatment with placebo or ponesimod 10, 20 or 40 mg for 24 weeks.
Patients were randomised by assignment of a unique randomisation number using an interactive voice or web response system, supplied by an independent service provider (ICON Clinical, Research, USA). Patient randomisation was stratified by centre using a block size of four for the first two blocks and eight thereafter. The primary investigator/treating neurologist, independent evaluating neurologist, physician evaluating cardiac safety assessments, care providers, patients and sponsor were blinded to the treatment. The investigators and sponsor were blinded to the lymphocyte count results and first-dose effects of ponesimod, unless alerted for safety reasons. All ponesimod doses and matching placebo were indistinguishable and identically packaged.
The dose range selected for this study was based on phase I data.12 ,13 Ponesimod 10 mg was selected as the lowest dose as it was associated with an approximate 50% reduction in peripheral lymphocyte counts,13 which was considered as the minimum reduction required for an immunomodulatory effect. Ponesimod 40 mg was selected as the highest dose as it was associated with an approximate 70% reduction in peripheral lymphocyte counts13; this degree of reduction has previously been shown to be associated with a significant therapeutic effect in patients with MS treated with fingolimod.18 Furthermore, phase I studies demonstrated that through the use of an up-titration dosing regimen, the first-dose effects of ponesimod 20 and 40 mg on heart rate and conductivity were reduced, and the safety and tolerability of this higher dose were considered acceptable.
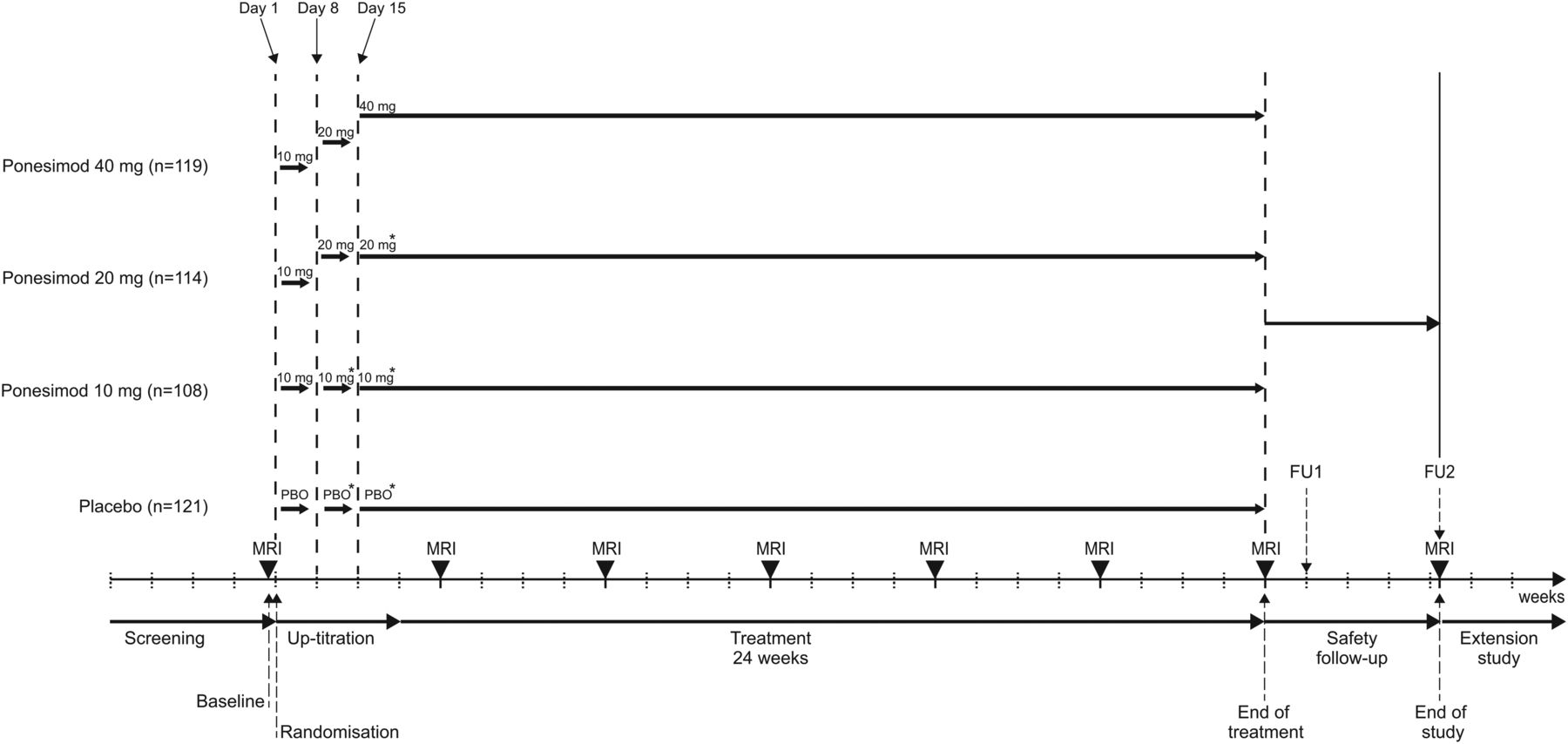
Study design is summarised in figure 1. All patients randomised to ponesimod initially received ponesimod 10 mg (days 1–7). On day 8, patients randomised to receive ponesimod 20 or 40 mg were up-titrated to the 20 mg dose and patients randomised to the 10 mg dose were mock up-titrated. On day 15, patients randomised to receive ponesimod 40 mg were up-titrated to the 40 mg dose; patients randomised to ponesimod 10 or 20 mg were mock up-titrated. Patients randomised to placebo were mock up-titrated on days 8 and 15 (figure 1).
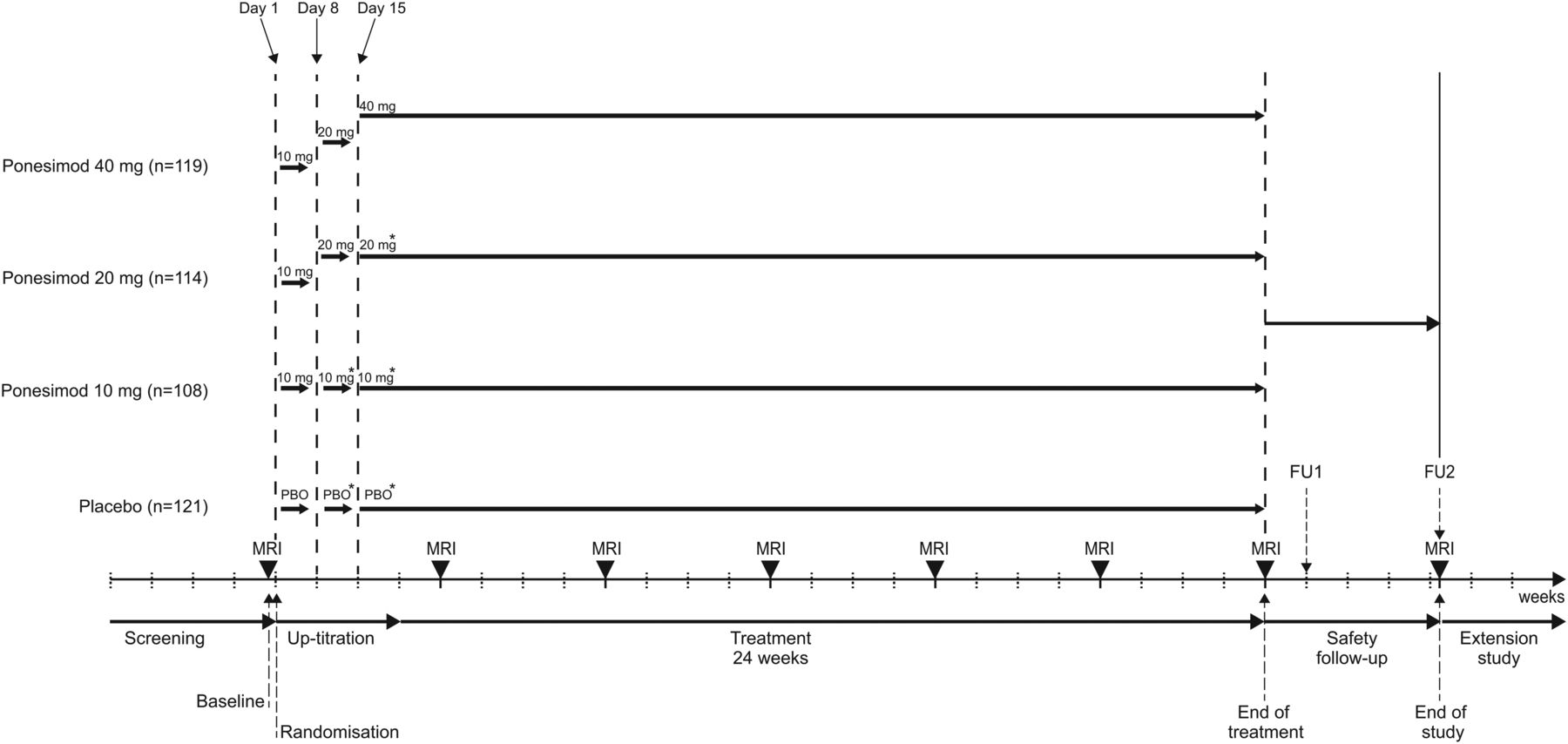
Study design. All patients randomised to ponesimod initially received ponesimod 10 mg (days 1–7). On day 8, patients randomised to receive ponesimod 20 or 40 mg were up-titrated to the 20 mg dose and patients randomised to the 10 mg dose were mock up-titrated. On day 15, patients randomised to receive ponesimod 40 mg were up-titrated to the 40 mg dose; patients randomised to ponesimod 10 or 20 mg were mock up-titrated. Patients randomised to placebo were mock up-titrated on days 8 and 15. Patient numbers are for the all-treated population. FU, follow-up.
Study visits took place at screening, baseline, on day 1 (randomisation), days 8 and 15 (up-titration), at week 4 and then every 4 weeks until the end of the 24-week treatment period or study drug discontinuation. Patients who completed treatment at week 24 were offered enrolment into an ongoing, long-term, dose-blinded extension study where all patients would receive ponesimod (ClinicalTrials.gov identifier: NCT01093326). For those patients who prematurely discontinued study treatment or who completed 24 weeks of treatment but chose not to enter the extension study, two safety follow-up visits were performed 7 and 30 days after the last dose of study drug (figure 1).
MRI scans were performed at baseline, every 4 weeks from week 4 to 24 (end of treatment (EOT)) and at the second safety follow-up visit for patients not entering the extension study. MRI scans were centrally evaluated in a fully blinded manner (Medical Image Analysis Centre, Basel, Switzerland). EDSS assessments were performed at screening, baseline, week 24, at follow-up visit 2 and at unscheduled visits in case of MS relapse by an independent neurologist not otherwise involved in patient care. The main pharmacodynamic variable was total lymphocyte count, which was centrally analysed at screening, baseline, on days 8 and 15, every 4 weeks thereafter until EOT and at the two safety follow-up visits.
On treatment initiation and dose up-titration days, cardiac monitoring was performed by an independent cardiologist. This included 12-lead ECG, blood pressure measurements over 6 h and Holter ECG monitoring over 24 h (subset of patients). All ECG and Holter measurements were centrally read in a blinded manner. Pulmonary function tests (PFTs) were performed at all study visits except day 1 (randomisation). Unscheduled PFTs were performed in the event of respiratory symptoms or decreased PFT parameters during treatment.
Adverse events (AEs) and serious AEs (SAEs) were collected at each visit. Laboratory assessments were performed at each visit except day 1 (randomisation). AEs of special interest were defined on the basis of preclinical and previous clinical safety findings for ponesimod. They were reported during the treatment and follow-up periods and grouped as follows: cardiovascular AEs, infection-related AEs, pulmonary AEs, hepatobiliary disorders/liver toxicity and macular oedema.
Study endpoints
The primary efficacy endpoint was the cumulative number of new Gd+ lesions per patient detected on T1-weighted MRI scans from weeks 12 to 24. MRI scans from weeks 4 and 8 were excluded from analysis of the primary endpoint due to the delayed anti-inflammatory effects evident with the S1P modulator fingolimod.18 In addition, the primary endpoint was aligned with that of another agent (dimethyl fumarate) with delayed onset of anti-inflammatory effects in patients with RRMS.19 Secondary efficacy endpoints were the annualised confirmed relapse rate (ARR) and time to first confirmed relapse within 24 weeks of ponesimod initiation. Relapse was defined as the occurrence of an acute episode of one or more new symptoms or a worsening of existing symptoms of MS, not associated with fever or infection, and lasting for at least 24 h after a stable period of at least 30 days. A confirmed relapse was defined as a relapse with an increase of ≥0.5 points from baseline in the EDSS score or an increase of one point in at least one functional system score (excluding bowel, bladder and mental functional systems) assessed within 7 days of onset by the independent neurologist.
Exploratory efficacy MRI endpoints included cumulative number of new or enlarging non-enhancing T2 lesions at weeks 12, 16, 20 and 24; cumulative number of combined unique active lesions (CUALs; sum of all new T1 Gd+ lesions and new or enlarging T2 lesions since previous MRI scan) at weeks 12, 16, 20 and 24; and percentage change from baseline to week 24 in brain volume as measured by MRI using the Structural Image Evaluation Using Normalisation of Atrophy (SIENA) program.20
Statistical analyses
The global null hypothesis was that none of the three ponesimod groups differed from the placebo group in the mean cumulative number of new T1 Gd+ lesions at weeks 12 to 24. The alternate hypothesis was that at least one of the ponesimod treatment groups differed from the placebo group. The test of the null hypothesis was based on a negative binomial regression model with treatment group as a four-level nominal covariate for the per-protocol analysis set. The null hypothesis was tested by a Wald χ2 test with a two-sided significance level of 5%. No adjustment was applied for multiple comparisons.
The per-protocol analysis set was defined as all randomised patients who received ≥80% of study drug from study drug initiation to the planned EOT and had ≥2 valid post-baseline MRIs at weeks 12–24. The dose–response relationship was explored for the primary endpoint and lymphocyte counts by means of multiple comparison procedures and modelling techniques, as previously described.21 Briefly, linear, linear in log-dose, emax, sigmoid emax, logistic and exponential models were evaluated using multiple comparisons, and the data were fitted using the model with either the smallest Akaike Information Criteria for normally distributed data or maxT for count data. To assess the precision of the model fitting, a bootstrap simulation was performed using 1000 iterations.
The cumulative number of new or enlarging non-enhancing T2 lesions at weeks 12–24 and the cumulative number of CUALs at weeks 12 to 24 were analysed using the same statistical model and in the same patient analysis set as for the primary endpoint. The time to first confirmed relapse was analysed using the Kaplan–Meier method. The treatment effect versus placebo was described by HRs and the 95% CIs derived from a proportional hazards model. Total lymphocyte counts were summarised for the all-treated analysis set (all randomised patients who received at least one dose of study drug); the absolute values and changes from baseline to scheduled visits and to EOT (last available value) were summarised by descriptive statistics. AEs and SAEs were reported and summarised throughout the treatment and safety follow-up periods for the all-treated analysis set.
The underlying assumption for the sample size calculation was a 50% decrease in the mean cumulative number of new Gd+ lesions from weeks 12 to 24 in at least one ponesimod treatment group compared with placebo and assuming a mean of eight lesions under placebo; the anticipated sample size of 90 evaluable patients per group had a 90% power to detect a significant difference between ponesimod groups and placebo.
Results
Patients
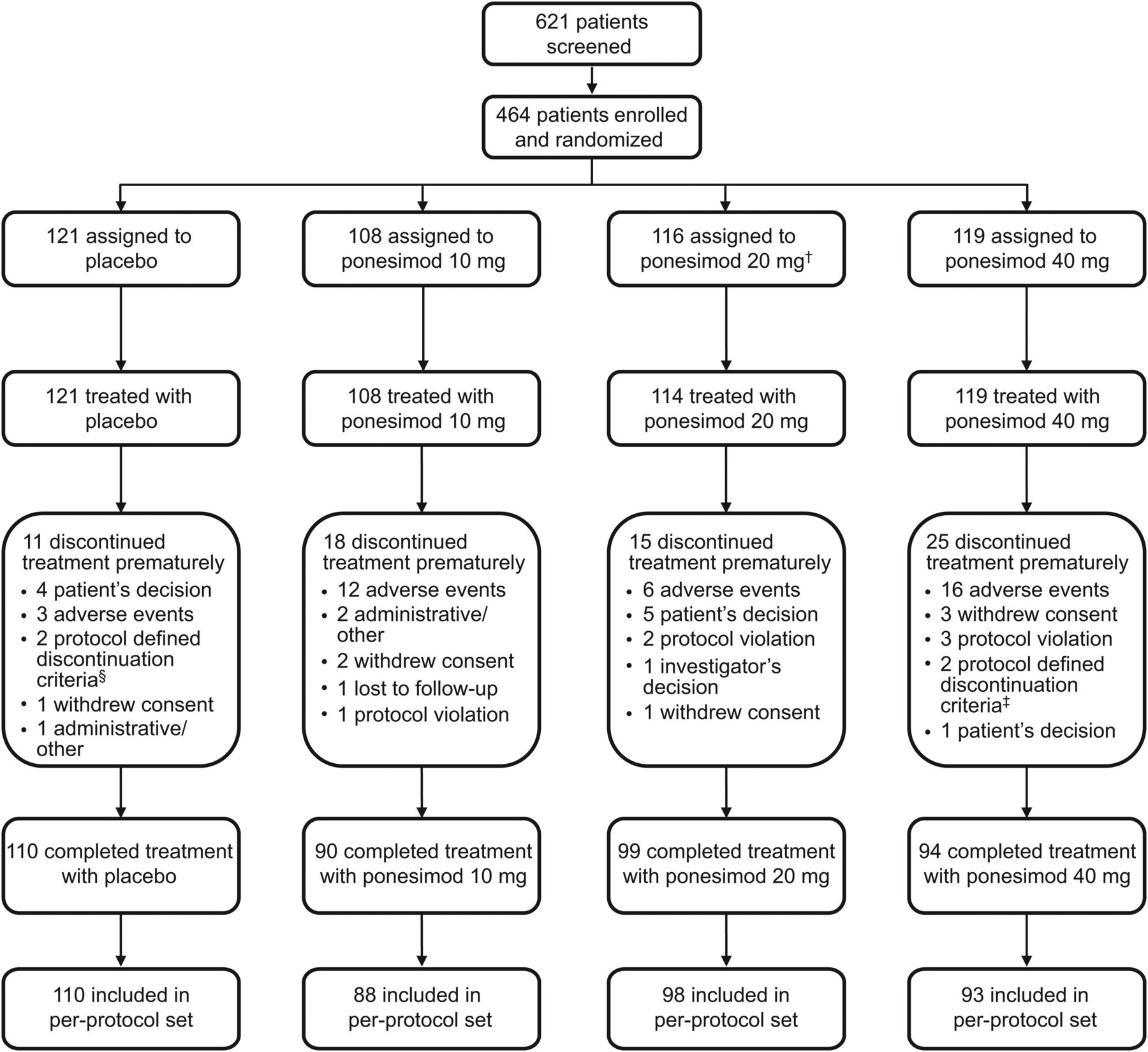
Of the 621 patients screened, 464 were randomised between October 2009 and November 2010 to receive ponesimod 10, 20 or 40 mg, or placebo. Patient disposition is summarised in figure 2. In the ponesimod 10, 20 and 40 mg groups, 16.7%, 13.2% and 21.0% of patients prematurely discontinued treatment, respectively, compared with 9.1% of patients receiving placebo. Demographic and baseline disease characteristics were generally similar across treatment groups (table 1).
Demographics and baseline patient characteristics (all-treated analysis set)
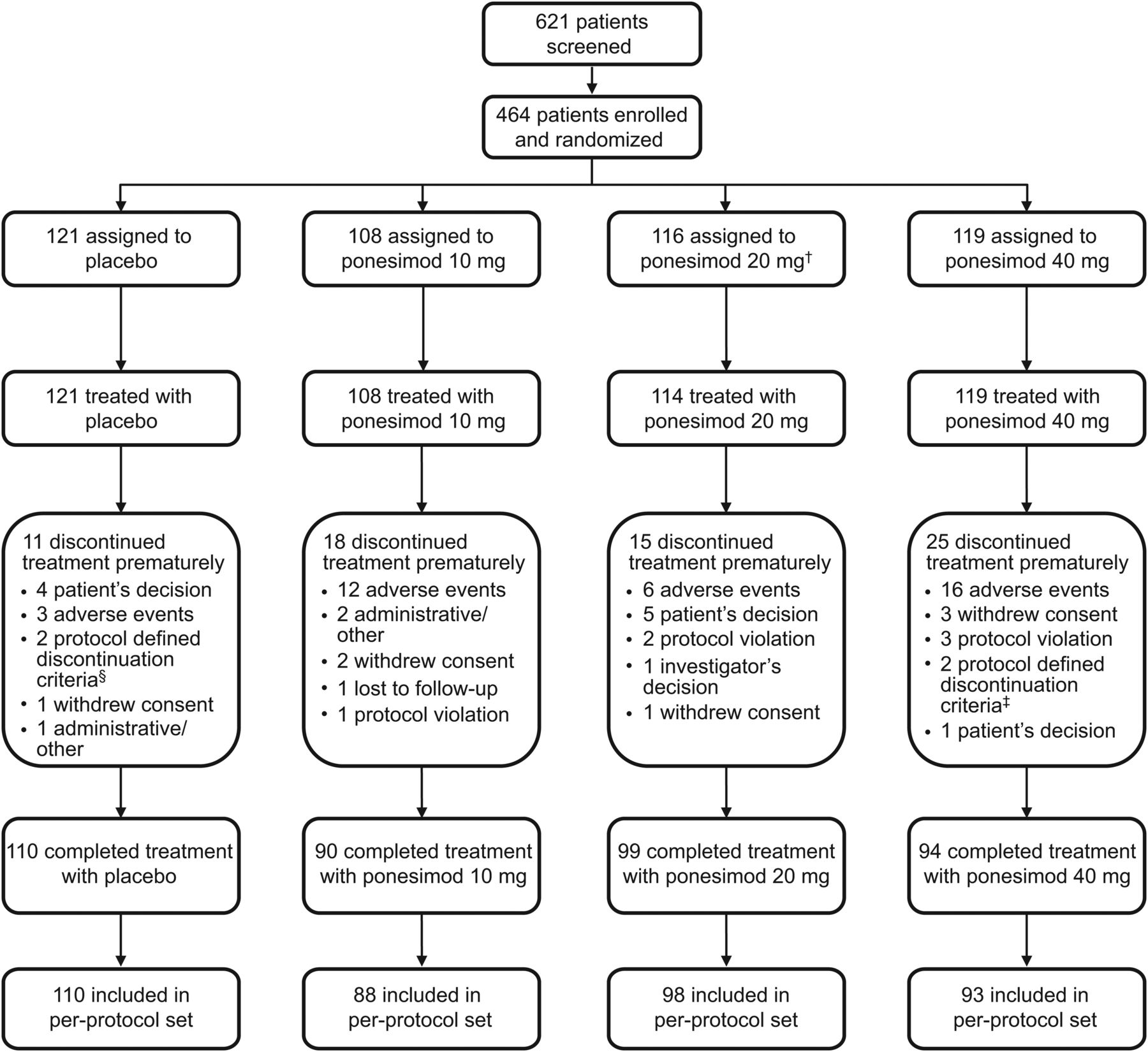
Patient flow. †Two patients randomised to the ponesimod 20 mg group did not start treatment due to safety findings that were noted after randomisation (low heart rate and abnormal Holter ECG readings). ‡Respiratory system criteria. §One patient discontinued treatment due to pregnancy, one patient discontinued treatment due to respiratory system criteria.
MRI and clinical outcomes
In the ponesimod groups, the mean cumulative number of new T1 Gd+ lesions at weeks 12–24 was lower in the ponesimod 10 mg (3.5), 20 mg (1.1) and 40 mg (1.4) groups compared with the placebo group (6.2) (figure 3A); the cumulative number of new T1 Gd+ lesions was significantly reduced by 43% with ponesimod 10 mg (treatment effect [ratio] 0.57, 95% CI 0.337 to 0.952; p=0.0318), by 83% with ponesimod 20 mg (treatment effect [ratio] 0.17, 95% CI 0.100 to 0.289; p<0.0001) and by 77% with ponesimod 40 mg (treatment effect [ratio] 0.23, 95% CI 0.133 to 0.384; p<0.0001) compared with placebo (figure 3A). Exploratory analyses showed a significant dose–response relationship (p<0.0001) for the primary endpoint (figure 3B). The ARR was numerically lower in each ponesimod group compared with placebo: the ARR was reduced by 37% with ponesimod 10 mg (p=0.1619), by 21% with ponesimod 20 mg (p=0.4420) and by 52% (p=0.0363) with ponesimod 40 mg (table 2).
Secondary, exploratory clinical and MRI endpoints

(A) Cumulative number of new T1 Gd+ lesions detected by magnetic resonance image scanning at weeks 12–24 (per-protocol analysis set). Graph shows mean+SE. The percentage reduction (95%CI) versus placebo is shown for each ponesimod treatment group. (B) Dose–response analysis for the cumulative number of new T1 Gd+ lesions from week 12 to 24 (per-protocol analysis set). Black dots represent the mean value for each dose and grey dots represent the fitted models obtained in the bootstrap process. *p<0.05; **p<0.0001. T1 Gd+, T1-weighted gadolinium-enhanced.
Ponesimod treatment increased the time to first confirmed relapse compared with placebo within 24 weeks of ponesimod initiation. Figure 4 shows the estimated proportion of patients experiencing their first confirmed relapse over time. The risk of first confirmed relapse was reduced by 58% in the ponesimod 40 mg group (HR 0.42, 95% CI 0.20 to 0.87), 21% in the ponesimod 20 mg group (HR 0.79, 95% CI 0.43 to 1.45) and 36% in the ponesimod 10 mg group (HR 0.64, 95% CI 0.33 to 1.22) compared with placebo.
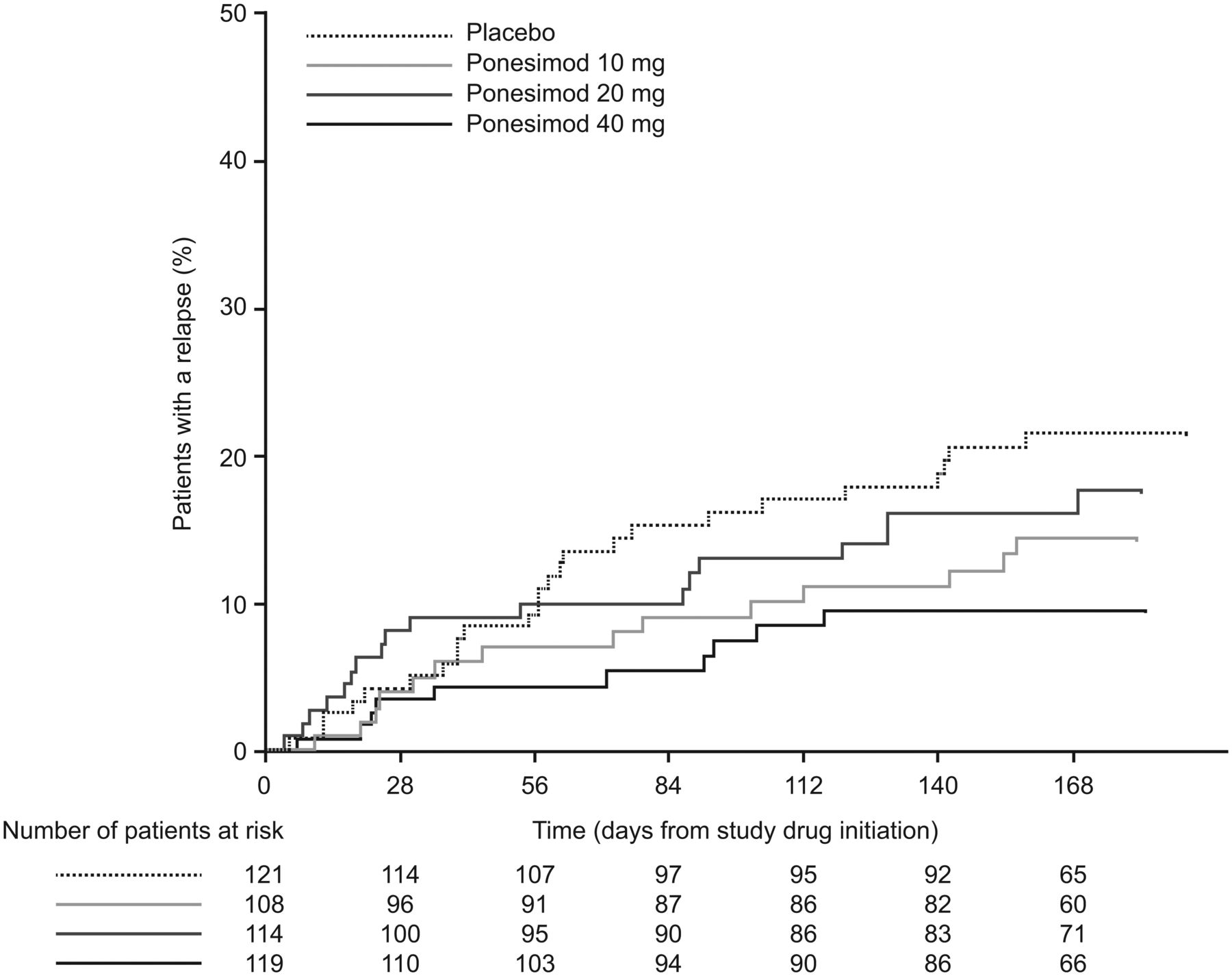
Kaplan–Meier estimate of the time to first confirmed relapse up to week 24 (all-treated analysis set).
Exploratory endpoints are shown in table 2. The mean cumulative number of new or enlarging non-enhancing T2 lesions at weeks 12–24 was reduced by 31% (p=0.2194), 56% (p=0.0208) and 34% (p=0.2194) with ponesimod 10, 20 and 40 mg, respectively. Compared with placebo, the mean cumulative number of CUALs at weeks 12–24 was reduced by 42% (p=0.0318), 80% (p<0.0001) and 73% (p<0.0001) with ponesimod 10, 20 and 40 mg, respectively. At week 24, a small mean increase in brain volume was observed with ponesimod (0.02%, 0.05% and 0.23% in the 10, 20 and 40 mg groups, respectively) compared with a mean decrease (–0.26%) with placebo.
Pharmacodynamic analysis
Lymphocyte counts were rapidly reduced with ponesimod treatment in a dose-dependent manner (figure 5A). Following initial treatment (up-titration) with ponesimod 10 mg, the mean decrease from baseline to day 8 (predose) in lymphocyte count ranged from 43% to 45% in all ponesimod groups and was 1% in the placebo group. Following up-titration to 20 mg on day 8, the mean decrease from baseline to day 15 (predose) in lymphocyte count was 62% for both the ponesimod 20 and 40 mg groups. Following up-titration to 40 mg on day 15, the mean decrease from baseline to week 4 in lymphocyte count was 67% in the ponesimod 40 mg group. Mean reductions from baseline to week 24 were 50%, 65% and 69% for ponesimod 10, 20 and 40 mg, respectively, and 3% in the placebo group.
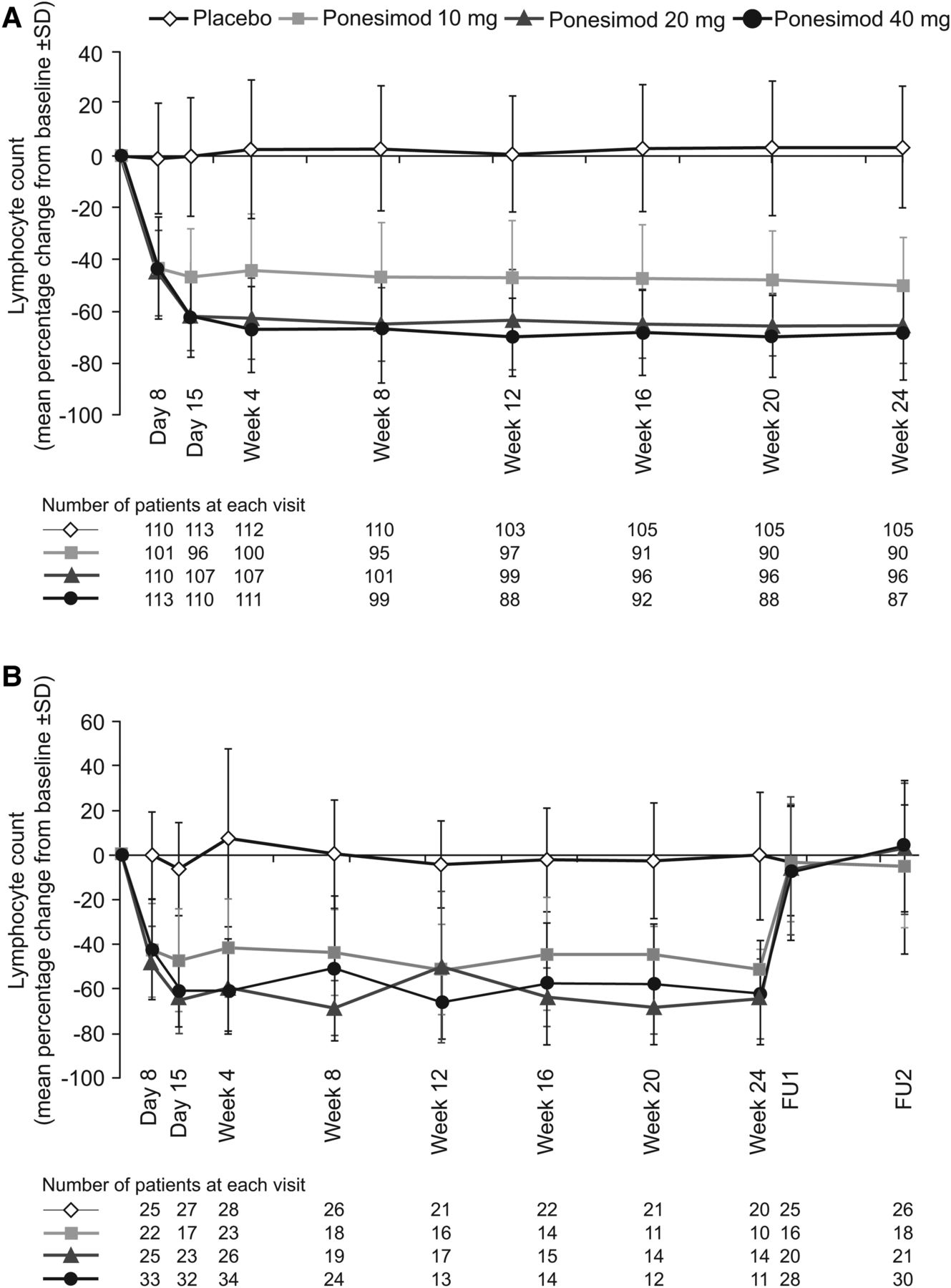
(A) Percentage change from baseline in lymphocyte counts up to week 24 (all-treated analysis set). (B) The group of patients who underwent safety follow-up (discontinued treatment prematurely or did not enter the extension study). FU1, follow-up visit 1 (end of treatment + 7 days); FU2, follow-up visit 2 (end of treatment+30 days).
In the subgroup of patients who discontinued treatment prematurely or did not enter the extension study at week 24 (n=122), mean lymphocyte count returned to baseline range within 1 week of treatment discontinuation (figure 5B). The proportion of patients with a lymphocyte count ≥0.8×109/L at the first safety follow-up visit was 100% (19/19 patients), 100% (24/24 patients) and 97% (31/32 patients) for ponesimod 10, 20 and 40 mg groups, respectively, and 96% (27/28) for the placebo group.
Safety and tolerability
The majority of AEs were mild or moderate in intensity, and the proportions of patients who had ≥1 AE during the treatment period were similar across all ponesimod groups (73.9–77.2%) and placebo (74.4%) (table 3). Frequently reported treatment-emergent AEs with a higher incidence in the three ponesimod groups compared with placebo were anxiety, dizziness, dyspnoea, increased alanine aminotransferase, influenza, insomnia and peripheral oedema. Incidences of dyspnoea and peripheral oedema appeared to be dose-related, with substantially more cases reported in the ponesimod 40 mg group compared with the ponesimod 10 and 20 mg groups.
AEs observed in ≥3% of patients in any treatment group and all serious AEs (all treated set)
During the treatment period, a total of 27 SAEs (excluding hospitalisations for MS relapse) were reported (table 3). Two malignancies were reported: one case of breast cancer in the ponesimod 10 mg group and one case of cervix carcinoma in the placebo group. There were no deaths during the study.
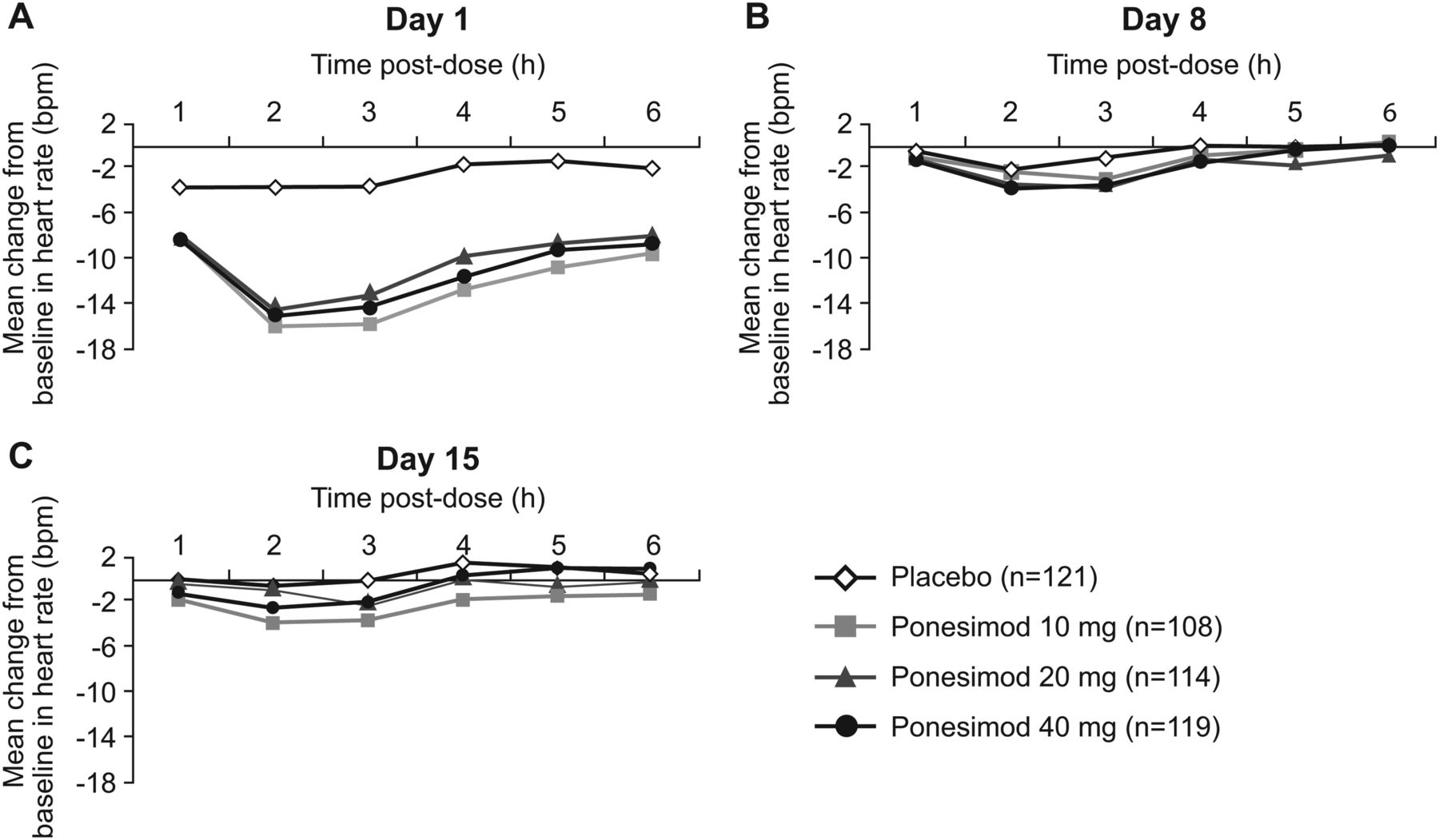
Cardiac AEs associated with ponesimod treatment initiation included first-degree (1.2%) and second-degree (0.9%; no cases of Mobitz type II) atrioventricular block and bradycardia (2%). All AEs relating to heart rate and rhythm occurred on day 1 when all patients randomised to ponesimod received a dose of 10 mg; there was no need for intervention and no recurrence of these AEs later during treatment. The reduction in heart rate on day 1 reached a maximum at 2–3 h postdose and returned close to predose values 6 h postdose (figure 6A); on up-titration days (days 8 and 15), heart rate changes with the higher doses of ponesimod were small and similar to those observed in the placebo group (figure 6B,C). Nine patients (2.6%) receiving ponesimod discontinued treatment due to cardiac AEs compared with none in the placebo group; in eight of these patients (two randomised to ponesimod 40 mg, two randomised to ponesimod 20 mg and four randomised to ponesimod 10 mg), the onset of cardiac AEs was on day 1, when these patients received ponesimod 10 mg, and ponesimod was discontinued early, usually during the first 2 weeks.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Mean change from baseline in heart rate (bpm) on day 1 (A), day 8 (B) and day 15 (C) (all-treated analysis set).
The proportion of patients with ≥1 infection-associated AEs was similar across the four groups (placebo 45.5%; ponesimod 10 mg, 37.0%; 20 mg, 32.5%; 40 mg, 36.1%). Of these, three discontinued treatment due to infections: one each in the placebo group (measles), the ponesimod 10 mg group (ear infection) and the ponesimod 40 mg group (sinusitis). There were no treatment discontinuations due to lymphopenia, nor was there a correlation between the incidence of infections and lymphocyte count reduction during the study.
The proportion of patients with ≥1 respiratory AE was higher in the ponesimod than in the placebo group (placebo, 6.6%; ponesimod 10 mg, 9.3%; ponesimod 20 mg, 16.7%; ponesimod 40 mg, 31.9%). In total, seven patients prematurely discontinued ponesimod because of dyspnoea (six in the 40 mg group). The onset of dyspnoea leading to treatment discontinuation in this group usually occurred within the first month of treatment and led to discontinuation during the second month; all cases resolved without sequelae. Three patients discontinued treatment due to protocol-defined respiratory criteria based on PFT results; two patients were in the ponesimod 40 mg group and one in the placebo group.
A dose-dependent decrease in forced expiratory volume in 1 s (FEV1) was observed with ponesimod treatment: the mean decrease in FEV1 from baseline to week 24 was 0.6%, 5.2%, 6.0% and 10.3% in the placebo and ponesimod 10, 20 and 40 mg groups, respectively. In the cohort of patients who underwent safety follow-up, FEV1 returned to within normal range within 1 week of treatment discontinuation.
Hepatic effects observed in the ponesimod treatment groups included liver transaminase increases >3× the upper limit of the normal range (ULN): ponesimod 10 mg, 2.8%; ponesimod 20 mg, 4.5%; ponesimod 40 mg, 4.2% compared with no cases in the placebo group. There were no cases of total bilirubin elevation ≥2× ULN and no cases of Hy's law.
Four macular oedema cases were reported, all starting within 3 months of treatment initiation. Two cases were reported as SAEs in the ponesimod 20 mg group (only one confirmed by optical coherence tomography) and resolved after treatment discontinuation, while the third non-serious AE on 20 mg resolved during study treatment. The fourth case (AE) of macular oedema was reported in the placebo group and resolved during study treatment.
Discussion
Once-daily treatment with ponesimod 10, 20 or 40 mg for 24 weeks significantly and dose-dependently reduced the cumulative number of new T1 Gd+ lesions in patients with RRMS compared with placebo. These findings were corroborated by observed reductions in the number of new or enlarging T2 lesions and the number of CUALs with ponesimod treatment compared with placebo. Treatment with ponesimod was also associated with a lower ARR and an increase in the time to first confirmed relapse. A clear dose–response relationship was observed for the primary endpoint; a similar dose–response relationship has also been reported for another S1P receptor modulator in development, siponimod.22 Although it is difficult to draw direct comparisons due to differences in study design, when compared with the phase II data from other available oral therapies for RRMS, ponesimod generally produced results that were of a similar magnitude. With regards to the primary endpoint of this study (the mean cumulative number of new T1 Gd+ lesions at weeks 12–24), ponesimod was comparable with dimethyl fumarate, fingolimod and teriflunomide.18 ,19 ,23 The magnitude of the treatment effect with ponesimod was also similar for other study endpoints, including ARR.18 ,19 ,23
Ponesimod was generally well tolerated at doses of 10 and 20 mg; the 40 mg dose showed signs of reduced tolerability with no additional benefit with respect to MRI or pharmacodynamic efficacy compared with the 20 mg dose. The reason for the lack of additional benefit with ponesimod 40 mg compared with ponesimod 20 mg is not entirely clear, although a lower mean T2 lesion volume at baseline as compared with other treatment groups may partly account for the fall-off in efficacy with regards to new or enlarging non-enhancing T2 lesions.
Ponesimod was associated with first-dose cardiac effects, similarly to other S1P receptor modulators such as fingolimod5 ,18 and siponimod22; with ponesimod, heart rate reduction reaches a maximum at 2–3 h after first dose and normalises approximately 6 h postdose, earlier than observed with fingolimod therapy.18 The up-titration dosing scheme was used to reduce the first-dose cardiac effects.
Consistent with ponesimod mode of action, lymphocyte counts were rapidly reduced upon treatment initiation, with the maximum effect established within 7–14 days for each dose level. Importantly, no patients discontinued treatment due to lymphopenia, and the decrease in lymphocyte count was not associated with an increased risk of infections.
As shown in preclinical9 ,10 and phase I studies,12 ,13 ponesimod-associated lymphocyte sequestration was rapidly reversed upon treatment discontinuation, reaching levels close to baseline within 1 week. By comparison, lymphocyte counts return to normal range within 1–2 months of stopping therapy with fingolimod.24 The difference in lymphocyte recovery between the two compounds may be attributed to their pharmacokinetic profiles; while ponesimod has a terminal half-life of approximately 32 h,13 ,14 the terminal half-life of fingolimod is approximately 8 days.25 Although no documented cases have yet been reported to suggest that the long half-life of fingolimod has been problematic, rapid drug elimination and effect reversibility may be of clinical benefit in cases of serious infection, vaccination and pregnancy. Furthermore, fingolimod undergoes a phosphorylation–dephosphorylation cycle in vivo, which is required for its activation/deactivation26; this step is not required for ponesimod and may contribute to its low pharmacokinetic/pharmacodynamic variability. Additionally, ponesimod is highly selective for the S1P1 receptor,10 while fingolimod is active on the S1P1 and S1P3–5 receptors.26 The clinical significance of this selectivity is not yet clear. Concerns surrounding non-selectivity primarily revolve around safety, particularly with regards to cardiac effects. At least three S1P receptors (S1P1–3) are present in the heart and vasculature, and the predominantly expressed receptor subtype varies with cell type. Specific S1P receptor subtypes may also mediate specific cardiac effects, such as hypertension and bradycardia.27 Notably, activation of the S1P3 receptor results in bradycardia in both mice and humans.28 Nevertheless, first-dose effects and decreased heart rate have been observed with ponesimod and fingolimod. Further investigation and long-term studies are needed to define the importance of receptor selectivity with regards to cardiac function. There are other effects arising from S1P receptor selectivity that deserve further investigation; evidence from in vitro studies suggests that fingolimod may have pro-fibrotic effects due to the stimulation of the S1P3 receptor, an effect that is not replicated with ponesimod.29 ,30 On the other hand, modulation of S1P3 and S1P5, in addition to S1P1, may enhance remyelination, which is critical in restoring electrical impulse conduction and preventing further degeneration or injury.31
The 24-week treatment duration of this study, although sufficient to establish the dose–response effect on MRI parameters, limits the provision of long-term safety and clinical efficacy data. To consolidate the observations reported in this study, an extension study of up to 5 years’ duration is ongoing. Further studies to confirm the efficacy and favourable benefit/risk profile of ponesimod for the treatment of patients with RRMS are under consideration.
Acknowledgments
The authors would like to thank the independent Data Monitoring Committee (CH Polman, L Kappos, I Steiner, J Behr, C Pratt, D DeMets) and the Advisors (J Antel, E Waubant, H-P Hartung, F Lublin, P O'Connor). The B201 Study Investigators were as follows: Australia: S Vucic; Austria: F Leutmezer, B Kepplinger; Belgium: E Bartholome, P Jacquerye; Bulgaria: I Tarnev, L Haralanovf, N Deleva, S Andonova-Atanasove; Canada: MS Freedman, G Vorobeychik; Czech Republic: V Ticha, O Zapletalova, I Rektor, M Vachova, P Kanovsky, O Skoda; Finland: M Kallela, E Kinnunen, J-P Erälinna, I Elovaara; France: W Camu; Germany: H-C Diener, H Tumani, L Harms; Hungary: M Satori, A Valikovics, A Csanyi, P Harcos, Z Szakacs, G Jakab; Israel: R Milo, A Achiron, S Flechter, A Karni; Italy: A Ghezzi, P Gallo, C Pozzilli, GL Mancardi, N De Stefano, G Comi; The Netherlands: R Hupperts, E Sanders, C Zwanikken; Poland: J Kamienowski, W Kozubsky, A Czlonkowska, J Zbrojkiewicz; Romania: A Campeanu, A Bulboaca, M Simu; Russia: I Stolyarov, A Skoromets, A Boyko, A Belova, F Stuchevskaya, E Yakupov, G Mishin, I Poverennova, R Magzhanov, I Sholomov; Serbia: E Dincic, S Vojinovic, S Miletic Drakulic; Spain: G Izquierdo, Ó Fernandez, B Casanova, JA Garcia, X Montalban; Sweden: T Olsson, A Svenningson, J Lycke; Switzerland: S Mueller, C Gobbi; Ukraine: T Kobys, S Kareta, L Dzyak, T Muratova; UK: E Silber, D Cottrell, J Zajicek; USA: J Wendt, D Mattson, V Simnad, S Lynch, V Rowe, R Hamill, M Melanson, M Agius, BJ Keller, J Dunn, K Carnes, P Coyle, S Staunton, K Edwards, A Boster, J Carter, S Kamin, S Cohan, D Huang.
References
Footnotes
-
Contributors MSM participated in study design and writing of the Protocol, and was involved in the conduct of the study. DB wrote the statistical analysis plan and was responsible for the statistical analyses of the study. OB and TS directed the study at Actelion. TO, AB, ÓF, MSF, CP and MM were study investigators and involved in patient recruitment, study conduct and data collection. E-WR was responsible for central review of MRI data. All authors reviewed and interpreted the data, agreed on manuscript content, and reviewed and revised all drafts. TO is the lead author and directed the manuscript content at each draft, supervising the medical writer. All authors approved the final version for submission.
-
Funding This study was sponsored by Actelion Pharmaceuticals Ltd, Switzerland. Medical writing assistance was provided by Sophie Yarwood at Medi Cine, funded by Actelion Pharmaceuticals Ltd.
-
Competing interests TO has received personal compensation from Biogen Idec, Genzyme, Sanofi, Novartis and Merck Serono. He has received financial support from Biogen Idec, Sanofi, Novartis and Merck for unrestricted multiple sclerosis research grants. AB has received personal compensation from Merck Serono, Biogen Idec, Tev, Medtronic, Genzyme and Novartis for consulting services. He has received financial support for research activities from Actelion, Accorda, Merck Serono, Biogen Idec, Teva Neuroscience, NMSS, Roche, Sunpharma and Novartis. ÓF has received honoraria as a consultant in advisory boards and as a chairman or lecturer in meetings from Bayer-Schering, Biogen Idec, Merck Serono, Teva, Novartis and Sanofi. He has participated in clinical and other research projects sponsored by Bayer-Schering, Biogen Idec, Merck Serono, Teva, Novartis, Sanofi, Actelion and Almirall. MSF has received compensation for activities with Actelion, Bayer HealthCare, Biogen Idec, Celgene, EMD Canada, Genzyme, Glycominds, Novartis, Opexa, Sanofi and Teva Canada Innovation. He has received financial support for research from Bayer HealthCare and Genzyme. CP has received personal compensation as a speaker in meetings and as a member of advisory boards from Merck Serono, Genzyme, Biogen Idec, Bayer, Novartis and Teva. He has received financial support for research from Biogen Idec, Novartis, Merck Serono, Bayer and Sanofi. E-WR has received personal compensation from Bayer Schering, Biogen Idec, Novartis and Merck Serono for consulting and speaking services. He has received financial support for research activities from Actelion, Basilea Pharmaceuticals Ltd, Biogen Idec, Merck Serono and Novartis. MM is a member of the Multiple Sclerosis Advisory Council. She has received compensation for speaking engagements and consultancy for Biogen Idec and compensation for speaking engagements from Teva, Pfizer, QuestCor, Novartis and Accorda. DB and OB are employees of Actelion Pharmaceuticals Ltd. MM is a former employee of Actelion Pharmaceuticals Ltd. TS is a former employee of Actelion Pharmaceuticals Ltd and current employee of Novartis.
-
Ethics approval This study was conducted with approval from an Independent Ethics Committee or Institutional Review Board at each participating study centre.
-
Provenance and peer review Not commissioned; externally peer reviewed.