Article Text

Abstract
Sudden unexpected death in epilepsy (SUDEP) is the most common cause of death in patients with intractable epilepsy. The substantial lifetime risk of SUDEP and the lack of a clear pathophysiological connection between epilepsy itself and sudden death have fuelled increased attention to this phenomenon. Understanding the mechanisms underlying SUDEP is paramount to developing preventative strategies. In this review, we discuss SUDEP population studies, case–control studies, witnessed and monitored cases, as well as human seizure cardiorespiratory findings related to SUDEP, and SUDEP animal models. We integrate these data to suggest the most probable mechanisms underlying SUDEP. Understanding the modifiable risk factors and pathophysiology allows us to discuss potential preventative strategies.
- EPILEPSY
- EPILEPSY, SURGERY
- RESPIRATORY MEDICINE
- CARDIOLOGY
- GENETICS
Statistics from Altmetric.com
Introduction
Epilepsy patients are dying unexpectedly, without warning and without a clear understanding of how or why.1 Surprisingly, epilepsy patients, their family members and even many physicians, are unaware of the mortality and risk of sudden death associated with epilepsy.2 Sudden unexpected death in epilepsy (SUDEP) refers to the unpredictable and unanticipated death of a reasonably healthy individual with epilepsy, where no cause of death can be found. SUDEP is used to denote a diagnostic category to which these types of unexplained deaths are ascribed. Criteria have been established that define SUDEP as the sudden, unexpected, witnessed or unwitnessed, non-traumatic and non-drowning death of a person with epilepsy with or without a seizure, excluding documented status epilepticus, and in whom postmortem examination does not reveal a structural or toxicological cause of death.3 The criteria for SUDEP are merely a description of the clinical findings surrounding death and reflect our lack of understanding of the underlying pathophysiology. SUDEP has recently garnered considerable attention in the scientific literature due to its shockingly high prevalence in chronic refractory and surgical epilepsy patients, and lack of any well-defined mechanisms.1 A better understanding of the cause(s) of SUDEP is essential to develop objective criteria and biomarkers, identify high-risk individuals and provide preventative measures.
In this review, we discuss proposed mechanisms involved in the pathophysiology of SUDEP by integrating data from SUDEP epidemiological studies, witnessed and monitored SUDEP cases, human epilepsy research and epilepsy animal models of SUDEP. Lastly, we discuss what steps are being taken to prevent SUDEP.
Epidemiology
Incidence
Epidemiological studies on SUDEP have been heterogeneous in their methodology. Therefore, the reported incidence of SUDEP varies widely depending on the type and size of the study, the population analysed, the definition of SUDEP and how the cause of death was determined.1 Additionally, identifying SUDEP cases has been a challenge. Although examination of death certificates and coroners’ cases are widely used, the term SUDEP is often not used to describe the cause of death when it is the appropriate diagnosis.4 Therefore, the incidence of SUDEP may be under-reported in some epidemiological studies.
Population-based studies reveal the lowest incidence as they consider all types of epilepsy, including very well-controlled epilepsy patients and patients in remission. The incidence of SUDEP is inversely related to remission of epilepsy.5 Ficker et al conducted a retrospective population-based study of 1535 epilepsy patients in Rochester, Minnesota from 1935 to 1994, which revealed an incidence of SUDEP that was 0.35 years/1000 person-years. Although the incidence was low in this study, the rate of sudden unexpected death was 24 times greater in young adults with epilepsy than in the general population.6 Including all population-based SUDEP studies, the incidence of SUDEP ranges from 0.09 to 2.3/1000-person years. In populations of individuals with more severe epilepsy, the incidence increases. The reported incidence in chronic refractory epilepsy patients ranges from 1.1 to 5.9/1000 person-years. The highest reported incidence of SUDEP occurs in epilepsy surgery candidates or in patients who continue to have seizures after surgery, and ranges from 6.3 to 9.3/1000 person-years.1
Risk of death from SUDEP
The low incidence of SUDEP in population-based studies belies the cumulative lifetime risk of death from SUDEP. In chronic refractory epilepsy patients who are seen in epilepsy referral centres, SUDEP is the leading cause of death, accounting for 10–50% of all deaths.1 In a cohort study, children with epilepsy were found to have a 7% cumulative risk of dying suddenly within 40 years. Over this 40-year period, SUDEP accounted for 38% of all deaths, almost all of which occurred in adulthood.7 The risk of death from SUDEP translates to a substantial health burden. Although the number of SUDEP deaths in comparison to many other neurological disorders is far smaller, SUDEP is second only to stroke in years of potential life lost.8
SUDEP risk factors
Examining details surrounding individual SUDEP cases has been crucial in identifying risk factors and revealing clues to the underlying mechanisms. These data come from large epidemiological studies, smaller case–control studies and observed cases. Most SUDEP cases are unwitnessed, however, circumstantial and physical evidence suggests that SUDEP usually follows a generalised tonic clonic seizure (GTCS).9 ,10 Supporting this, most witnessed SUDEP cases have occurred after a GTCS.11
Case–control studies of SUDEP and non-SUDEP epilepsy deaths have determined risk factors specific for SUDEP. Although risk factors vary due to the heterogeneity of studies, some risk factors are common to many of the studies.12 SUDEP occurs in all ages, and the ages of reported cases have ranged from 8 months to 83 years.5 Typically, the mean age at death in most studies is between 25 and 39 years, and, in general, ages between 20 and 40 years are considered to be associated with the highest risks for SUDEP.5 Ethnicity and gender are unlikely to significantly affect SUDEP risk. An increase in duration of epilepsy and earlier age of onset of epilepsy are often-cited risk factors for SUDEP.12 Whether these are independent risk factors is unclear. One of the strongest and most often cited risk factor for SUDEP is an increased frequency of GTCS.5 ,12 This provides supportive evidence that SUDEP results from a seizure.
Although one would presume that better control of seizures through antiepileptic drugs (AEDs) would lead to a decrease in SUDEP, some case–control studies have identified AED polytherapy,12 as well as lamotrigine12 ,13 and carbamazepine,14 ,15 as risk factors for SUDEP. However, these findings are difficult to interpret since polytherapy can be a surrogate for more severe epilepsy. Additionally, across epidemiological studies, these findings are inconsistent.12 ,16 ,17 Increasing evidence from meta-analyses and randomised controlled studies suggests that monotherapy and polytherapy at efficacious doses reduce SUDEP.18 Although it appears that AEDs are not associated with an increased risk for SUDEP on a population level, some epilepsy patients may be susceptible to effects of AEDs through direct or indirect effects on the heart and autonomic system.19
Although identifying risk factors for SUDEP has been extremely important, many patients with these risk factors do not die, suggesting that other major causal mechanisms are involved.
Witnessed and monitored SUDEP cases
Witnessed SUDEP cases and cases from epilepsy monitoring units (EMUs) have provided the greatest insight to understanding the key contributory mechanisms underlying SUDEP.
Witnessed SUDEP cases
Witnessed SUDEP cases are infrequent. In a study of 15 witnessed SUDEP cases, 12 occurred in association with a GTCS.11 In two cases, the patient was postictal, and in one case, the patient was suspected to have an aura preceding death. In 12 cases, witnesses commented that the individuals had difficulty breathing, suggesting a respiratory component to the cause of death. Seven of the 15 cases occurred while in bed, suggesting that sleep and the patient's level of arousal and the position of the face, mouth and chest may predispose epilepsy patients to SUDEP.11
Video EEG-monitored SUDEP cases
Fortunately, EMU SUDEP cases are extraordinarily rare. In two cases, death occurred after GTCS triggered respiratory dysfunction, hypoxaemia and cardiac events.20 ,21 In another case, seizures followed by asphyxia while in the prone position led to death.22 In other cases, postictal cessation of cerebral function was followed by cardiorespiratory failure.23 ,24 Although these case reports are limited, their conclusions were supported by the MORTality in Epilepsy Monitoring Unit Study (MORTEMUS),25 a retrospective review of SUDEP and near-SUDEP cases from 147 EMUs across the world. This study detailed the events surrounding 16 SUDEP cases, of which 11 had VEEG at the time of death. In all 11 monitored cases, GTCS immediately preceded death. Of the 14 SUDEP cases for which position of the patient could be assessed, 13 were in the prone position at the time of cardiorespiratory arrest, often with the face partly tilted to one side.
It was not possible to evaluate cardiac or respiratory activity during the fatal seizure for any of the patients. Among the 11 VEEG monitored cases, there were nine patients in whom heart rate and breathing movements could be assessed by an expert panel from the time the GTCS ended until death occurred. Seizures were followed by a short period of normal or increased heart and respiratory rates, after which there was “early postictal parallel collapse of respiratory and cardiac rates, which was observed in every patient during the first 3 min postictally”. This was accompanied by postictal generalised EEG suppression (PGES). The early cardiorespiratory collapse was terminal in three of the patients. In the remaining six patients, there was transient restoration of cardiac function associated with abnormal and possibly ineffective respiration, likely aggravated by the prone position. Respiration then progressively deteriorated until terminal apnoea occurred, followed by terminal asystole. In every case, terminal apnoea occurred first, and the heart rate continued for a variable period of time before terminal asystole (figure 1). Analysis of individual cases reveals that in many patients breathing was markedly abnormal or absent for long periods during which there was a heart rate that could often be normal for a significant period of time (figure 2). These data must be interpreted with the understanding that blood pressure was not measured and it is possible that there was some ventilation that occurred but was not seen on videotape.
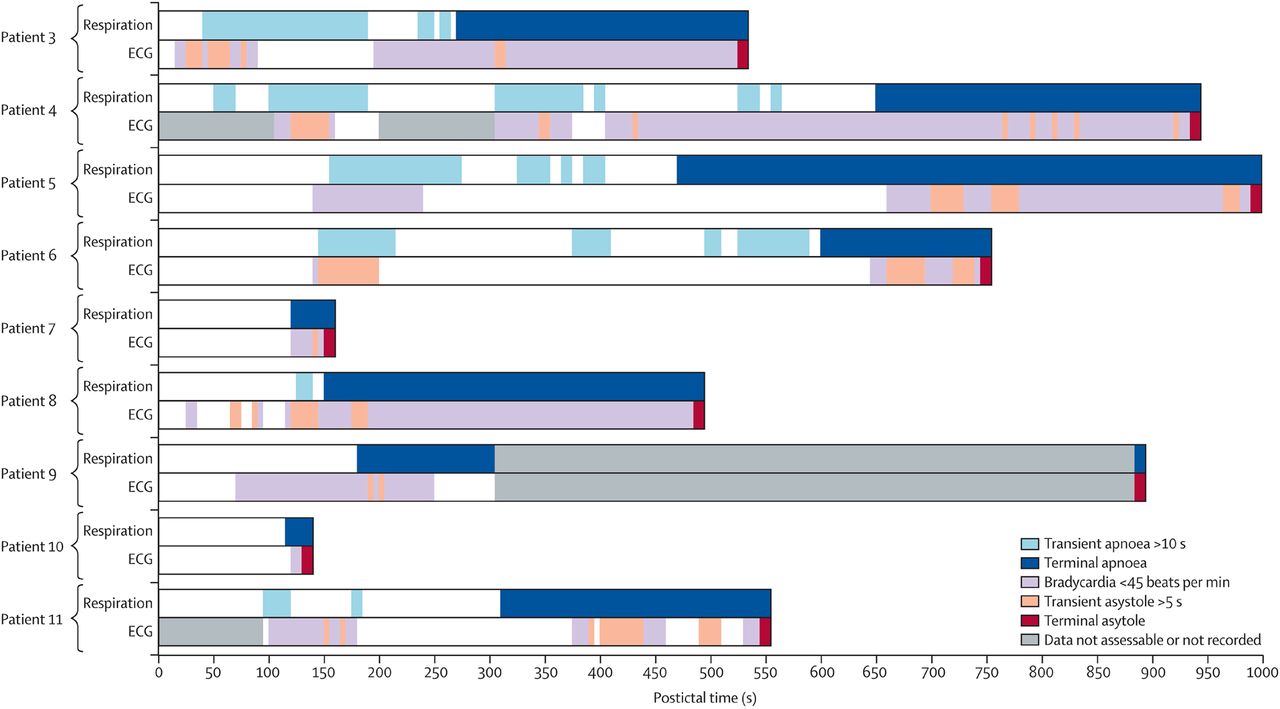
Patterns of postictal cardiorespiratory functions in video EEG-monitored sudden unexpected death in epilepsy (SUDEP) cases. Nine cases of SUDEP demonstrating the individual patterns of postictal cardiorespiratory function before death are illustrated. Transient episodes of apnoea (>10 s), asystole (>5 s) and bradycardia (<45 bpm) are shown, together with terminal apnoea and asystole. Cardiorespiratory abnormalities were always observed during the first 3 min postictally, often concomitantly. In each case, terminal apnoea proceeded terminal asystole for varying lengths of time. From Ryvlin, Lancet Neurology, 2013.25

Cardiorespiratory function in video EEG-monitored sudden unexpected death in epilepsy (SUDEP) cases. Individual patterns of postictal cardiorespiratory functions from SUDEP cases, starting from the end of each fatal seizure, in patients with video EEG-monitored SUDEP for which both respiration and heart rates could be measured by an expert panel. The black arrows indicate the early postictal collapse of respiratory and cardiac rates during the first 3 min postictally, leading to immediate death in three patients (A–C). A similar pattern was observed in another case (D) for which transient arrest of recording occurred. In other cases (E–I), cardiorespiratory functions were transiently and partially restored, until terminal apnoea occurred, followed by terminal asystole. From Ryvlin, Lancet Neurology, 2013.25
Pathophysiology
The combination of SUDEP risk factors, clues from witnessed SUDEP cases, EMU monitoring of SUDEP cases, physiological data obtained from non-fatal seizures in epilepsy patients and data from animal models have led to many proposed pathophysiological mechanisms underlying SUDEP. Most SUDEP cases occur after a GTCS.9 Therefore, it is unlikely that SUDEP results strictly from something inherent or intrinsic to epilepsy and unrelated to seizure events, and it suggests that seizures themselves induce pathophysiological changes that play a major role in SUDEP. However, as will be discussed, seizure-induced cardiorespiratory changes are likely only one aspect to the pathophysiology of SUDEP, which appears complex and multifactorial, and may involve a variety of other factors such as loss of arousal, time of day, position of the patient during the seizure, intrinsic pulmonary and cardiac dysfunction, and genetic mutations that predispose epilepsy patients to seizure-induced cardiorespiratory dysfunction.
Respiratory
Seizure-induced respiratory changes
In 1899, Hughlings Jackson reported that, with “slight epileptic attacks, les petits maux, the patient ‘turns blue’ or ‘purple’”.26 Over the following century, the notion that seizures could impair breathing was supported by scattered reports describing hypoventilation (including apnoea) and oxygen desaturation in patients with either generalised or focal seizures.27–29 The frequency with which seizures result in respiratory dysfunction was not fully appreciated until recently, because hypoventilation (including apnoea) can easily go unnoticed unless respiratory parameters are directly measured. Respiratory monitoring has rarely been employed in EMUs. For example, none of the SUDEP cases in the MORTEMUS study included any measurements of breathing, even SaO2. Recently, however, investigations that have employed respiratory monitoring techniques used in polysomnography, including the analysis of respiratory effort (using respiratory inductance plethysmography belts), oronasal airflow (using nasal pressure transducers and oronasal thermistry), SaO2 and end tidal CO2 (ETCO2), have shed light on the frequency and severity of peri-ictal respiratory dysfunction.
It is clear from these studies that ictal respiratory dysfunction is common during seizures and can be severe. In a study by Nashef et al30, 10 of 17 (59%) patients undergoing long-term video EEG monitoring exhibited apnoea concurrent with seizures, with SaO2 falling below 85% in six. More recently, Bateman et al31 analysed 304 seizures in 56 patients with intractable localisation-related epilepsy. In this cohort, SaO2 decreased below 90% in 33.2% of seizures and below 70% in 3.6% of seizures. Desaturation was not limited to generalised seizures: indeed, the SaO2 decreased below 90% in 34% of partial seizures. Central apnoea or hypopnoea occurred with 50% of 100 seizures in which nasal airflow and respiratory effort were measured, while mixed or obstructive apnoeas were present in 9% of seizures. Analysis of ETCO2 levels in this study31 and in a subsequent one32 by the same investigators, revealed that seizures were frequently associated with severe and prolonged increases in ETCO2 levels, to a level exceeding 50 mm Hg in 11 of 33 patients and in 35 of 94 recorded seizures. Other investigators have reported a similar frequency of ictal hypoxaemia in adults and children with epilepsy.33–35
Intrinsic pulmonary dysfunction
Interestingly, hypercapnia and oxygen desaturation appear in many patients to extend well into the postictal period, during which time respiratory effort has been reported to be qualitatively preserved or even increased, suggesting the possibility of intrinsic pulmonary dysfunction.32 ,36 Conceivably, seizures may cause ventilation/perfusion mismatch through alterations in pulmonary blood flow and/or venous return. In the absence of direct measurements of minute ventilation or blood gases, however, the evidence in support of the hypothesis that intrinsic pulmonary dysfunction is a major contributor to peri-ictal hypoxaemia, or to SUDEP itself, is currently circumstantial.
Pulmonary oedema of varying severity has been found on autopsy in many SUDEP cases.25 ,37–39 However, in the majority of cases, the oedema is mild and not felt to be significant enough to cause death.25 The aetiology of the oedema is not clear. Some have postulated that the oedema is a consequence of pulmonary vascular causes and terminal cardiac arrest.1
Prone positioning and postictal loss of arousal
Given the frequency and severity of seizure-induced respiratory abnormalities, it is perhaps surprising that death does not occur more often. If peri-ictal respiratory dysfunction is capable of causing death, what additional factors are required for SUDEP to occur? It may be that ictal respiratory dysfunction is only dangerous in certain circumstances. As noted above, patients who die of SUDEP are frequently found prone in bed.11 ,25 In such a position, the mouth and nose may be wholly or partly occluded, and it may take more muscular effort to expand the chest, increasing the risk of rebreathing or asphyxia if the patient fails to achieve arousal. As noted in the MORTEMUS study, “patients who died of SUDEP were not seen to take any corrective action to optimise their position and remained in the same position from seizure end until death”. The epilepsy patient has therefore apparently lost the normal arousability from hypercapnia,40 which should occur from seizure-induced hypoventilation. Possible mechanisms for this include suppression of brainstem arousal centres and loss of consciousness that occurs in GTCS and complex partial seizures. Serotonergic neurons in the midbrain contribute to the ascending arousal system, whereas serotonergic neurons in the medulla stimulate breathing.40–42 Both are activated by hypercapnia. Postictal depression of serotonergic activity could impair breathing and reflexive repositioning if the mouth and nose are obstructed by bedding.
Sleep and breathing
The temporal clustering of deaths at night25 also suggests the possibility that SUDEP risk is influenced by circadian or sleep-wake processes that influence breathing and vigilance.43 Breathing during sleep differs from that during wakefulness in a variety of ways that have the potential to increase risk. Breathing is inherently unstable at sleep onset as respiratory stimuli related to wakefulness and behavioural influences are withdrawn.44 Many otherwise healthy participants exhibit hypoventilation or frank apnoea during this transition. Sleep is associated generally with a reduction in the ventilatory response to hypoxia and hypercapnia as well as with a reduced response to ventilatory loading.45–47 Upper airway dilator tone also decreases during sleep (particularly rapid eye movement sleep), leading to obstructive apnoeas in susceptible individuals.48 Interestingly, recent studies suggest that sleep apnoea may be more common in patients with epilepsy49 ,50 and may be linked to poor control of seizures.51 Finally, circadian influences may also play a role in SUDEP. A circadian propensity to seizure occurrence has been shown in animals,52 while seizures themselves affect sleep-wake processes differently depending on their circadian timing.53 In addition, the circadian timing system plays an important role in modulating vigilance and control of breathing.54 ,55 To date, this area has received little attention.
Secondary cardiac effects from pulmonary dysfunction
Rather than being viewed as entirely ‘respiratory’ or ‘cardiac’ in aetiology, some deaths may reflect individual cardiac susceptibilities to peri-ictal respiratory dysfunction. Peri-ictal hypoxaemia has been shown to be associated with QT prolongation and shortening,56 suggesting that in some cases abnormalities in cardiac repolarisation may be induced by respiratory dysfunction. Similarly, hypoxaemia reliably leads to bradycardia when the patient is apnoeic.57 ,58 In a study performed in the 1940s,59 participants who fainted in response to acute hypoxia exhibited pronounced bradycardia, muscle vasodilation and apnoea, when compared with those who did not, providing an early illustration of individual differences in the response to acute hypoxia.60 Conceivably, the association between certain cardiac gene mutations and SUDEP risk may be partly mediated by cardiac susceptibility to peri-ictal respiratory dysfunction.
Collectively, these data indicate that peri-ictal respiratory dysfunction is common and can be severe, at least among patients admitted to inpatient EMUs.25 Little is known about the mechanisms responsible for this dysfunction. Peri-ictal hypoxaemia is associated with EEG evidence of contralateral spread61 as well as with PGES.36 Conceivably, these associations may reflect the spread of seizures into respiratory control centres of the brainstem.2 In the 1950s, it was shown that apnoea could be produced by the intraoperative stimulation of various targets within the frontal and temporal lobe.62 Whether a similar mechanism is responsible for peri-ictal respiratory dysfunction is not known.
Animal models of SUDEP and serotonin dysfunction as a molecular mechanism
Additional evidence supporting a respiratory mechanism for SUDEP comes from animal models and the study of the molecular mechanisms underlying the control of breathing. Some mouse species and genetically altered mice develop spontaneous or provoked seizures, which leads to death. These mouse models have allowed researchers to examine the cardiorespiratory events proceeding death. In both DBA/1 and DBA/2 mice, audiogenic seizures lead to respiratory arrest and death.63–65 In DBA/1 mice, seizure-induced death is prevented by resuscitation using a polyethylene tube placed over the nostrils of the mice and connected to a rodent respirator.63 In DBA/2 mice, similar findings are observed as oxygenation prevents death after seizure.64 Fluoxetine, which is a selective serotonin reuptake inhibitor, has been shown to reduce the incidence of respiratory arrest in DBA/166 and DBA/2 mice65 ,67 after audiogenic seizures, suggesting serotonin (5-HT) may play a role in the molecular mechanisms underlying seizure-induced hypoventilation. Additionally, 5-HT receptor expression in DBA mice exhibits abnormal levels, including reductions of 5-HT2C levels.68
5-HT neurons found in the medullary raphe act as central chemoreceptors that regulate PCO2 and pH and project to key respiratory neurons throughout the brainstem to stimulate breathing.69 Lmx1bf/f/p mice, which lack >99% of 5-HT neurons in the central nervous system by genetic deletion in utero, exhibit high rates of apnoea and death during the early postnatal period along with defects in arousal to an increase in ambient CO2 during sleep.40 ,70 Compared to littermate controls, Lmx1bf/f/p mice have a lower seizure threshold and increased seizure-induced mortality, suggesting that serotonin neurons raise the seizure threshold and decrease seizure-related mortality.71 Breathing ceased during most seizures without recovery, whereas cardiac activity persisted for up to 9 min before terminal arrest. The mortality rate of the Lmx1bf/f/p mice as well as the littermate controls was reduced by mechanical ventilation during the seizure. Both of these findings suggest that death ensued first from respiratory failure, followed by terminal asystole. In another study, deletion of 5-HT2c receptors in mice led to recurrent seizures and sudden death.72 These various rodent models suggest serotonin dysfunction may play a role in SUDEP. Translating these findings to humans, in a retrospective study of patients with medically refractory partial epilepsy, serotonin reuptake inhibitors were found to be associated with reduced severity of ictal hypoxaemia.73 Imaging findings help support a role for brainstem respiratory dysfunction in the pathophysiology of SUDEP. Compared to controls, MRI revealed greater volume loss in the mesencephalon in two individuals who died from SUDEP compared to 30 individuals with temporal lobe epilepsy (TLE).74
Summary
There is growing evidence supporting respiratory dysfunction as a cause of SUDEP. Although some have been long-time proponents of a respiratory dysfunction theory for SUDEP,30 this is a major divergence from the previous majority, which held that SUDEP was primarily due to cardiovascular dysfunction. Although serotonin has been implicated in mouse models of SUDEP, many questions remain regarding the potential mechanisms and triggering factors involved in peri-ictal hypoventilation that leads to death.
Cardiac
Seizure-induced cardiac changes
In the general population, sudden cardiac death has garnered considerable scientific and public attention. Unlike many sudden cardiac deaths, no structural cause of death can be found in SUDEP.10 However, seizures do induce a variety of transient cardiac effects. These include changes in heart rate, arrhythmias, asystole and various other ECG abnormalities, some potentially lethal. Additionally, epilepsy may predispose patients to cardiac autonomic dysfunction. Genetic diseases known to affect the heart may also result in epilepsy, predisposing certain epilepsy patients to SUDEP.
Seizures induce various transient heart rate changes. In a study by Opherk et al,75 of 102 seizures in 41 patients, 99% of the seizures led to an increase in heart rate. In this study, sinus tachycardia (HR>100 bpm) occurred in 100% of generalised seizures and 73% of non-generalised seizures. In a separate study by Leutmezer et al,76 of 145 seizures in 58 patients, tachycardia was observed in 86.9% of seizures. This was significantly more common with mesial TLE than with non-lesional TLE or extra-TLE. Including all studies, an increased heart rate has been recorded in 64–100% of seizures.76 Other than sinus tachycardia, ECG abnormalities have been observed in 21.5% of seizures and 37% of patients.34 These changes are mostly benign. Seizure-induced bradycardia is an infrequent event. In the study by Leutmezer et al,76 bradycardia was observed in only 1.4% of seizures. Overall, it occurs in less than 2% of all seizures. Ictal asystole (commonly defined as RR intervals longer than 3–5 s) is extremely rare. Only 5 of 1244 patients exhibited asystole concurrent with an ictal event.77 A retrospective study of 6825 patients undergoing long-term video EEG monitoring reported that ictal asystole occurred in only 0.27% of patients.78 The rarity of asystole does make it a rational candidate for a mechanism of SUDEP or at least a subset of SUDEP cases.
Seizures can induce other ECG abnormalities in addition to changes in heart rate. In a study of 102 seizures and 41 patients, only 6 seizures (6%) were associated with potentially serious ECG abnormalities occurring in only 4 patients (10%).75 These included ST segment depression and T-wave inversion. The benign abnormalities included premature atrial depolarisations, atrial bigeminy, premature ventricular depolarisations, ventricular couplets and first degree and second degree atrioventricular block, Mobitz I. In a study of 43 patients and 105 seizures, clinically significant ictal prolongation of the QTc interval using Bazett's formula was found in 16.2% of seizures, whereas shortened QTc intervals were observed in 4.8% of seizures.34
Unfortunately, many of these studies lacked measurements of respiratory rate, tidal volume, alveolar ventilation, oxygen saturation or other information about breathing. Therefore, it is unclear whether changes in heart rate, arrhythmias, asystole and other ECG abnormalities were secondary to seizure-induced respiratory changes. Hypoxaemia, hypoventilation and/or apnoea as well as other changes in breathing have known effects on heart rate and rhythm, likely through secondary autonomic changes. As discussed in the respiratory section, seizure-induced QT changes have been associated with seizure-induced hypoventilation and oxygen desaturation.56 Further supporting the suggestion that cardiac changes can sometimes be secondary to seizure-induced respiratory effects, in a study of over 250 seizures in 56 patients, one case of peri-ictal bradycardia was reportedly followed by asystole.31 In this case, the patient had concurrent oxygen desaturation to less than 50%, suggesting that bradycardia and asystole may have been secondary to hypoxaemia due to hypoventilation.
Heart rate variability
Heart rate variability (HRV) is a measure of the beat-to-beat variability of the heart and results from modulation of the sinoatrial node by the autonomic nervous system.79 Reduced HRV has been associated with increased cardiac mortality and sudden cardiac death.79 Studies have shown that HRV is reduced in people with poorly controlled epilepsy compared with well-controlled participants and normal controls.80 ,81 A separate study showed that TLE was associated with reduced HR variability, which was more pronounced during the night than during the day.82 However, unlike previous studies, the alteration in autonomic regulation of HRV was similar in patients with both refractory and well-controlled TLE.82 Another study, although limited by small sample size, compared seven SUDEP cases with seven age-matched controls and found no difference in HRV.83 Further study is needed to determine whether HRV plays a role in the pathophysiology of SUDEP or whether it may be a biomarker for SUDEP.
The cause of seizure-induced transient cardiac changes and reduced HRV is unclear, but is likely due to modulation of autonomic output. One possible mechanism is that seizures activate pathways that project to neurons in the brainstem that control autonomic output to the cardiovascular system.84 Others have suggested that transient cardiac effects and possibly reduced HRV are secondary to the response of the autonomic nervous system to apnoea and oxygen desaturation frequently seen in seizures.2 Further study of epilepsy patients and animal models is needed to define the mechanisms of seizure-induced cardiorespiratory effects, and the importance of these autonomic changes in the pathophysiology of SUDEP.
Molecular mechanisms—ion channels
Genetic mutations in ion channels that are expressed in the brain and heart may result in epilepsy. Evidence for this mostly comes from human genetic studies and rodent models containing these ion-channel mutations. In some of these mouse models, epilepsy occurs and seizures result in cardiac dysfunction that leads to death. Examples of these channelopathies include congenital long QT syndrome (LQTS), Dravet Syndrome and hyper-SUMOylation of Kv7 channels present in the brain.
LQTS is an inherited cardiac disorder resulting in abnormal cardiac ventricular repolarisation characterised by QT interval prolongation and abnormal T-waves.85 LQTS patients are susceptible to polymorphic ventricular tachycardia and torsades de pointes, resulting in syncopal episodes and even sudden cardiac death in young, otherwise healthy individuals. A total of 15 genes have so far been implicated in LQTS pathogenesis.86 However, 90% of the genetically identified mutations are found in three genes and their encoded proteins. Two are potassium channels, Kv7.1 (KCNQ1; type 1 LQTS (LQT1)) and Kv11.1 (KCNH2; type 2 LQTS (LQT2)) and one is a sodium channel, Nav1.5 (SCN5A; type 3 LQTS (LQT3)).86
These ion channels are expressed in the heart, and their mutations were first identified as causing cardiac pathology, but many of these proteins are also expressed in the brain. Some patients with congenital LQTS, especially LQT2, have syncopal episodes that appear seizure-like, suggesting that some patients may actually have epilepsy as well.87 In a study of 610 patients with LQTS, 10 (1.6%) patients were diagnosed with a seizure disorder by an epileptologist on the basis of clinical findings of seizures and EEG studies.86 Seven of the 10 patients had LQT2. Goldman et al88 studied a LQTS mutation in Kv7.1 in mice and found that this potassium channel was expressed in the heart as well as neurons in the brain. EEG recordings from these mice revealed epileptiform discharges and appeared to have spontaneous seizures.
In summary, patients with congenital LQTS have increased risk of sudden death, and some patients with LQTS have been found to have epilepsy.86 However, it is unclear whether patients with congenital LQTS and epilepsy are at increased risk of SUDEP. Additionally, it is uncertain how many patients in previous SUDEP reports had concurrent congenital LQTS.
Dravet syndrome is a rare disorder that results in severe childhood-onset epilepsy characterised by febrile and afebrile, generalised and unilateral, clonic or tonic–clonic seizures that occur in the first year of life.89 As the children age beyond 1 year, they develop myoclonus, atypical absence and partial seizures. Unfortunately, all seizure types are resistant to AEDs and therefore risk of SUDEP is high, probably more than other infantile epilepsies, often occurring in young children (<5 years) but also in adults. Developmental delay becomes apparent within the second year of life and is followed by cognitive impairment and personality disorders.89 80% of patients with Dravet syndrome have a mutation in SCN1A, the gene that encodes the sodium channel Nav1.1 that is present in the brain and heart.
A mouse model of Dravet syndrome (Scn1a heterozygous KO) recapitulates components of the human phenotype including epilepsy, and provides an avenue for understanding the pathophysiology of SUDEP.90 These mice have suppressed interictal resting HRV, episodes of ictal bradycardia that correlate with tonic phases of GTCS and death after prolonged ictal-onset bradycardia. Similar studies in conditional KO mice demonstrated that brain, but not cardiac, KO of Scn1a produced cardiac and SUDEP phenotypes similar to those found in the Scn1a heterozygous DS mice. Muscarinic antagonists decrease bradycardia and seizure-induced death, suggesting that vagal parasympathetic output may cause lethal bradycardia in SUDEP.90 However, because the study of this mouse model lacked respiratory monitoring during seizures, it is unclear if this mutation led to worsening respiratory dysfunction, and, additionally, whether these cardiac findings were secondary to seizure-induced hypoventilation and/or apnoea and oxygen desaturation. As discussed previously, apnoea associated with oxygen desaturation can lead to bradycardia.
Other ion channel modifications that result in epilepsy in mice have been discovered. Qi et al91 showed that deletion of SENP2 in the mouse brain resulted in hyper-SUMOylation of Kv7 channels. These mice developed spontaneous seizures, which resulted in cardiac abnormalities and death. Again, respiratory monitoring was not performed during seizures and the period of time proceeding death. Therefore, it is unclear whether apnoea and oxygen desaturation resulted in these cardiac changes, or whether the apnoea and oxygen desaturation was more severe because of the mutation.
Summary
Cardiac changes occur during seizures. Increasing evidence suggests genetic mutations in ion channels, present in the brain and heart, may increase the risk of epilepsy and SUDEP. Although rodent models with these genetic mutations demonstrate cardiac changes during seizures prior to death, it is unclear whether these cardiac events are caused by hypoventilation and oxygen desaturation during seizures. Future studies using a combination of cardiorespiratory monitoring in SUDEP animal models, similar to the study by Buchanan et al,71 will help clarify these issues.
Electrocerebral shutdown
PGES is diffuse flattening of the EEG in the postictal period and likely the result of a GTCS.92 Some have suggested ‘electrocerebral shutdown’, or prolonged PGES, as a causal mechanism of cardiorespiratory dysfunction and SUDEP.93 However, it is unclear if prolonged PGES can directly cause cardiorespiratory dysfunction. Studies are inconsistent on whether the presence or duration of PGES is an independent risk factor for SUDEP.92 ,93 More research is needed to understand how PGES plays a role, if any, in the pathophysiology of SUDEP.
Genetic susceptibility
Genetic conditions such as Tuberous Sclerosis complex and Dravet syndrome contain mutations, which lead to medically refractory epilepsy and frequent seizures with an increased rate of SUDEP. However, it is unclear whether these mutations result in more frequent seizures that lead to an increased likelihood of SUDEP or whether these mutations result in an altered cardiorespiratory response to seizures. With future genetic studies and a better understanding of the pathophysiological mechanisms of SUDEP, we will begin to parse out the genetics of SUDEP susceptibility.
Other molecular mechanisms
We previously discussed the role of serotonin dysfunction in the pathophysiology of SUDEP in the respiratory section as well as how ion-channel mutations expressed in the heart and brain may contribute to SUDEP in the cardiac section. Other molecular mechanisms may also play a role in cardiorespiratory changes. Adenosine has received a significant amount of attention since it was one of the first molecular mechanisms identified as a possible contributor to SUDEP.94
Adenosine
Adenosine is a modulator of synaptic transmission and neuronal activity, exerting most of its functions via activation of the excitatory and inhibitory adenosine receptors.95 Additionally, adenosine may have effects on brainstem mediated cardiorespiratory function.94 In a study by Shen et al,94 mice were treated with pharmacological inhibitors responsible for metabolising adenosine, after which seizures were induced. These mice were protected from seizures for the first 15 min, but eventually developed seizures and died. If the mice were treated with caffeine (an adenosine receptor antagonist) at seizure onset, however, their survival time was significantly increased compared with their untreated counterparts. Shen and colleagues concluded that adenosine may contribute to seizure-induced death. More research is required on the effects of caffeine on cardiorespiratory function in the peri-ictal period in mouse models.
Summary of SUDEP pathophysiology
SUDEP likely occurs through multiple mechanisms (figure 3). However, evidence from witnessed11 and monitored EMU SUDEP cases25 suggests that a majority of patients have substantial respiratory dysfunction at the initial stages of the terminal event. Prolonged respiratory difficulty is often not an early symptom of sudden cardiac death, but is instead typically followed by an increase in ventilation and then gasping (figure 4).96 Therefore, it is unlikely for SUDEP to be caused principally by a primary cardiac mechanism in which seizure-induced autonomic dysfunction of the heart leads to a lethal arrhythmia. Additionally, the fact that many patients are found prone in bed suggests that this position plays a role in the pathophysiology. The prone position where the face may be covered by pillows or blankets likely predisposes to worsening ventilation and oxygenation during respiratory dysfunction. This position also suggests a loss of arousal by patients, as they do not sense the alarm of rising CO2 levels. Therefore, we suggest SUDEP may commonly occur by seizure-induced hypoventilation and/or apnoea with oxygen desaturation and loss of arousal, where being prone worsens oxygenation, resulting in secondary autonomic cardiac dysfunction and lethal bradyarrhythmias. Although yet to be found, genetic mutations may predispose epilepsy patients to worsening oxygen desaturation during seizures. Additionally, cardiac arrhythmias and dysfunction may be more likely to occur during periods of seizure-induced oxygen desaturation in patients with congenital LQTS or other genetic mutations, including ion-channel mutations. Other causes of SUDEP may include primary cardiac arrhythmias from seizure activated autonomic pathways.

Pathophysiological mechanisms underlying sudden unexpected death in epilepsy (SUDEP). SUDEP often results from a generalised tonic-clonic seizure, which leads to inhibition of specific midbrain and medulla mediated effects via an unknown pathway. Other factors shown may predispose these patients to SUDEP. LQTS, long QT syndrome; PGES, postictal generalised EEG suppression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
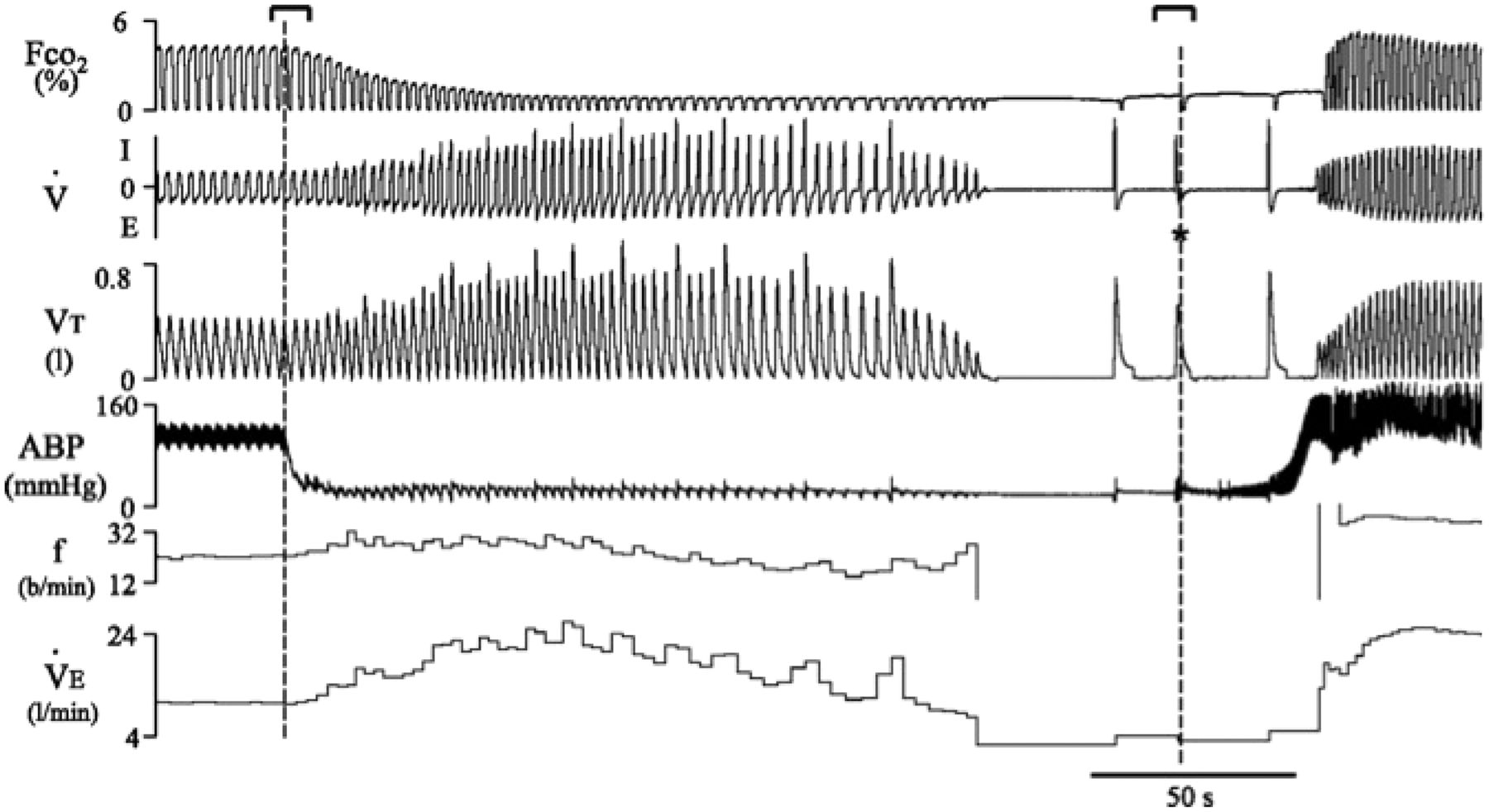
Respiratory response to sudden cardiac arrest. Recording of respiratory/ventilatory and circulatory responses to ventricular fibrillation (VF)-induced cardiac arrest (CA) in a sheep. From top to bottom, breath-by-breath respired PCO2, respiratory flow (inspiration (I) and expiration (E)), tidal volume (VT), carotid blood pressure (ABP), respiratory rate in breaths per minute (bpm) (f), and minute ventilation (VE) are shown. When VF was triggered (first vertical dotted line), blood pressure decreased to minimal levels, but ventilation remained elevated for 2 min, with an increase in both tidal volume and breathing frequency. During this period, alveolar PCO2 fell dramatically, and several augmented breaths can be observed. Apnoea then occurred followed by typical gasps. *Respiratory event triggered by the defibrillation procedure. Modified from Haouzi et al.96
Prevention
There is no definitive way to prevent SUDEP, since the pathophysiological mechanism is uncertain, other than probably completely preventing seizures. However, the modifiable risk factors and proposed mechanisms allow us to consider what treatments may be preventative or decrease the risk of SUDEP (table 1).
SUDEP preventative strategies
Seizure control
Complete seizure control in epilepsy patients has always been the primary goal of treatment, and it is becoming evident that seizure control appears important not only for quality of life but life itself. SUDEP is often triggered by a GTCS and increased frequency of GTCS is the most commonly cited risk factor for SUDEP. Therefore, seizure control is likely to be the most direct way of preventing SUDEP.
Using one or a combination of the more than 20 different AEDs currently available is the initial treatment for seizure control in epilepsy. Additionally, in the majority of epilepsy patients, AEDs decrease the risk of SUDEP. In a meta-analysis, adjunctive AED treatment appeared to reduce the risk of SUDEP more than sevenfold in patients with uncontrolled seizures.18
Up to 30% of epilepsy patients have persistent seizures and develop medically intractable epilepsy.97 Attempting to continually treat patients whose seizures do not respond to the first few AEDs with all the various combinations of multiple AEDs is impractical and places the patient at undue risk. Medically intractable epilepsy patients are those who are at most risk of SUDEP. Early identification of patients who are refractory to two or more AEDs may be an important strategy to reduce their risk.97–99
Early surgical evaluation and intervention in adults and in children with medically intractable epilepsy has been shown to control seizures better than medical therapy alone.99 In addition, surgical treatment may also decrease SUDEP risk by decreasing seizure frequency or by other mechanisms. TLE surgery has been shown to significantly decrease SUDEP risk if seizures are eliminated.100 After TLE surgery, there is a reduction in sympathetic cardiovascular modulation and baroreflex sensitivity.101 This enhanced cerebrovascular stability after surgery may improve autonomic balance in epilepsy patients.101 ,102 Further study is needed to determine whether other surgical epilepsy procedures such as extratemporal lobe surgery, corpus callosotomies and hemispherectomies, among many others, are as effective. Although some of these surgical procedures may not lead to seizure freedom, surgical disruption of the neuronal circuitry in the brain may prove beneficial in disrupting the pathophysiological cardiorespiratory pathways thought to contribute to SUDEP. Further study is needed to determine which surgical epilepsy treatments reduce SUDEP risk. Not only would this provide additional treatment strategies, but may increase the pathophysiological understanding of SUDEP.
Unfortunately, many patients who undergo a presurgical evaluation are not found to be good candidates for resective surgical therapy intended to make them seizure-free. These patients are at the highest risk for SUDEP. Patients can then be offered additional AEDs or drug combinations, dietary therapy, vagal nerve stimulation (VNS) or palliative surgical therapy. Focal resection can be considered as a palliative surgery when seizure freedom is not otherwise expected, for example, in patients with bilateral seizure foci.
SUDEP may be the end result of autonomic effects on the heart (figure 1). Inhibiting these autonomic changes may decrease SUDEP risk. VNS is effective at decreasing seizure frequency in medically intractable epilepsy103 ,104 and has been shown to have autonomic effects on the heart.105 In fact, VNS is an active area of study in heart failure.105 Additionally, long-term VNS in rats increases 5-HT levels.106 Given that VNS can decrease seizure frequency, has autonomic effects on the heart, and may affect 5-HT levels, further study is needed to determine whether VNS could decrease SUDEP risk.
Improvements in AEDs will hopefully decrease seizure frequency and SUDEP risk with decreased pharmacological side effects. Surgical treatment of epilepsy is continuously evolving. Complication rates have decreased significantly over the past 30 years and permanent neurological deficits are rare compared with the long-term risks of intractable epilepsy.107 Advancements in keyhole craniotomy procedures and minimal access approaches have allowed for decreased hospital stay and recovery time.108
Prevention of respiratory changes
Preventing or minimising seizure-induced apnoea, hypoventilation and oxygen desaturation may preclude the secondary autonomic response, cardiac abnormalities and death. As discussed previously, fluoxetine and several other, but not all selective serotonin reuptake inhibit (SSRIs), reverse respiratory arrest in a mouse model of epilepsy.65–67 Translating this finding to humans, Bateman et al73 found that seizure-induced oxygen desaturation was significantly reduced in 87 seizures in 16 epilepsy patients taking SSRIs compared to 409 seizures in 57 epilepsy patients not taking SSRIs. Further study is needed to determine whether taking SSRIs is effective at preventing SUDEP. Supervision at night may be effective in decreasing SUDEP,14 presumably by allowing repositioning during seizures, inducing arousal and therefore stimulating breathing. In a retrospective study of 39 patients with 105 generalised convulsions, early peri-ictal nursing intervention was associated with a reduced duration of respiratory dysfunction and PGES.109 The institution of routine respiratory monitoring may prevent in-hospital deaths and ultimately lead to the identification of high-risk patients. Wearable and portable pulse oxygen saturation seizure alarms may provide a similar type of benefit, allowing family, friends or others to be alerted and to therefore reposition the patient for optimal breathing. The development of a diaphragmatic pacemaker110 that becomes activated during periods of prolonged apnoea or oxygen desaturation could also be of benefit. Treating obstructive sleep apnoea using a continuous positive airway pressure device111 or with an upper airway implantable stimulation device112 could also decrease SUDEP risks by mitigating a contributing SUDEP risk factor for epilepsy patients.
Prevention of cardiac changes
The events leading to cardiac arrhythmias, asystole and cardiac failure are uncertain, but as discussed previously, occur either secondary to respiratory dysfunction or from primary seizure-induced autonomic cardiac effects. Regardless, preventing the bradyarrhythmias, asystole or tachyarrhythmias associated with these autonomic changes, may prevent death. In patients presenting initially with concerns of seizures and possibly epilepsy, ECG evaluation is critical as syncopal episodes from LQTS can be misdiagnosed as seizures. Additionally, as noted, some patients with congenital LQTS have epilepsy as well. These patients may be at high risk for SUDEP. Treating the LQTS may prevent sudden death in these patients. Multiple case reports have implanted cardiac pacemakers after finding substantial periods of ictal asystole. However, no large study of epilepsy patients with an implanted automatic implantable cardioverter defibrillator (AICD) has been conducted demonstrating prevention of SUDEP. It would not be prudent to implant cardiac pacemakers and cardiac defibrillators in all epilepsy patients. Further knowledge is needed to refine which patients are at highest risk, as these patients may benefit from an AICD. However, if the initiating and primary event that leads to cardiac dysfunction is respiratory dysfunction, cardiac pacing or defibrillation will not correct the oxygen desaturation leading to autonomic changes. In these cases, if oxygen saturation is not improved, maintaining a normal heart rhythm may prove challenging. An on-demand diaphragmatic pacemaker may be an effective way to prevent the hypoventilation that can occur in some patients after a seizure.
Seizure alarms
Using seizure alarms to alert family members, medical personnel or others that an epilepsy patient is having a seizure may be beneficial in reducing respiratory dysfunction and oxygen desaturation as well as allowing for resuscitatory measures, thereby preventing SUDEP. As discussed above, early peri-ictal nursing intervention was associated with a reduced duration of respiratory dysfunction109 and supervision at night was protective against SUDEP.14 Seizure alarms studied include a wireless wrist accelerometer sensor that detects GTCS with high sensitivity and specificity.113 Another seizure alarm that has been studied is an under-mattress device,114 which is triggered by rhythmic motor activity of a specifiable duration, frequency and intensity using a quasi-piezoelectric material sensitive to changes in mattress pressure.
Conclusion
The pathophysiology underlying SUDEP appears complex. Analysing and incorporating the various studies on SUDEP have helped elucidate cardiorespiratory mechanisms and predisposing factors. Ultimately, understanding the intricate mechanisms involved in SUDEP will lead to development of effective preventative strategies.
References
Footnotes
Contributors BJD, BKG and GBR wrote and edited the manuscript.
Competing interests None declared.
Provenance and peer review Commissioned; externally peer reviewed.