
Abstract
Interleukin-1 beta (IL-1β) is an inflammatory mediator which may contribute to the pathophysiology of rheumatoid arthritis (RA) and type 2 diabetes mellitus (T2DM). Population pharmacokinetics (PK) of LY2189102, a high affinity anti-IL-1β humanized monoclonal immunoglobulin G4 evaluated for efficacy in RA and T2DM, were characterized using data from 79 T2DM subjects (Study H9C-MC-BBDK) who received 13 weekly subcutaneous (SC) doses of LY2189102 (0.6, 18, and 180 mg) and 96 RA subjects (Study H9C-MC-BBDE) who received five weekly intravenous (IV) doses (0.02–2.5 mg/kg). Frequency of anti-drug antibody (ADA) development appears dose-dependent and is different between studies (36.7% in Study H9C-MC-BBDK vs. 2.1% in Study H9C-MC-BBDE), likely due to several factors, including differences in patient population and background medications, administration routes, and assays. A two-compartment model with dose-dependent bioavailability best characterizes LY2189102 PK following IV and SC administration. Typical elimination and distribution clearances, central and peripheral volumes of distribution are 0.222 L/day, 0.518 L/day, 3.08 L, and 1.94 L, resulting in a terminal half-life of 16.8 days. Elimination clearance increased linearly, yet modestly, with baseline creatinine clearance and appears 37.6% higher in subjects who developed ADA. Bioavailability (0.432–0.721) and absorption half-life (94.3–157 h) after SC administration are smaller with larger doses. Overall, LY2189102 PK is consistent with other therapeutic humanized monoclonal antibodies and is likely to support convenient SC dosing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Interleukin-1 beta (IL-1β) is a member of interleukin-1 (IL-1) family of cytokines and is an important mediator of inflammatory response, cell proliferation, differentiation, and apoptosis. IL-1β is implicated in a variety of autoinflammatory diseases, including rheumatoid arthritis (RA) and type 2 diabetes mellitus (T2DM) (1). Anakinra, an analogue of the endogenous IL-1 receptor antagonist that blocks IL-1α and IL-1β activity, is approved for the reduction in signs and symptoms of RA and slowing the progression of structural damage in moderately to severely active RA (2,3). Furthermore, anakinra and XOMA 052, an anti-IL-1β monoclonal immunoglobulin (Ig)G2, have demonstrated a significant reduction of hemoglobin A1c (HbA1c) in T2DM patients (4–6), supporting earlier reports suggesting the involvement of IL-1β in the pathophysiology of T2DM (7–10).
LY2189102 is an anti IL-1β humanized monoclonal IgG4 that binds to IL-1β with high affinity (2.8 pM) and neutralizes its activity in preclinical in vitro (IC50 < 2 pM) and in vivo models (data on file, Eli Lilly and Company). Initial clinical evaluation of LY2189102 was focused on RA and T2DM. This report describes a nonlinear mixed effects analysis of the PK of LY2189102 using pooled data from multiple SC dosing in subjects with T2DM (clinicaltrials.gov identifier: NCT00942188) (11,12) and multiple IV dosing in subjects with RA (clinicaltrials.gov identifier: NCT00380744). A fit for purpose population PK model for LY2189102 was developed to characterize the drug exposure in the two patient populations included in the analysis dataset for use in separate pharmacokinetic/pharmacodynamic analyses (12). Additionally, the influence of subject descriptors, such as demographics and immunogenicity, on LY2189102 PK variability was evaluated.
METHODS
Study Designs, Dosing Regimens, and Subjects
Data used to perform this population analysis were collected from two clinical trials, Study H9C-MC-BBDE (hereafter, referred to as BBDE) and Study H9C-MC-BBDK (hereafter, referred to as BBDK). Study BBDE was a Phase 1b/2, multicenter, placebo-controlled, randomized, double-blind, two part, and modified dose-escalation study. Subjects enrolled in this study had been diagnosed with RA and had been taking methotrexate on a regular basis for at least 3 months (with stable doses for at least 2 months) at the time of study entry. Study BBDE consisted of two parts: Part A, an initial dose-escalation phase, and Part B, a parallel dose group monitoring phase. In both parts, LY2189102 was administered on Day 0 as an IV loading dose (equal to twice the maintenance dose amount) followed by four weekly IV maintenance doses administered on Days 7, 14, 21, and 28. The maintenance dose levels tested in Part A were 0.1, 0.3, 1, and 2.5 mg/kg; those tested in Part B were 0.02, 0.15, 1, and 2.5 mg/kg. Pharmacokinetic samples were collected prior to (Part A only) and within 3 min of termination of the first infusion (Day 1), 48 h after the start of the first infusion (Part A only), prior to and within 3 min of termination of infusions on Day 14 and Day 28, and at Week 5 (Part A only) and Week 9.
Study BBDK was a Phase 2, randomized, double-blind, placebo-controlled, parallel design study of the safety, PK, and efficacy of LY2189102 in subjects with T2DM. Subjects included in this study had been diagnosed with T2DM at least 3 months prior to enrollment and exhibited baseline HbA1c between 7 and 10%, and baseline high sensitivity C-reactive protein greater than or equal to 2 mg/L. Subjects were maintained on diet and exercise alone or together with concomitant anti-diabetic medications (except for thiazolidinediones and insulin products). It was recommended that subjects be taking background statin therapy per National Cholesterol Education Program Adult Treatment Panel III guidelines (13). LY2189102 was administered as weekly SC injections of 0.6, 18, or 180 mg for 13 weeks. Pharmacokinetic samples were collected prior to each dose, at 24 h and between 72 and 96 h after the first dose, and 1, 6, and 12 weeks after the last dose of LY2189102. It should be noted that different expression systems were developed to produce the LY2189102 batches used in Study BBDE (insect cells) and Study BBDK (mammalian cells).
All protocols and consent forms were reviewed and approved by the institutional review board of each of the research sites. Before participating in the studies, all subjects were informed about the risks of the studies and signed an informed consent form, according to the recommendations of the Declaration of Helsinki.
Bioanalytical Method
Serum was analyzed for LY2189102 using a validated, specific, and quantitative enzyme-linked immunosorbent assay (ELISA) method, with lower and upper limits of quantification of 4.0 and 256.0 ng/mL. The inter-assay accuracy (% accuracy) during validation ranged from 80.8 to 97.5%. The inter-assay precision (% relative standard deviation) during validation ranged from 6.3 to 12.6%.
Anti-LY2189102 Antibody Monitoring
Serum was analyzed for the presence of anti-drug antibodies (ADA) using specifically developed ELISA methods. Study BBDE used a qualitative bridging ELISA in which ADA were captured on an LY2189102-coated ELISA plate and detected by enzyme-labeled LY2189102. The assay sensitivity was 31.2 ng/mL. The drug interference level was not established. Study BBDK used a qualitative assay using biotin extraction and acid dissociation with electrochemiluminescent detection. The assay sensitivity was 188.8 ng/mL. Drug tolerance was established at both high and low concentrations of the positive control for ADA (i.e., monkey hyperimmune antiserum at 1:500 and 1:4000 dilutions, respectively). The assay was tolerant up to LY2189102 concentrations of 100 and 10 μg/mL in the presence of high and low ADA concentrations, respectively. Both assays could detect IgG, IgA, and/or IgM antibodies specific for LY2189102, as well as the corresponding antibody titers. In Study BBDE, samples were evaluated at baseline and 2, 5, 9, and 14 weeks after the first dose of study drug. In Study BBDK, samples were evaluated at baseline and 1, 6, and 12 weeks after the last dose of study drug. For this analysis, subjects having at least one ADA-positive sample collected after the first administration of LY2189102 were considered positive for their entire PK profile.
PK Modeling Methodology
A population model for LY2189102 PK was developed using nonlinear mixed effects modeling. The first-order conditional estimation with interaction method of the computer program NONMEM, Version 6.2.0, was used at all stages of the model development process (14). Model selection was based upon the goodness-of-fit plots, successful outcomes of the estimation and covariance routines, and the reasonableness and precision of the parameter estimates. Initially, inter-individual variability (IIV) was assumed to be log-normally distributed, and exponential variability models were combined in a simple diagonal matrix form (i.e., all covariance terms fixed to 0). Various residual variability (RV) models were tested, including constant coefficient of variation, additive plus constant coefficient of variation, and logarithmic models.
Model development was performed in two stages due to the availability of the data. In the first stage, a structural model for LY2189102 PK after IV and SC administration was developed on an interim dataset containing more than 95% of the final data. A covariate analysis then explored the effects of the following variables on the base model parameters: age, gender, body weight, baseline creatinine clearance (CrCL, calculated using the Cockcroft and Gault method (15), modified by the use of ideal body weight (16), and capped at 160 mL/min to be within physiological limits), and concomitant anti-diabetic medications consolidated in a single dichotomous variable.
Covariate analysis was performed using a stepwise forward selection followed by a backward elimination process. In the stepwise forward selection, each covariate was added univariately to each LY2189102 PK parameter associated with IIV in the base model using centered linear, exponential, or power relationships for continuous covariates, and proportional shift for categorical covariates. Covariates contributing at least a 3.84 change in the objective function value (OFV) and resulting in a decrease in IIV in the PK parameter of interest were considered significant, but only the covariate contributing the most significant change in the OFV (smallest P value < 0.05) was included in the model at each step. This process was repeated until there were no further covariates that produced significant changes in the OFV. In the backward elimination process, each covariate was removed from each parameter equation separately. A covariate was still considered significant if it resulted in at least a 10.83 change in the OFV when removed from the model. The most non-significant covariate (the highest P value greater than 0.001) was removed from the model at each step. The backward elimination procedure was repeated until all remaining covariates were significant at α = 0.001.
In the second stage of model development, the reduced model including the significant covariates identified at the first stage was reevaluated using the final dataset, which included a larger number of subjects and the ADA data. The influence of outlier points was also tested. A second round of covariate analyses was conducted to explore the influence of ADA on LY2189102 PK variability. The process described above was applied; however, only the influences of ADA status (considered as positive if the subject ever developed ADA) and maximum ADA titer (excluding baseline titer) were considered. The effects of these two variables were tested on all parameters for which IIV was estimated and on parameters associated with LY2189102 bioavailability.
Additional model refinements (addition of IIV terms; modification of the covariance matrix, e.g., correlation between random effects; and modification of the RV model) were tested between the two stages of analysis, between forward selection and backward elimination, and at the stage of the final model.
The final model was then evaluated using a simulation-based prediction-corrected visual predictive check (VPC) method. Due to the inclusion of various dose groups in the analysis dataset and the nonlinear nature of the final PK model, the prediction-corrected VPC, as suggested by Bergstrand et al., was utilized (17). This technique provides an enhanced ability to diagnose possible model misspecification by removing the variability introduced in an ordinary VPC when binning across influential covariates (17). Conditional on the final model point estimates, 1,000 replicates of the analysis dataset were simulated with NONMEM. The 5th, 50th (median), and 95th percentiles were calculated for the distributions of the simulated concentration values corrected using the median of population prediction within the associated time bin. These percentiles were then plotted versus time since previous dosing with the original observed dataset (also corrected using the median of population prediction within the associated time bin) overlaid to visually assess concordance between the model-based simulated data and the observed data.
Assessment of Covariate Effects on the LY2189102 Exposures
Stochastic simulations (N = 10,000) were conducted to predict the effect of significant covariates on LY2189102 exposures after SC administration over a clinically relevant range of doses. Exposure was assessed by the steady-state area under the LY2189102 concentration curve, AUCτ,SS, calculated as follows:
where F is the bioavailability after SC injection as compared to that following IV infusion, Dose is the dose received by SC injection, TVCL is the typical elimination clearance estimated from the final model (see Eq. 3 below), and ηCL is the estimated IIV in CL.
The simulation and all graphical representations were performed using R Version 2.12 (18).
RESULTS
Data Description and Subject Characteristics
A total of 1,603 LY2189102 measurable concentrations were available from 96 subjects enrolled in Study BBDE (582 concentrations) and 79 subjects enrolled in Study BBDK (1,021 concentrations). Thirty-five concentrations were below the limit of quantification and were excluded during modeling.
Subjects included in the final analysis dataset were predominantly adult women (67.4% of the overall population), between 23 and 76 years of age (mean age: 52.6 years), having baseline CrCL between 7.3 and 160 mL/min (mean value: 88.6 mL/min), and exhibiting mostly normal renal function or mild renal impairment (only 13 subjects had moderate to severe renal impairment). Subject characteristics were similar in both studies (Table I). However, mean body weight was 14% higher in Study BBDK than in Study BBDE. A higher proportion of female subjects in Study BBDE likely contributed to this difference.
The percentage of subjects developing ADA was also considerably higher in Study BBDK (36.7%) than in Study BBDE (2.1%). Of the ADA-negative subjects in Study BBDK, two subjects had drug concentrations above potentially interfering levels at all available post-dosing time points and, therefore, may be potential false negatives. In Study BBDK, the fraction of positive ADA appeared to increase with LY2189102 dose (23.1% at 0.6 mg, 34.6% at 18 mg, and 51.9% at 180 mg), but there was no consistent relationship between LY2189102 dose and maximum ADA titer. Most ADA-positive subjects (75.9%) in Study BBDK developed a maximum ADA titer lower than 50. The number of subjects who developed ADA in Study BBDE was too low (i.e., 1 at 0.02 mg/kg and 1 at 1 mg/kg) to allow any evaluation of the relationship between dose and ADA status.
Exploratory Data Analysis
In both studies, LY2189102 dose-normalized pharmacokinetics exhibited a degree of variability that is commonly observed in other IgG monoclonal antibodies (mAbs; Fig. 1a) (19). In Study BBDE, the disposition of LY2189102 after IV dosing was characterized by a shallow biexponential decline. In Study BBDK, LY2189102 concentrations slowly increased after SC dosing and reached a peak more than 1 week after the previous dose, suggesting that the compound was slowly absorbed from the injection site. After reaching peak, LY2189102 concentrations exhibited an apparent monophasic decline, which may be an artifact of the paucity of data between 400 and 800 h after SC dosing (i.e., 16.7 to 33.3 days) or the consequence of the slow rate of absorption. Weekly SC dosing of LY2189102 produced a slow increase in trough concentrations before a plateau was reached between 1,000 and 1,250 h (approximately 6 and 7.5 weeks) after the first dose (Fig. 1b).

LY2189102 dose-normalized serum concentrations versus time since the previous dose stratified by study and dose a, and LY2189102 concentration versus time since first dose in Study BBDK stratified by antidrug antibody status b
In all three BBDK dose groups, multiple measurements were observed below the range of the remaining data. All measurements corresponded to data collected in either ADA-positive or negative subjects who stopped receiving LY2189102 or in compliant ADA-positive subjects and were kept in the dataset. However, five of the seven subjects who developed an ADA titer greater than or equal to 50 presented substantial and abrupt decreases, and occasionally increases, in their concentration versus time profile (not shown in Fig. 1) despite perfect compliance, and were excluded from the analysis to avoid model convergence issues. All other ADA-positive subjects exhibited expected PK behavior based upon individual dosing history.
Dose-normalized concentrations were reasonably well superimposed after IV dosing (Fig. 1a, Study BBDE), suggesting that LY2189102 disposition was dose-proportional within the range of tested doses (0.02–2.5 mg/kg). In contrast, the range of dose-normalized concentrations appeared to decrease with increasing dose after SC dosing, while the slope of elimination did not seem markedly modified (Fig. 1a, Study BBDK). These decreases in dose-normalized concentrations at medium and high doses were observed in both ADA-negative and ADA-positive subjects, suggesting that, although LY2189102 bioavailability after SC administration appears to be dose-dependent, these changes are likely to be independent of the increase in ADA rate at higher doses. Finally, the ranges of concentrations observed in the absence or presence of approved concomitant anti-diabetic medications in Study BBDK were similar (data not shown).
LY2189102 Pharmacokinetic Model
The structural model selected for LY2189102 pharmacokinetics after repeated IV infusions and SC injections was an open two-compartment model with linear distribution and elimination and dose-dependent bioavailability after SC dosing. The extent of absorption was modeled with a hyperbolic function on the logit scale defined as follows:
where FA and FS are the asymptote and slope of the hyperbolic function. The rate of absorption (ka) was indirectly related to the dose, by the estimation of a correlation factor (α) between F and the half-life of absorption (Tka) into the central compartment, which is equal to log(2)/ka. The interim dataset supported the estimation of IIV in the elimination clearance (CL), the central volume of distribution (Vc), the peripheral volume of distribution (Vp), and in Tka. Residual variability was modeled using a separate log-error model for each study.
Covariates found to be statistically significant after forward selection were the linear effect of baseline CrCL on CL and the power effects of body weight on CL and Vc. However, after further refinement of this multivariable model by estimation of the covariance between the IIV in CL and Vc and between the IIV in CL and Tka, only the linear effect of baseline CrCL on CL remained in the model after backward elimination. Additional model refinements such as the addition of IIV in the distribution clearance (Q) or the estimation of a full covariance matrix were not supported by the interim data.
Once the final dataset (including ADA assessment) became available, the interim PK model was reevaluated. Attempts were made to include the five ADA-positive subjects previously excluded as outliers due to substantial and abrupt decrease in their concentration versus time profiles. However, the data from those subjects induced a high correlation between CL and Q estimates, and inflated some typical and RV parameter estimates as much as twofold. Consequently, those five subjects remained excluded for further analysis of ADA influence on LY2189102 PK.
Following the second forward covariate selection, ADA status, introduced as proportional shift in CL, was found to significantly improve the model fit (ΔOFV = −17.69, P = 2.6⋅10−5). Additional/alternative effects of maximum ADA titer on CL and ADA status on FA were also found to significantly decrease the OFV, but were not included due to the poor precision of the estimate of the covariate effect or the numerous high correlations introduced between model parameter estimates. Given the P value of the unique covariate introduced, a second backward elimination process was not necessary.
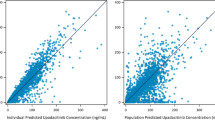
No further refinements of the model were found to improve the fit of the model. Overall, the structure of the final model allowed a good estimation of all parameters except α (Table II) and provided reasonably good fit of the data (Fig. 2). The prediction-corrected VPC indicated that the central tendency and the variability of the LY2189102 concentrations in the study population were well predicted by the final model, although the variability at low concentrations was slightly over-predicted (Fig. 3).

goodness-of-fit plots for the final model for LY2189102 pharmacokinetics

Prediction-corrected visual predictive check of the final model for LY2189102 pharmacokinetics
Assessment of Covariate Effects on the Final Model Exposures
The typical individual CL value, TVCLj, was predicted from the final PK model using the following equation:
where CrCLj is the CrCL value in the jth subject and ADAj indicates whether the jth subject was ADA-positive (1) or negative (0).
The influence of CrCL on LY2189102 steady-state exposure after SC dose was evaluated by stochastic simulations at CrCL values of 47.8, 83.2, and 145.6 mL/min, that is, at the 5th, 50th, and 95th percentiles of the CrCL distribution in the study population. Figure 4 suggests that exposure linearly increases with dose for doses greater than 0.5 mg, and that there was a fair amount of overlap in the predicted exposures for subjects with low (47.8 mL/min) or high (145.6 ml/min) CrCL.

Influence of creatinine clearance on steady-state LY2189102 exposures. Lines delimit the 80% prediction intervals of the steady-state exposure versus dose relationship predicted from the final LY2189102 model at the 5th, 50th, and 95th percentiles of creatinine clearance in the study population. Inset represents the same data for the 0 to 3 mg dose range
Although ADA status was another statistically significant descriptor of TVCLj, its influence on LY2189102 steady-state exposure was not evaluated for reasons discussed below.
DISCUSSION
Consistent with what is commonly observed for IgG mAbs (20), the PK of LY2189102 were characterized by slow absorption after SC administration and slow elimination, as illustrated by the time to peak concentration of approximately 7 days and the terminal half-life of 16.8 days (calculated using the model parameters).
During model development, it was found that a two-compartment model, consistent with typical IgG PK behavior, provided a better fit to the data than a one-compartment structure. However, the PK of LY2189102 exhibited nonlinearity over the range of tested doses. Initial models based on linear absorption and disposition showed dose-dependent biases in the fit that suggested incomplete and dose-dependent LY2189102 bioavailability after SC administration. While overlapping ranges of doses were administered by IV infusions (0.96–373 mg) or SC administrations (0.6–180 mg), no dose-dependent trend could be observed in the fit of the data collected after IV infusion. This finding was consistent with the pattern shown in Fig. 1a and indicated that the bioavailability of LY2189102 after SC administration, unlike its disposition, was dose-dependent. The extent of bioavailability decreased while the rate of absorption increased with dose when these parameters were specifically estimated at each studied SC dose level. Such relationships were adequately captured with a more parsimonious empirical model that defines F as a hyperbolic function of dose on a logit scale (Eq. 2) and Tka as a proportional function of F. Several models describing bioavailability and/or rate of absorption using alternative empirical functions of dose (e.g., linear, power, Weibull, or logistic functions in the linear or logit scale) were tested but rejected due to optimization problems, poor parameter precision, poor descriptive performance of the data, or lack of improvement over the proposed final model despite an increase in the number of required parameters. More complex absorption models were not tested to mechanistically characterize the saturable absorption process due to the relative paucity of the data available during the absorption phase following SC administration. The final model (including covariate effects) predicted that the typical value of F was 0.721, 0.442, and 0.432 and that the typical value of Tka was 157, 96.3, and 94.3 h after SC administration of 0.6, 18, and 180 mg LY2189102, respectively, indicating that the dose effect achieved a plateau by 18 mg. Alternative models based upon dose-dependent or nonlinear elimination clearance or distribution of LY2189102 did not compare favorably with the proposed final model and did not correct the biases observed in the initial model fit. This pattern of dose-dependent bioavailability is consistent with the decrease in rituximab bioavailability from 0.31 to 0.18 over a dose range of 10 to 40 mg/kg in rats (21).
The bioavailability of subcutaneously administered drugs can be reduced by presystemic catabolism, the extent of which is determined by the duration of exposure of the drug to local degradation mechanisms and thus by the relative rates of systemic absorption and local catabolism. The absorption of mAbs after SC administration is influenced by several processes, including convective transport through lymphatic vessels into the blood and diffusion across blood vessels near the site of injection, although there remains much uncertainty regarding the primary determinants of these processes (22). Presystemic catabolism may depend on rates of extracellular degradation, endocytosis, intracellular degradation, and recycling through the neonatal Fc receptor (FcRn) (23,24). The role of FcRn in reducing the systemic clearance of mAbs is well established. Binding to FcRn in the acidic environment of lysosomes protects endocytosed mAbs from degradation, which are then recycled and released in the more neutral extracellular environment (25). FcRn interaction results in low systemic clearance and long half-life for IgG and IgG-based mAbs (26). A growing body of literature is also suggesting a significant, if not major, role of FcRn in SC absorption of IgG-based mAbs. In addition to protecting mAbs from intracellular catabolism at the site of action, FcRn may also play a role in transporting IgGs across the vascular endothelium (22). In support of this hypothesis, FcRn-deficient mice were reported to have threefold lower bioavailability of an IgG1 antibody compared to wild-type mice (22). Similarly, increased affinity of mAbs to FcRn at pH 6.0 (akin to lysosomal pH), but not pH 7.4, was associated with greater SC bioavailability in mice (27,28), while coadministration of rituximab with IgG (and thus saturating the FcRn interaction) reduced its bioavailability in mice (29). In contrast, engineering a series of five IgG4 variants for enhanced FcRn interaction failed to consistently improve SC bioavailability in cynomolgus monkeys (30), although a trend was suggested for those associated with lower starting bioavailability. The same report, however, found a reduction in absorption rates when FcRn interactions were enhanced, suggesting that FcRn recirculation may slow the transfer of IgGs through the tissue at the site of injection, and hence slow the delivery to systemic circulation. Thus, we hypothesize that higher SC doses of LY2189102 may saturate FcRn interaction at the site of injection, resulting in an overall increase in presystemic catabolism, and hence lower extent of bioavailability, while increasing the fraction of LY2189102 that is transferred to systemic circulation through faster FcRn-independent absorption mechanisms. Further investigation is warranted to elucidate the mechanisms underlying the dose-dependent absorption kinetics of LY2189102.
Overall, the final model predicted the central tendency and the variability of the LY2189102 concentrations reasonably well, even during the absorption phase after SC administration (Fig. 3). Furthermore, the absorption and disposition parameter estimates of the final model were consistent with those reported for other monoclonal antibodies (19). For instance, Vc was small and close to the typical plasma volume (~3 L), as expected for the distribution of IgG; CL was low and within the typical range of 0.2–2 L/day; F was also moderate to low and within the typical 40–80% range (19,20).
Route of administration, the expression system by which LY2189102 was produced, and disease status are confounded factors in this analysis. Therefore, it was not possible to assess whether the lower bioavailability after SC administration in Study BBDK was only related to SC injections. Disposition was assumed to be similar in both populations of subjects, and study-specific effects on disposition parameters were not evaluated. This assumption is strongly supported by the good predictive performance of the selected model, as assessed by inspection of the VPC plots (Fig. 3) and the lack of dose-dependent or study-dependent trends in the distribution of random effects for CL, Vc, and Vp.
Covariate analysis identified both baseline CrCL and ADA status as significant factors altering LY2189102 elimination clearance. No other subject descriptors were statistically significant predictors of LY2189102 PK. While ADA can commonly alter elimination of IgG (31), it is not readily apparent why CrCL was identified as the most statistically significant descriptor of CL variability. The molecular size of LY2189102 (~145 kDa) greatly exceeds the cutoff weight for glomerular filtration (70–100 kDa) (19,20); accordingly, renal filtration should only amount to a very small portion of LY2189102 elimination and, thus, should minimally influence LY2189102 CL variability (32). It is possible, however, that CrCL or some of the metrics used in its computation (i.e., gender, age, ideal body weight, and serum creatinine; Eq. 3) are correlated with one or more significant subject descriptors that were not monitored in Studies BBDK and BBDE. The baseline CrCL was estimated to modestly increase LY2189102 CL, resulting in a 50% reduction in LY2189102 CL in the context of severe renal impairment (CrCL of 20 mL/min). Given the small number of subjects with moderate or severe impairment included in our dataset and the large overlap in the model-predicted AUCτ,SS for hypothetical subjects with various CrCL levels (Fig. 4), the effect of CrCL on LY2189102 CL should be interpreted with caution, and its implication on dose adjustment can only be judged in the context of the exposure–response relationship.
Caution should also be exercised in interpreting the ADA status effect on CL quantified in the final model. Several elements of study design and analysis results contributed to the interpretation of this estimate as a hybrid value. Large differences between Study BBDE and Study BBDK were observed in the frequency of positive ADA status (2.1 versus 36.7%) and the range of maximum ADA titer (0 to 16 versus 0 to 6,250), and may be accounted for by several factors. First, background treatment with methotrexate, an immunosuppressive agent, was administered in all subjects from Study BBDE and could have reduced immunogenic response to LY2189102 in this population. Second, the SC route of injection used in Study BBDK is generally reported as more immunogenic than the IV route of injection (33,34), since skin and local lymph nodes exposed to antigens (i.e., drugs) after SC injections constitute a richer environment for antigen processing and presentation than the spleen, which is the first lymphoid organ reached after IV injection. Longer exposure to LY2189102 in Study BBDK, between-study differences in the assay platforms used for ADA detection, lack of understanding of potential interference of free drug on ADA detection in Study BBDE, and modifications of the expression systems used for production of the LY2189102 batches administered in Study BBDE and Study BBDK may also have resulted in higher immunogenic response in subjects enrolled in Study BBDK.
Additionally, the lack of detailed longitudinal ADA data in both studies prevented the evaluation of a time-varying effect on CL, making the final model estimate an average over time of the real effect. A richer sampling schedule for ADA measurement may theoretically support finer assessment of an ADA effect on CL. However, this approach should be carefully considered in light of the drug tolerance of the ADA assay, as true positive ADA reactions may be masked by high drug concentrations of therapeutic proteins at early time points if these concentrations exceed the drug tolerance of the assay. The estimate of ADA effect on CL was also affected by the exclusion of the five subjects with high ADA titer who exhibited substantial abrupt changes in LY2189102 concentrations despite documented compliance with treatment which prevented the quantification of the ADA effect at the higher end of the ADA titer spectrum. Finally, the covariate analysis suggested that the increase in maximum ADA titer was statistically as strong a predictor of the increase in CL as ADA status, but showed that this linear effect of the titer could not be precisely estimated. Overall, with respect to the effect of ADA on LY2189102 PK, the results of this covariate analysis indicate that ADA has a clear impact on LY2189102 disposition and suggested that the magnitude of this effect likely increases with ADA titer.
LY2189102 dose levels were fixed in Study BBDK and body weight-based in Study BBDE. The effects of body weight on CL and Vc included during the covariate forward selection process were not significant enough to remain in the model after backward elimination. Therefore, the current dataset does not provide support for weight-based dosing. However, given the relatively small number of subjects included in the current evaluation, one cannot draw a definitive conclusion regarding the need for weight-based dosing until the PK model is refined using a larger study population.
CONCLUSION
LY2189102 exhibits typical IgG-like PK. A fit for purpose population model was successfully developed to characterize the LY2189102 concentration data following repeated IV and SC dosing in patients with RA and T2DM; LY2189102 PK was best described with a two-compartment structural model with dose-dependent bioavailability. The elimination clearance of LY2189102 increases linearly, yet modestly, with baseline CrCL and is higher in subjects with positive ADA status. The extent of bioavailability decreases while the absorption rate increases with larger SC doses.
REFERENCES
Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. 2009;27:519–50.
Kineret (anakinra) [package insert]. Stockholm, Sweden: Swedish Orphan Biovitrum AB; 2012. http://www.kineretrx.com/fileadmin/user_upload/kineretus/documents/Kineret_Full_Prescribing_Information.pdf. Published Dec 2012. Accessed 20 Feb 2014.
Kineret (anakinra). Summary of product characteristics. 2002. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000363/WC500042310.pdf. Accessed 20 Feb 2014.
Donath MY, Weder C, Whitmore J, Bauer RJ, Der K, Scannon PJ, et al. XOMA 052, an anti–IL–1β antibody, in a double-blind, placebo-controlled, dose escalation study of the safety and pharmacokinetics in patients with type 2 diabetes mellitus—a new approach to therapy. Diabetologia. 2008;51 Suppl 1:S7.
Larsen CM, Faulenbach M, Vaag A, Vølund A, Ehses JA, Seifert B, et al. Interleukin–1–receptor antagonist in type 2 diabetes mellitus. N Engl J Med. 2007;356(15):1517–26.
Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, Mandrup-Poulsen T. Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care. 2009;32(9):1663–8. doi:10.2337/dc09-0533.
Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, et al. Glucose–induced beta cell production of IL–1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest. 2002;110(6):851–60.
Maedler K, Spinas GA, Lehmann R, Sergeev P, Weber M, Fontana A, et al. Glucose induces beta–cell apoptosis via upregulation of the Fas receptor in human islets. Diabetes. 2001;50(8):1683–90.
Maedler K, Storling J, Sturis J, Zuellig RA, Spinas GA, Arkhammar PO, et al. Glucose–and interleukin–1beta induced beta–cell apoptosis requires Ca2+ influx and extracellular signal–regulated kinase (ERK) 1/2 activation and is prevented by a sulfonylurea receptor 1/inwardly rectifying K+ channel 6.2 (SUR/Kir6.2) selective potassium channel opener in human islets. Diabetes. 2004;53(7):1706–13.
Welsh N, Cnop M, Kharroubi I, Bugliani M, Lupi R, Marchetti P, et al. Is there a role for locally produced interleukin–1 in the deleterious effects of high glucose or the type 2 diabetes milieu to human pancreatic islets? Diabetes. 2005;54(11):3238–44.
Sloan-Lancaster J, Abu-Raddad E, Polzer J, Miller JW, Scherer JC, De Gaetano A, et al. Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1β antibody, in patients with type 2 diabetes. Diabetes Care. 2013;36(8):2239–46.
Abu-Raddad E, DeGaetano A, Bihorel S, Fiedler-Kelly J, Sloan-Lancaster J. Pharmacokinetic (PK) and pharmacodynamic (PD) modeling of subcutaneous (SC) LY2189102, a neutralizing IL–1β antibody, in patients with type 2 diabetes mellitus. Diabetologia. 2011;54 Suppl 1:S366.
Grundy SM, Cleeman JI, Merz CN, Brewer Jr HB, Clark LT, Hunninghake DB, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation. 2004;110(2):227–39.
NONMEM [computer program]. Version VI. Ellicott City, MD: ICON Development Solutions; 2006.
Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31–41.
Peck CC, Conner DP, Murphy MG. Bedside clinical pharmacokinetics: Simple techniques for individualized drug therapy. Vancouver: Applied Therapeutics, Inc; 1989.
Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction-corrected visual predictive checks for diagnosing nonlinear mixed-effects models. AAPS J. 2011;13(2):143–51. doi:10.1208/s12248-011-9255-z.
R Development Core Team (2010). R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. ISBN 3-900051-07-0, URL http://www.R-project.org.
Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(10):633–59. doi:10.2165/11535960-000000000-00000.
Keizer RJ, Huitema ADR, Schellens JHM, Beijnen JH. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49(8):493–507. doi:10.2165/11531280-000000000-00000.
Kagan L, Turner MR, Balu-Iyer SV, Mager DE. Subcutaneous absorption of monoclonal antibodies: Role of dose, site of injection, and injection volume on rituximab pharmacokinetics in rats. Pharm Res. 2012;29(2):490–9. doi:10.1007/s11095-011-0578-3.
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58. doi:10.1038/clpt.2008.170.
Porter CJ, Charman SA. Lymphatic transport of proteins after subcutaneous administration. J Pharm Sci. 2000;89(3):297–310.
Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 2001;50(1–2):3–20.
Israel EJ, Wilsker DF, Hayes KC, Schoenfeld D, Simister NE. Increased clearance of IgG in mice that lack beta 2-microglobulin: Possible protective role of FcRn. Immunology. 1996;89(4):573–8.
Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004;93(11):2645–68.
Deng R, Meng YG, Hoyte K, Lutman J, Lu Y, Iyer S, et al. Subcutaneous bioavailability of therapeutic antibodies as a function of FcRn binding affinity in mice. MAbs. 2012;4(1):101–9. doi:10.4161/mabs.4.1.18543.
Deng R, Loyet KM, Lien S, Iyer S, DeForge LE, Theil FP, et al. Pharmacokinetics of humanized monoclonal anti-tumor necrosis factor-α antibody and its neonatal Fc receptor variants in mice and cynomolgus monkeys. Drug Metab Dispos. 2010;38(4):600–5. doi:10.1124/dmd.109.031310.
Kagan L, Mager DE. Mechanisms of subcutaneous absorption of rituximab in rats. Drug Metab Dispos. 2013;41(1):248–55. doi:10.1124/dmd.112.048496.
Datta-Mannan A, Witcher DR, Lu J, Wroblewski VJ. Influence of improved FcRn binding on the subcutaneous bioavailability of monoclonal antibodies in cynomolgus monkeys. MAbs. 2012;4(2):267–73.
Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14(2):296–302. doi:10.1208/s12248-012-9340-y.
Meibohm B, Zhou H. Characterizing the impact of renal impairment on the clinical pharmacology of biologics. J Clin Pharmacol. 2012;52 Suppl 1:S54–62. doi:10.1177/0091270011413894.
Food and Drug Administration. Guidance for industry: immunogenicity assessment for therapeutic protein products (draft guidance). http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM338856.pdf. Published Feb 2013. Accessed 22 May 2013.
Goldsby RA, Kindt TJ, Osborne BA, Kuby J. Antigens. In: Goldsby RA, Kindt TJ, Kuby J, Osborne BA, editors. Immunology. 5th ed. New York: WH Freeman & Co Publishers; 2002. p. 57–75.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Bihorel, S., Fiedler-Kelly, J., Ludwig, E. et al. Population Pharmacokinetic Modeling of LY2189102 after Multiple Intravenous and Subcutaneous Administrations. AAPS J 16, 1009–1017 (2014). https://doi.org/10.1208/s12248-014-9623-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-014-9623-6