Article Text

Abstract
Objective To identify the genetic characteristics in a large-scale of patients with Charcot-Marie-Tooth disease (CMT).
Methods From May 2012 to August 2016, we collected 1005 cases with suspected CMT throughout Japan, whereas PMP22 duplication/deletion were excluded in advance for demyelinating CMT cases. We performed next-generation sequencing targeting CMT-related gene panels using Illumina MiSeq or Ion Proton, then analysed the gene-specific onset age of the identified cases and geographical differences in terms of their genetic spectrum.
Results From 40 genes, we identified pathogenic or likely pathogenic variants in 301 cases (30.0%). The most common causative genes were GJB1 (n=66, 21.9%), MFN2 (n=66, 21.9%) and MPZ (n=51, 16.9%). In demyelinating CMT, variants were detected in 45.7% cases, and the most common reasons were GJB1 (40.3%), MPZ (27.1%), PMP22 point mutations (6.2%) and NEFL (4.7%). Axonal CMT yielded a relatively lower detection rate (22.9%), and the leading causes, occupying 72.4%, were MFN2 (37.2%), MPZ (9.0%), HSPB1 (8.3%), GJB1 (7.7%), GDAP1 (5.1%) and MME (5.1%). First decade of life was found as the most common disease onset period, and early-onset CMT cases were most likely to receive a molecular diagnosis. Geographical distribution analysis indicated distinctive genetic spectrums in different regions of Japan.
Conclusions Our results updated the genetic profile within a large-scale of Japanese CMT cases. Subsequent analyses regarding onset age and geographical distribution advanced our understanding of CMT, which would be beneficial for clinicians.
- charcot-marie-tooth disease
- molecular epidemiology
- next generation sequencing
- gene panel
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0
Statistics from Altmetric.com
Introduction
Charcot-Marie-Tooth disease (CMT) is the most common phenotype of inherited peripheral neuropathy (IPN), the latter of which also encompass hereditary sensory and autonomic neuropathy, hereditary neuropathy with liability to pressure palsy and hereditary motor neuropathy (HMN).1 In terms of median motor nerve conduction velocity (MNCV), CMT can be further classified into demyelinating CMT (MNCV<38 m/s) and axonal CMT (MNCV ≥38 m/s).
CMT is typically characterised by progressive motor and sensory polyneuropathy, but it may also present with significant clinical heterogeneity. CMT disease-causing genes, such as GARS (CMT2D and distal HMN5A), HSPB1 (CMT2F and distal HMN2B) or IGHMBP2 (CMT2S and spinal muscular atrophy with respiratory distress type 1), often produce other IPN phenotypes.2–4 To date, approximately 100 different genes have been linked to CMT-like phenotypes (https://neuromuscular.wustl.edu/). Owing to its clinical complexity and genetic diversity, the clinical subtyping of CMT is always laborious and difficult.
The development of next-generation sequencing (NGS) technology allows us to conduct gene panel sequencing simultaneously the targeting of numerous genes. Within approximately 4 years, using two NGS systems successively, we have completed genetic assessment in more than 1000 Japanese cases with suspected CMT, which enables us to describe the genetic and clinical features of these cases.
Materials and methods
From May 2012 to August 2016, we collected blood or DNA samples from 1005 apparently unrelated patients throughout Japan with suspected CMT. These cases were examined by their local neurologists or paediatricians and were referred to our genetic laboratory for diagnostic genetic test. Duplication/deletion mutation of PMP22 was pre-excluded in all cases suspected with demyelinating CMT, using fluorescence in situ hybridisation or multiplex ligation-dependent probe amplification.
On the basis of their family history, the included cases were grouped into sporadic (n=570, 56.7%), autosomal dominant (AD) or X-linked (n=341, 33.9%), autosomal recessive (AR; n=72, 7.2%) or with an unknown inheritance pattern (no clinical data, n=22). All cases were further categorised as demyelinating CMT (n=282), axonal CMT (n=682) or unclassified type with no MNCV data or MNCV=0 (n=41) referring to their records of electrophysiological examination.
Genomic DNA was extracted from peripheral blood using a Gentra Puregene Blood kit (Qiagen, Valencia, California, USA), according to the manufacturer’s instructions. The protocol was reviewed and approved by the Institutional Review Board of Kagoshima University. All cases and their family members provided written informed consent to participate in this study.
Targeted gene panel sequencing
Primers were designed to cover the coding regions and exon/intron junctions of genes in our CMT panel. Beginning in May 2012, we conducted mutation screening targeting 60 genes (online supplementary table 1) with the Illumina MiSeq platform (Illumina, San Diego, California, USA). We used the same methodology as the one employed in a previous study.5 We completed genetic analysis in 437 cases with this system, concluding in July 2014.
Supplemental material
In September 2014, a custom Ion AmpliSeq gene panel targeting 72 IPNs disease-causing or candidate genes (online supplementary table 1) was designed and introduced. This panel consisted of 1800 amplicons divided into two primer pools. Library and template preparation was performed according to the manufacturer’s instructions, and then run on the Ion Proton (Thermo Fisher Scientific, Waltham, Massachusetts, USA) applying the Ion PI Chip kit v2/v3 BC (Thermo Fisher Scientific, Carlsbad, California, USA). We used the same methodology as the one employed in a previous study.6 Using this platform, we executed genetic assessment in 568 cases until August 2016.
Data analysis and variant interpretation
We confirmed all previously reported pathogenic mutations with reference to the Human Gene Mutation Database Professional 2017.3 (https://portal.biobase-international.com/hgmd/pro). Moreover, we checked all variants against global databases, including the 1000 Genomes (http://www.internationalgenome.org), the Exome Sequencing Project (http://evs.gs.washington.edu/EVS) and the Exome Aggregation Consortium (http://exac.broadinstitute.org/), as well as against Japanese databases, including the integrative Japanese Genome Variation Database (https://ijgvd.megabank.tohoku.ac.jp) and the Human Genetic Variation Database (http://www.hgvd.genome.med.kyoto-u.ac.jp). We also checked the variants against our in-house whole-exome sequencing database of individuals with non-IPNs. In silico analyses of variants were performed using SIFT (http://sift.jcvi.org), PolyPhen2 (http://genetics.bwh.harvard.edu/pph2), PROVEAN (http://provean.jcvi.org/index.php), Mutation Assessor (http://mutationassessor.org) and Condel (http://bg.upf.edu/fannsdb). We completed the annotation process using the CLC Genomic Workbench software and an in-house R script. All suspected variants were validated using Sanger sequencing and interpreted according to the American College of Medical Genetics and Genomics standards and guidelines.7
Results
Genetic profile
Among the 1005 cases with suspected CMT, we detected pathogenic or likely pathogenic variants in 301 cases (30.0%). The most common genetic causes in the mutation-positive cases were GJB1 and MFN2, and each accounted for 21.9% (66 cases). Within MFN2, 40 types of reported and three novel variants (two pathogenic and one likely pathogenic) were identified. The following genetic causes were MPZ (n=51, 16.9%), HSPB1 (n=14, 4.6%), PMP22 point mutations (n=13, 4.3%), GDAP1 (n=9, 3.0%), NEFL (n=9, 3.0%), MME (n=8, 2.7%), BSCL2 (n=6, 2.0%), MARS (n=6, 2.0%), DNM2 (n=5, 1.7%), SETX (n=5, 1.7%), SH3TC2 (n=5, 1.7%), PRX (n=4), GARS (n=3), IGHMBP2 (n=3), LRSAM1 (n=3), AARS (n=2), ARHGEF10 (n=2), FGD4 (n=2), SACS (n=2), SBF2 (n=2), TRPV4 (n=2) and TTR (n=2). Pathogenic or likely pathogenic variants were also detected in COA7, DCTN1, DHTKD1, EGR2, FBLN5, GALC, GAN, HARS, HSPB3, HSPB8, INF2, KARS, MTMR2, PRPS1, RAB7A and SOX10 in single cases (figure 1). Additionally, digenic variants were identified in five cases, which were variants in SETX (likely pathogenic) and ARHGEF10 (likely pathogenic); SH3TC2 (biallelic, likely pathogenic) and SACS (biallelic, likely pathogenic); LRSAM1 (likely pathogenic) and MARS (likely pathogenic); HARS (likely pathogenic) and ARHGEF10 (likely pathogenic); and MFN2 (reported, pathogenic) and PMP22 (reported pathogenic).

Genetic spectrum of 301 cases with pathogenic or likely pathogenic variants. The following genes are indicated: GJB1 (21.9%), MFN2 (21.9%), MPZ (16.9%), HSPB1 (4.6%), PMP22 point mutation (4.3%), GDAP1 (3.0%), NEFL (3.0%), MME (2.7%), BSCL2 (2.0%), MARS (2.0%), DNM2 (1.7%), SETX (1.7%), SH3TC2 (1.7%), PRX (1.3%), GARS (1.0%), IGHMBP2 (1.0%), LRSAM1 (1.0%), AARS (0.7%), ARHGEF10 (0.7%), FGD4 (0.7%), SACS (0.7%), SBF2 (0.7%), TRPV4 (0.7%), TTR (0.7%), COA7 (0.3%), DCTN1 (0.3%), DHTKD1 (0.3%), EGR2 (0.3%), FBLN5 (0.3%), GALC (0.3%), GAN (0.3%), HARS (0.3%), HSPB3 (0.3%), HSPB8 (0.3%), INF2 (0.3%), KARS (0.3%), MTMR2 (0.3%), PRPS1 (0.3%), RAB7A (0.3%) and SOX10 (0.3%).
In terms of sporadic cases, detection rate was 21.9% (125/570), comprising 108 monoallelic and 18 biallelic variants. Molecular diagnosis was accomplished in 44.6% (152/341) cases with AD or X-linked inheritance and in only 25.0% (18/72) of cases with AR. In demyelinating CMT cases, 45.7% (129/282) received a genetic diagnosis, and mutations in GJB1 (40.3%) and MPZ (27.1%) were the most common reasons, accounting for 67.4% of all mutation-positive cases. Among cases with axonal CMT, mutation detection rate was 22.9% (156/682), with MFN2 as the most frequent causative gene, accounting for 37.2% of all mutation-positive cases, followed by MPZ (9.0%), HSPB1 (8.3%), and GJB1 (7.7%), GDAP1 (5.1%) and MME (5.1%) (figure 2).

Detection rate and proportional detection of variants in cases with demyelinating and axonal CMT. *PMP22 point mutation.
Onset age analysis
We analysed the onset age in the mutation-positive CMT cases and attempted to specify their onset features. The CMT onset age of all the patients was determined to be when either of their parents noticed any motor abnormalities in their children, or when the patients themselves began to be aware of their motor or sensory dysfunctions. In cases with demyelinating CMT, 104/282 (36.9%) cases developed clinical symptoms in the first decade of life, and pathogenic or likely pathogenic variants were identified in 57 of these cases (54.8%). In the same age group, 58 out of 190 cases (30.5%) with axonal CMT received a molecular diagnosis, which also yielded the highest diagnostic rate. Unexpectedly, cases with demyelinating CMT with onset in the seventh decade demonstrated the highest diagnostic rate of 66.7% (6/9) (figure 3A,B).
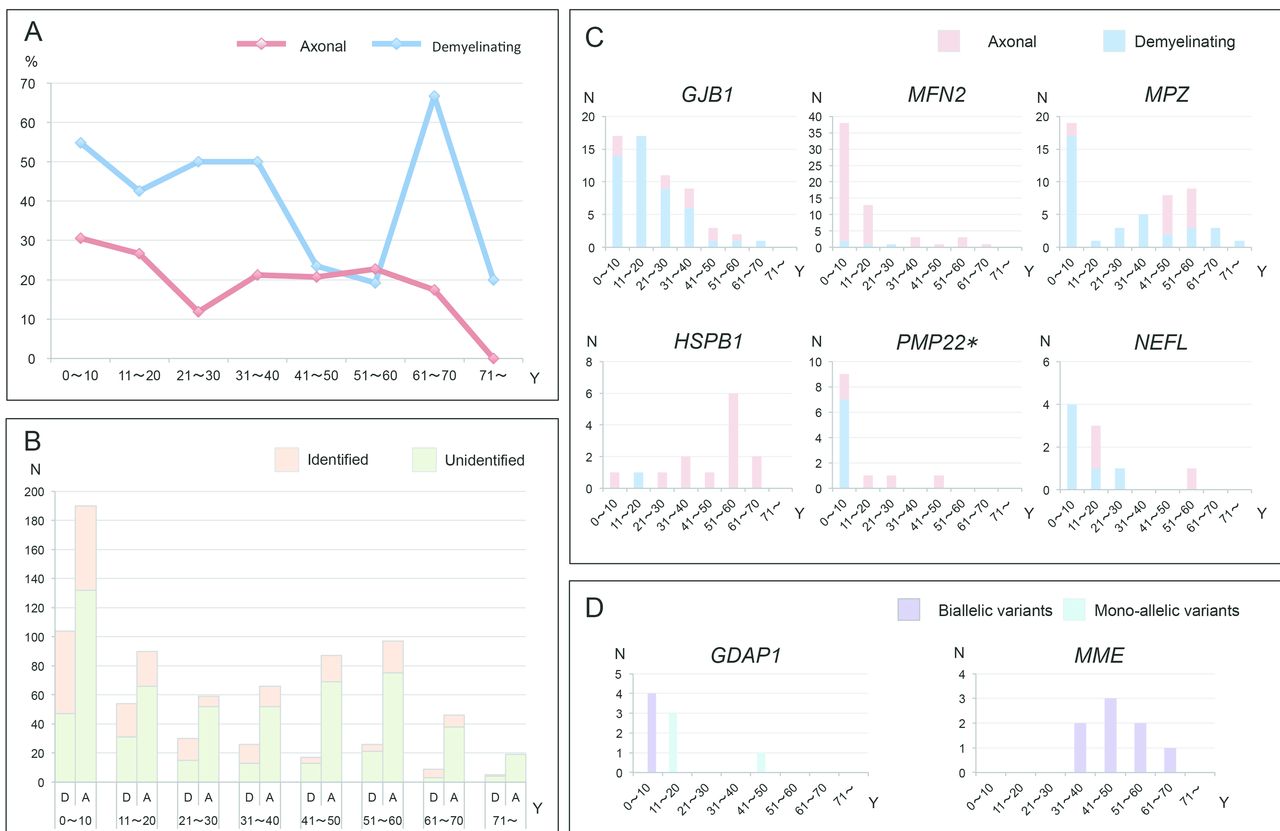
Onset age analyses of mutation-positive cases. (A and B) Curve graph and column diagram of varied onset age and diagnostic rate of axonal or demyelinating CMT. (C) Diagram of disease onset features in cases with GJB1, MFN2, MPZ, HSPB1, PMP22 and NEFL variants. (D) Diagram of disease onset features of cases with monoallelic or biallelic variants of GDAP1 and MME genes. A, axonal type; D, demyelinating type; N, number; Y, year. *PMP22 point mutation.
The majority (48/58cases) of cases with MFN2 variants were manifested with an early-onset (0~20 years) axonal polyneuropathy. Demyelinating neuropathy was predominant in cases with GJB1 variants, with case numbers decreasing with age. A two-peak pattern was observed in cases with MPZ variants, consisting of a peak of demyelinating type with onset age in the first decade, and a second peak of axonal type in cases with disease onset during the fifth and six decades. The most common onset age of cases with HSPB1 variants was during the six decade. Most cases with PMP22 point variants (7/13 cases) developed demyelinating neuropathy in the first decade, and the majority of cases with NEFL variants presented with a demyelinating phenotype at a younger age (figure 3C).
Regarding two genes linked to AR-CMT, GDAP1 and MME, we noted that cases with biallelic variants of GDAP1 developed clinical manifestations earlier than those with monoallelic variants; cases with biallelic variants in MME commonly presented with late-onset axonal neuropathies (figure 3D).
Geographic distribution analysis
We conducted a geographic distribution analysis on the basis of all available medical records but without further validation of their familial place of origin. Here, Japan was separated into eight regions. Variants in GJB1, MFN2 and MPZ were identified to be the top three causative genes in six regions of Japan. Therein, MFN2 was found as the most common cause of CMT in northern (Hokkaido and Tohoku) and southern Japan (Chugoku and Kyushu/Okinawa), while GJB1-related CMT was more prevalent in middle Japan (particularly in Kanto and Kinki). In Hokkaido region (figure 4A), MFN2 variants accounted for more than half of all mutation-positive cases, and no case with MPZ variant was identified. In Shikoku (figure 4G), a characteristic high incidence of NEFL variants was observed (figure 4).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Geographic analysis of genetic spectrum of CMT in Japan. Japan is divided into eight regions (A–H), and axonal/demyelinating type and the causative genes are indicated in different colours. Mutation-positive and total numbers of each region are indicated around the pie chart. NE, not examined.
Discussion
Using two targeted gene panel sequencing systems, we genetically analysed 1005 cases with suspected CMT to demonstrate their genetic profile. To the best of our knowledge, this is the largest Asian study to date. The total diagnostic rate of our study was 30.0% (301 cases), and remarkable genetic heterogeneity was recognised that pathogenic or likely pathogenic variants were detected from 40 genes. We diagnosed 27.7% cases with the Illumina MiSeq targeting 60 genes and 31.7% cases with the Ion Proton targeting 72 genes. Cases with demyelinating CMT received a much higher diagnostic rate (45.7% vs 22.9%) and lower genetic diversity than axonal type.
To date, genetic spectrum studies of CMT have been completed in multiple countries (table 1). The PMP22 duplication/deletion mutations, accounting for 23.3% of demyelinating CMT in Japan,8 were not involved in this study and were removed from the original data of previous studies to facilitate comparison. Consequently, we found that three genes, GJB1, MFN2, and MPZ, were the leading reasons in the present study, using data from Germany,9 USA,10 UK,11 Norway12 and Denmark13 studies; however, these results differ from previous reports from Japan,8 Spain,14Italy,15 Korea16 and a cross-country study.17 Particularly, in the other Japanese study,8 regarding other genes with mutation frequency higher than 1%, PMP22 (3.3%), NEFL (2.7%) and PRX (1.7%) have been reported, whereas we detected HSPB1 (1.4%) and PMP22 (1.3%) in the present study. A sampling bias should be considered to have contributed to these differences.
Mutation detection rates of patients with CMT in various populations
In cases with monoallelic variants, GJB1 (n=66), MFN2 (n=65) and MPZ (n=51) were the top three genes, accounting for 68.7% of all mutation-positive cases. Clinically, the majority of cases with MFN2 variants (n=58) showed axonal phenotype, making MFN2 as the most common reason of axonal CMT. Twelve cases with GJB1 variants exhibited axonal phenotype, whereas the other 52 cases exhibited the demyelinating phenotype. Cases with MPZ variants also exhibited both axonal (n=14) and demyelinating (n=35) phenotypes.
In cases with biallelic variants, MME accounted for 22.2% (n=8) of all mutation-positive cases, followed by SH3TC2 (n=5, 13.9%). In 2016, we have reported that MME gene, which encodes neprilysin, is responsible for a late-onset AR-CMT type 2T.6 Shortly thereafter, monoallelic rare variants have been reported to be associated with axonal polyneuropathies or dominant spinocerebellar ataxia with neuropathy.18 19 In the present study, monoallelic variants were detected in family members of four cases with AR-CMT2T, none of whom developed notable clinical manifestations. We also identified digenic variants from five cases (approximately 0.5%), the majority of which were likely pathogenic (8/10); further study is required to identify whether the combinatorial effect of these variants contributes to the phenotypic variability as disease burden.
The onset age distribution of our CMT cases suggested an evident clustering at the first decade, regardless of axonal or demyelinating phenotype. Our targeted gene panel sequencing yielded a significantly high diagnostic rate of axonal (30.5%) and demyelinating (54.8%) CMT for individuals diagnosed within the first decade of life. This result is not surprising given that genetic factors are more common than acquired factors in cases with early-onset polyneuropathies. Early onset is also a typical feature of cases with GJB1, MFN2, MPZ (demyelinating type), PMP22 (point variants), NEFL and GDAP1 (biallelic) variants. In contrast, MPZ (axonal type) and HSPB1 variants always produce a late-onset phenotype. For patients with late-onset demyelinating CMT with an onset age between the fifth and sixth decade, a significant decline of diagnostic rate was observed, which might be due to non-inherited factors or to undiscovered genetic causes. Interestingly, however, demyelinating CMT cases with onset at the seventh decade, yielded the highest diagnostic rate (6/9), which could be a coincidence owing to the mutations detected in various genes. Taken together, because the number of case of demyelinating CMT with onset age older than 40 years was limited, more samples should be collected for validating these unexpected results in the future.
We performed a geographic distribution analysis to elucidate the effect of geography on the genetic spectrum of our cases. GJB1, MFN2 and MPZ were the top three causative genes associated with CMT throughout the most regions in Japan. Although our case numbers were limited, we observed an unexpectedly high frequency of NEFL variants in Shikoku region, which was indicated in a previous study reporting that NEFL variants accounted for a much higher proportion of CMT cases (2.7%)8 Unfortunately, geographic distribution data were not available in the previous report. This unusually high frequency of NEFL variants was unlikely to be caused by a founder effect, because all these variants were completely different. High frequency of MFN2 variants and the absence of the MPZ variant were identified in Hokkaido. Our findings suggest that geographic regions could give rise to the variable genetic spectrum and diagnostic rate of CMT, and future well-powered analyses will be helpful to clarify these findings.
Within our CMT-related gene panels, the role of several genes still require further validation. Therein, six variants were detected in MARS, consisting of a previously reported P800T mutation (four cases) and two novel likely pathogenic variants. In two of the four pedigrees with P800T, cosegregation of genotype and phenotype was identified. For the two novel variants, further study is required to validate their pathogenicity. Besides, we also found two heterozygous variants, G585S and W426G in GALC gene (NM_000153.3), from one patient. GALC was known as the responsible gene for Krabbe disease; however, peripheral neuropathy was the original and predominant symptom of our patient, which is comparable with a former Japanese patient with isolated peripheral neuropathy.20
Recently, a number of new causative genes, such as those encoding MORC family CW-type zinc finger 2 (MORC2), minichromosome maintenance complex component 3 associated protein, neurofilament protein heavy polypeptide, diacylglycerol O-acyltransferase 2, dystrophin-related protein 2, cytochrome c oxidase subunit VIa polypeptide 1 and peripheral myelin protein 2 (PMP2) were associated with CMT phenotypes.21–26 These genes were not involved in any of our gene panels, but a high frequency (2.7%) of MORC2 variants in axonal CMT was revealed by whole-exome sequencing in our laboratory.27 These genes should be included in the upcoming version of our gene panel, and the diagnostic rate would be increased.
In conclusion, using targeted gene panel sequencing, we demonstrated the genetic features and geographical differences in a nationwide group of cases with CMT in Japan. Together with results of onset age analysis, our findings advanced the understanding of this intractable disease. Our sequencing strategy was proved effective, exempting for complicated and undefined subtyping in clinic. A limitation of this study is that our library could not yield either the non-coding region or the structural variant of these genes.
Acknowledgments
The authors would like to thank all patients and their families for participating in this study and appreciate the collaboration of their physicians. Additionally, the authors would like to thank Aya Ebina and Tomoko Onishi for their excellent technical assistance. The authors would also like to thank Enago (www.enago.jp) for the English language review. We would like to thank the Joint Research Laboratory, at the Kagoshima University Graduate School of Medical and Dental Sciences, for the use of their facilities.
References
Footnotes
Contributors AY, J-HY, and MA contributed to laboratory data acquisition, analysis or interpretation of data. AY, J-HY and HT contributed to drafting or revising of the manuscript. AH, YH and YO contributed to clinical data analysis or interpretation. TN contributed to nerve conduction study analysis. MN and HT contributed to study conception and supervision.
Funding This study is supported by grants from the research on the Nervous and Mental Disorders and Research Committee for Charcot–Marie–Tooth Disease, Neuropathy, and Applying Health and Technology of Ministry of Health, Welfare and Labour, Japan (201331010B, 201610002B). This research is also supported by the Research program for conquering intractable disease from Japan Agency for Medical Research and development (AMED) (201442014A, 201442071A and 17929553) and Japan Society for the Promotion of Science (26461275, 18H02742).
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.