
Abstract
Direct-acting antiviral agents (DAAs) are established as the standard of care for chronic hepatitis C virus (HCV) infection. One of the newest additions to the HCV arsenal is an oral three-DAA combination therapy (i.e., the 3D regimen) that does not require concomitant use of pegylated interferon. The clinical development program for the 3D regimen has yielded a robust dataset that is inclusive of various dosing schemes and a diverse patient population. Using data from nine phase 1b/2a/2b studies that enrolled patients with HCV genotype 1 infection, population pharmacokinetic models were developed for each component of the 3D regimen (ombitasvir, paritaprevir, ritonavir, and dasabuvir) and for ribavirin, an adjunctive therapy used to enhance therapeutic efficacy in some populations. Formulation effects, accumulation, relative bioavailability, and interactions between DAAs were assessed during model development, and demographic and clinical covariates were identified and evaluated for their effects on drug exposures. Proposed models were assessed via goodness-of-fit plots, visual predictive checks, and bootstrap evaluations. Population pharmacokinetic models adequately described their respective plasma concentration-time data with precise and reliable model parameter estimates and with good predictive performance. Covariates, including age, sex, body weight, cytochrome P450 2C8 inhibitor use, non-Hispanic ethnicity, and creatinine clearance, were associated with apparent clearance and/or apparent volume parameters; however, the magnitude of effect on drug exposure was modest and not considered to be clinically significant. No patient-related or clinical parameters were identified that would necessitate dose adjustment of the 3D regimen in patients with HCV genotype 1 infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Direct-acting antiviral agents (DAAs) have become a mainstay of hepatitis C virus (HCV) infection treatment. They mark an improvement over the previous standard of care, pegylated interferon (peginterferon) and ribavirin, which was burdened by a poor sustained virologic response rate, lengthy duration of treatment, and treatment-limiting toxicities for many patients (1). Newer additions to the DAA treatment armamentarium are moving toward interferon-free therapy that is suitable for a broad range of patient types. One such combination, a recently approved, oral three-DAA combination therapy (i.e., the 3D regimen), has proven to be effective and well tolerated for the treatment of HCV genotype 1 infection in clinical trials that enrolled diverse patient groups, including difficult-to-treat populations, such as non-responders to previous treatment with peginterferon and ribavirin, patients with cirrhosis, and liver transplant recipients (2–8).
The 3D regimen consists of ombitasvir (formerly called ABT-267), a novel non-structural protein 5A inhibitor; paritaprevir (formerly called ABT-450), a potent non-structural protein 3/4A protease inhibitor; and dasabuvir (formerly called ABT-333), a non-nucleoside, non-structural protein 5B polymerase inhibitor. To enhance the exposure of paritaprevir, a cytochrome P450 (CYP) 3A4 substrate, it is administered with ritonavir (the combination denoted as paritaprevir/r), a potent CYP3A4 inhibitor. A two-DAA combination of ombitasvir, paritaprevir, and ritonavir (i.e., the 2D regimen) has also been evaluated for patients with HCV genotype 4 infection (9).
Phase 1b and 2a/b studies of 3D regimen components have included a variety of dosing regimens and combinations, a large number of both treatment-naive and treatment-experienced patients and patients representing a range of demographic and clinically important subgroups (10–13). This varied cross section allows for the assessment of parameters that influence drug exposures, which could ultimately affect the efficacy and safety profile of the regimen. In this analysis, pharmacokinetic models were developed for paritaprevir, ombitasvir, dasabuvir, ritonavir, and ribavirin using data from nine phase 1b and 2a/b studies. As part of the model-building process, formulation effects, accumulation, relative bioavailability, and interactions between DAAs were characterized. In addition, demographic, clinical covariates, and concomitant medication classes or enzyme/transporter inhibitor or inducer categories that influenced apparent clearance (CL/F) and/or apparent volume parameters were identified and evaluated for their effects on drug exposures. These data were used to inform drug dosage recommendations for phase 3 studies and to provide safety and efficacy margins that can be applied to clinical practice.
MATERIALS AND METHODS
Study Population
Adult men and women with chronic HCV genotype 1 infection who were enrolled in a phase 1b, 2a, or 2b study of paritaprevir/r, ombitasvir, and/or dasabuvir with or without ribavirin were included in this analysis. Patients were 18 to 70 years of age with a body mass index (BMI) in the range of 18 to 38 kg/m2 and were required to be in generally good health without evidence of cirrhosis and to have negative test results for HIV and hepatitis B infection. Seven of the studies excluded patients who had previously been treated with an investigational or commercially available anti-HCV drug. The remaining two studies included both treatment-naive patients and non-responders to previous treatment with peginterferon and ribavirin. Non-response was defined as treatment for ≥12 weeks that did not result in a decrease of ≥2 log10 IU/mL in HCV RNA load. All study participants provided written informed consent.
Study Design
Data were extracted from two phase 1b, six phase 2a, and one phase 2b studies. Study design features and treatment regimens for these studies are described in Table I. Study treatments included combinations of DAAs (paritaprevir/r, ombitasvir, and dasabuvir) or DAA monotherapy given with or without peginterferon and ribavirin or ribavirin alone. Drug dosing regimens ranged from 50/100 to 250/100 mg once daily for paritaprevir/r, 5 to 200 mg once daily for ombitasvir, and 100 mg once daily to 800 mg twice daily for dasabuvir. Ribavirin dose was determined by body weight: patients weighing <75 kg received 1000 mg daily and patients weighing ≥75 kg received 1200 mg daily per ribavirin prescribing information; both regimens were given in divided doses.
Two different formulations of paritaprevir were used in the studies: a spray-dried dispersion (SDD) tablet and a hard gelatin capsule. Data from phase 1 studies indicate differences in bioavailability between the two formulations; the formulation effect was fixed to the phase 1 population pharmacokinetic model (see Eq. 3) (14). Two different formulations for ombitasvir, SDD and hot-melt extrusion (HME) tablets, were used in study 1 (Table I). All other included studies used the HME tablet of ombitasvir. The relative bioavailability of ombitasvir from SDD tablets compared with HME tablets was estimated in the ombitasvir population pharmacokinetic model. Capsule and tablet formulations of dasabuvir used in phase 2 clinical trials have equivalent bioavailability, and thus, an effect was not estimated in the dasabuvir population pharmacokinetic model.
Study protocols were institutional review board approved, and study procedures were conducted in accordance with the ethical principles set forth in the Declaration of Helsinki, the International Conference on Harmonisation Good Clinical Practice guidelines, and local regulations.
Assessments
In the phase 1b/2a studies (Table I), dosing times for the intensive sampling period (i.e., study days 1–3) were based on observed study drug administration. For all other pharmacokinetic samples, drug dosing times were collected for the last dose and second-to-last dose, and pharmacokinetic times were calculated based on these dosing times. For the phase 2b study, which represents the majority of subjects (571/828; 69%) in the datasets, study drug doses on day 1 were administered on-site; therefore, exact dosing times were recorded. Thereafter, the medication event monitoring system (MEMS) was utilized. This method provides a detailed profile of patient’s adherence behavior and exact dosing times and is a well-accepted measure for adherence (15). The actual blood sample collection times were used for population pharmacokinetics analyses.
Blood samples for the assessment of paritaprevir, ombitasvir, dasabuvir, ritonavir, and ribavirin were processed and assayed using validated liquid chromatography methods with tandem mass spectrometric detection. Sample collection times varied according to the protocol specifications for each study; the duration of sampling is summarized in Table I. Lower limits of quantification ranges for each analyte were 0.500 to 0.644 ng/mL (paritaprevir), 0.139 to 0.993 ng/mL (ombitasvir), 0.100 to 4.58 ng/mL (dasabuvir), 4.74 to 5.00 ng/mL (ritonavir), and 91 to 100 ng/mL (ribavirin).
Pharmacokinetic Model Development and Statistical Analyses
Population pharmacokinetic models were built using non-linear mixed-effects models in NONMEM 7.3 (ICON Development Solutions, Hanover, MD). A first-order conditional estimation method with η-ε interaction (FOCE-INT) was used for model building. A base model, including the models for the inter-individual and residual variabilities, was developed first that defined the structural model, followed by investigation of relevant covariate-parameter relationships. Model development was guided by goodness-of-fit plots, likelihood ratio tests (significance level of p < 0.01 for base model development), physiologic reasonability, precise parameter estimates, and previous knowledge of drug pharmacokinetics.
Covariates were tested using the forward inclusion and backward elimination procedure. For CL/F, the tested covariates included age, sex, race (black versus non-black), ethnicity (Hispanic/Latino versus non-Hispanic/Latino), body weight (BWT), body surface area, BMI, HCV subtype 1a or 1b, baseline creatinine clearance (CrCL), fibrosis (no/minimal, moderate, or severe), treatment experience (naive or peginterferon/ribavirin experienced), use of ribavirin, and co-medications. For apparent volume parameters, the tested covariates included age, sex, race, ethnicity, BWT, body surface area, and BMI. Covariate effects were added in the model in a multiplicative manner. Continuous covariates were normalized to a reference value (median value of the covariate) and were included in the model with a power function:
where θ is the population estimate of the pharmacokinetic model parameter (TVP). COV i refers to the ith individual continuous covariate value (e.g., BMI), COVmedian is the median value for the study population, and θ COV refers to the estimated covariate effect.
Categorical covariates were tested with a multiplicative model to obtain the fractional difference in the pharmacokinetic parameters between the tested categorical groups:
where θ is the population estimate of the pharmacokinetic model parameter (TVP) and θ levelcat was set to 1 for the reference population and to an estimated parameter for other levels of the categorical covariate.
Inferences about the clinical importance of covariate effects were made based on the magnitude and precision of covariate parameter estimates. The magnitude of covariate effect was assessed by estimating steady-state maximum plasma concentration (C max) and area under the plasma concentration-time curve (AUC) values for each significant covariate. Exposure data were calculated for the approved doses of paritaprevir (150 mg once daily), ombitasvir (25 mg once daily), dasabuvir (400 mg twice daily formulation used in the phase 2 studies; it is equivalent to 250 mg twice daily approved formulation), ritonavir (100 mg once daily) (16), and ribavirin (1000 or 1200 mg/day based on weight) (17,18) in patients with HCV genotype 1 infection. For the stepwise covariate selection, significance levels of α = 0.01 and α = 0.001 (approximate χ 2 distribution) were implemented in forward inclusion and backward elimination procedures, respectively. Several criteria were used to evaluate improvement in model performance and to select the final model. The likelihood ratio test was used to discriminate among alternative nested models. One additional model parameter, corresponding to 1 degree of freedom in the higher-order model, was considered significant when it lowered the objective function value by more than 6.63, corresponding to p < 0.01. For 2 degrees of freedom, the required reduction in objective function value was 9.21.
An extensive list of drug classes and enzyme/transporter inhibitor or inducer categories of co-medications was evaluated for effects on CL/F. Components of the 3D regimen are substrates or inhibitors for a number of metabolic enzymes and transport proteins, including various CYP family members, P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), organic anion transporting polypeptides 1B1 and 1B3 (OATP1B1/B3), and uridine diphosphate glucuronosyltransferase (UGT) 1A1 (19). Co-medication use was defined as receipt for ≥6 weeks (approximately half the treatment duration [12 weeks] of a typical HCV DAA regimen) during the treatment period. The potential for a drug class or category to influence the pharmacokinetics of DAAs, ritonavir, or ribavirin was determined by calculating dose-normalized steady-state exposures (AUC during a 24-hour dosing interval at steady state [AUC24,ss]) from the base models for patients receiving each drug class or inhibitor/inducer category and comparing these values with control groups (i.e., patients receiving no co-medications or co-medications that do not represent the tested drug inhibitor/inducer category). For paritaprevir, only the post hoc AUC24,ss values (from the base model) representing the approved 150-mg dose and SDD tablets were considered based on the drug’s known dose- and formulation-dependent non-linear bioavailability (14). Co-medication drug classes/categories that included ≥15 patients and met the base model AUC24,ss ratio criteria (≤0.5 or ≥2.0 for drug classes, ≤0.5 for inducer categories, or ≥2.0 for inhibitor categories) were included in covariate model building.
The population pharmacokinetic model for paritaprevir was developed in two steps. In step 1, non-linearity in bioavailability (dose- and formulation- dependent) and accumulation after multiple doses were characterized by using intensive pharmacokinetic data from phase 1 studies in healthy volunteers (14). In step 2, only the phase 2 pharmacokinetic data from patients were characterized by fixing formulation effect and accumulation factor to the estimates from the model fit of the phase 1 data. The dose- and formulation-dependent non-linear paritaprevir relative bioavailability (FREL) was described by
Formulation effect (θ capsule) was fixed to the phase 1 population pharmacokinetic model estimate of 1.27 during phase 2 model development (14). The parameter θ bio that represents the fold increase in paritaprevir bioavailability of the SDD tablet for each 50-mg increment in dose relative to bioavailability of the 50-mg paritaprevir SDD tablet was estimated. In addition, the effect of dasabuvir on paritaprevir bioavailability was implemented in a categorical manner to obtain the fractional difference in paritaprevir bioavailability with or without dasabuvir. The effect of ombitasvir on paritaprevir bioavailability was tested in a phase 1 pharmacokinetic model and was found to be not significant (14). Therefore, testing the effect of ombitasvir was not pursued in this analysis.
Model Evaluation
Methods used in model evaluation included goodness-of-fit plots (data not shown), visual predictive checks, and bootstrap evaluations. For visual predictive checks, 1000 simulated replicates of the model predictions were generated using NONMEM. Relevant visual predictive checks included plots of observed and predicted concentrations versus time. Observed medians and 5th and 95th percentiles were plotted against the 90% prediction interval obtained from these simulations for every unique observation time point. In addition to visual predictive checks, 1000 bootstrap replicates were constructed by randomly sampling (with replacement) N patients from the original dataset, where N is the number of patients in the original dataset. The final model was used to estimate population parameters for each bootstrap replicate, and the resulting values were used to estimate medians and 2.5th and 97.5th percentiles. Final model parameter estimates based on the original dataset were compared against the bootstrap results.
RESULTS
Data Sources
Paritaprevir and ritonavir assessments were based on 7698 and 7655 concentration observations, respectively, that were derived from 676 patients enrolled in five phase 1b/2a/2b studies. Five phase 1b/2a/2b studies contributed to the 6813 concentration observations from 601 patients available for ombitasvir. Data for dasabuvir were obtained from six phase 1b/2a/2b studies that yielded 6479 concentration observations from 560 patients. Ribavirin pharmacokinetic information was extracted from six phase 1b/2a/2b studies that included 6977 concentration observations from 670 patients.
Patient Demographic Characteristics
Demographic characteristics were generally consistent among populations used to evaluate the various study drugs (Table II). The majority of patients were male (55–58%) and non-black (86–88%). Hispanic/Latino ethnicity was self-described by 10 to 13% of patients. Moderate or severe liver fibrosis was present in fewer than half of the patients. Approximately one fifth of the patients were treatment experienced, having demonstrated a poor response to previous treatment with peginterferon and ribavirin. The dominant HCV genotype was 1a, representing 67 to 70% of the evaluated study population.
Co-medication Analyses
More than 750 co-medications belonging to 15 drug classes (non-opioid analgesics, antihypertensives, antidepressants, proton pump inhibitors, antihistamines, opioids, hormone replacement therapies, steroids, anti-infectives, antidiabetics, antiepileptics, statins and lipid-lowering agents, antipsychotics, hormonal contraceptives, and phosphodiesterase type 5 inhibitors) and 20 enzyme (CYP isoenzymes 3A4/5/7, 2C8, 2D6, 1A2, 2C9, 2C19, 2B6, and UGT) or transporter (P-gp, multidrug resistance-associated protein 2, BCRP, OATP1B1/B3) inhibitor/inducer categories were used concomitantly in phase 1b/2a/2b trials. After applying informed selection criteria (n ≥ 15 and AUC24,ss ratio of ≤0.5 or ≥2.0 for drug classes, ≤0.5 for inducer categories, or ≥2.0 for inhibitor categories), only co-medications representing the drug class of hormone replacement therapy (n = 18; AUC24,ss ratio, 2.10) and the category of CYP2C8 inhibitors (n = 16; AUC24,ss ratio, 2.17) were included in stepwise covariate model building for paritaprevir. None of the other drug classes or categories met the selection criteria for ombitasvir, dasabuvir, ritonavir, or ribavirin.
Paritaprevir Population Pharmacokinetics
A one-compartment model with first-order absorption and elimination adequately described paritaprevir concentration-time data. The non-linearity in bioavailability, the accumulation compared with the first dose, and the effect of dasabuvir on paritaprevir bioavailability that was identified in a previous analysis of phase 1 data (14) were implemented as part of the phase 2 pharmacokinetic model.
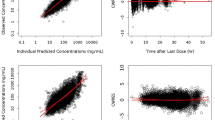
The bioavailability of paritaprevir increased supraproportionally and was formulation dependent (Fig. 1). Exposures from SDD tablets for the 150-mg dose (approved dose) of paritaprevir would be approximately 6.3-fold of exposures from the 50-mg dose and approximately 71% lower than exposures from the 250-mg dose. On average, co-administration of dasabuvir with paritaprevir increased paritaprevir bioavailability by 21% in HCV genotype 1-infected patients. Although this effect was not significant, it was retained in the model based on results of phase 1 pharmacokinetic modeling (14).

Dose-related changes in relative bioavailability of paritaprevir for the spray-dried dispersion (SDD) tablet and hard gelatin capsule (HGC) formulations
Model-derived population pharmacokinetic parameter estimates and significant covariates are presented in Table III. Sex and CYP2C8 inhibitor use were found to be covariates significantly associated with CL/F, and ethnicity was a significant covariate for apparent volume of the central compartment (Vc/F). Because of the known influence of dasabuvir on paritaprevir bioavailability, co-administration of dasabuvir was included in the assessment of predicted paritaprevir exposures in covariate subgroups. Among patients receiving CYP2C8 inhibitors (n = 39), CL/F was 32% lower than patients not on any inhibitor or inducer of metabolic enzyme(s) or transporter(s). Females had 19% lower CL/F than males, and patients representing non-Hispanic/Latino ethnicity had 35% higher Vc/F than patients of Hispanic/Latino ethnicity. Changes in predicted paritaprevir steady-state C max and AUC24 based on sex, ethnicity, use of CYP2C8 inhibitors, or co-administration of dasabuvir were generally modest (≤50%) (Fig. 2). The greatest putative AUC elevation (50%) was estimated for use of a concomitant CYP2C8 inhibitor.

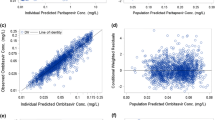
Population pharmacokinetic evaluations. Model-derived estimates of steady-state maximum plasma concentration (C max ) and area under the plasma concentration-time curve (AUC) for covariates identified as significantly associated with apparent clearance or apparent volume parameters for paritaprevir (150 mg once daily), ombitasvir (25 mg once daily), dasabuvir (400 mg twice daily), ritonavir (100 mg once daily), and ribavirin (600 mg twice daily). A ratio of 1.0 indicates similar values between the subgroup analyzed and the rest of the patient population. Error bars represent the 95% confidence intervals. Weight groups for ombitasvir analyses were stratified as 89 and 69 kg, and age was plotted for 58 and 38 years. Weight groups for ribavirin analyses were stratified as 89 and 69 kg, and creatinine clearance (CrCL) was plotted for 65 and 120 mL/min. BWT body weight, CYP2C8 cytochrome P450 2C8
Ombitasvir Population Pharmacokinetics
A two-compartment model with first-order absorption and elimination adequately described ombitasvir plasma concentration-time data. The bioavailability of ombitasvir from an SDD tablet relative to a HME tablet in patients with HCV genotype 1 infection was 41%. Age, sex, and BWT were found to be significant covariates for CL/F, and sex was found to be the sole significant covariate for volume parameters (Vc/F and apparent volume of the peripheral compartment [Vp/F]; Table III). On average, females had 30% lower CL/F and 17% lower apparent steady state volume of distribution (Vss/F) than males. Changes in ombitasvir steady-state Cmax and AUC24 values based on age (±10 years change from reference age [48 years] in the model), sex, and BWT (±10 kg change from reference BWT [79 kg] in the model) were modest (<50%) (Fig. 2). The most notable difference was between males and females, with 34% higher C max and 42% higher AUC24 values estimated for females compared with males.
Dasabuvir Population Pharmacokinetics
A one-compartment model with first-order absorption and elimination adequately described dasabuvir plasma concentration-time data. Sex was the only covariate that had a significant effect on CL/F; Vc/F was not influenced by any of the tested covariates (Table III). Females had 15% lower CL/F than males. Predicted changes in dasabuvir steady-state C max and AUC24 values based on sex were minimal (<20%) (Fig. 2).
Ritonavir Population Pharmacokinetics
A one-compartment model with first-order absorption and elimination adequately described ritonavir plasma concentration-time data. Ethnicity was the only covariate tested that demonstrated a significant effect on Vc/F (p < 0.01; Table III). Vc/F was 79% higher in non-Hispanic/Latino patients compared with Hispanic/Latino patients. However, Hispanic versus non-Hispanic ethnicity only minimally affected (≤10% change) ritonavir steady-state C max and AUC24 values (Fig. 2).
Ribavirin Population Pharmacokinetics
A three-compartment model with first-order absorption and elimination adequately described ribavirin plasma concentration-time data. Sex and baseline CrCL were found to be significant covariates for CL/F, and sex and BWT for the volume parameters (Table III). On average, females had 11% lower CL/F and 22% lower Vss/F than males. The covariates of baseline CrCL, sex, and BWT altered ribavirin steady-state Cmax and AUC24 values by less than 20% (Fig. 2).
The low computed η-shrinkage values (≤10%) for CL/F and volume parameters in the final models for paritaprevir, ombitasvir, dasabuvir, ritonavir, and ribavirin indicate that sufficient information about the parameter estimates was available from the drug concentration-time data. No substantial ε-shrinkage (≤5%) was computed, indicating the adequacy of the models. Goodness-of-fit plots (not shown), visual predictive checks (Fig. 3), and bootstrap analyses (Table III) indicate that the data have been well described by the pharmacokinetic models for paritaprevir, ombitasvir, dasabuvir, ritonavir, and ribavirin, with good predictive performance for each.

Visual predictive checks for population pharmacokinetic models. Filled circles represent observed median values (5th and 95th percentile error bars). The simulated median is represented by a solid line and the associated 90% interval of the simulated median by grey shading. The simulated 5th and 95th percentiles are represented by dashed lines and the associated 90% interval of the simulated 5th and 95th percentile by grey shading. The profile for ribavirin starts 2 weeks into the treatment period, thus illustrating steady-state
DISCUSSION
Direct-acting antiviral combinations of ombitasvir and paritaprevir/r with or without dasabuvir are established treatment options in the HCV armamentarium. Clinical trial data demonstrate high sustained virologic response rates across a variety of patient populations, including those who are treatment naive, treatment experienced, cirrhotic, non-cirrhotic, and/or liver transplant recipients (2–7,9). With the potential for broad use of the 3D and 2D regimens, it is important to establish that the therapy would be comparably effective and well tolerated regardless of patient-related variables, such as the use of concomitant medications and demographic characteristics. Given the large dataset and rich pharmacokinetic sampling of the 3D development program, these insights could be generated through the development of population pharmacokinetic models for ombitasvir, paritaprevir, dasabuvir, and ritonavir, as well as the adjunctive medication ribavirin. Data from our assessment show that population pharmacokinetic models adequately describe their respective plasma concentration-time data with precise and reliable model parameter estimates and with good predictive performance.
One of the observations that arose from the population modeling was the comparable pharmacokinetics of paritaprevir, ombitasvir, dasabuvir, ritonavir, and ribavirin in HCV treatment-naive and treatment-experienced patients. Treatment history was included as a potential covariate but was not identified in either the univariate analysis or the subsequent refinement process as significantly associated with CL/F for any component of the 3D regimen or ribavirin. These findings are robust as the study population included substantial representation of treatment-naive and treatment-experienced patients. Analogous pharmacokinetics are consistent with the comparable efficacy between treatment-naive and treatment-experienced patient populations that have been reported for both the 3D and 2D regimens (4,9,12). In clinical trials, the drug dosage regimen and treatment duration were the same, regardless of patient treatment history.
The modeling analyses also revealed no difference in the pharmacokinetics of paritaprevir, ombitasvir, dasabuvir, ritonavir, or ribavirin between patients with HCV genotype 1a or 1b infection. HCV subtype was not identified as a significant covariate in any of the models. Clinical trials of the 3D regimen have shown high rates of sustained virologic response in patients with either HCV genotype 1a or 1b infection (3–7), and 3D treatment is indicated for both HCV subtypes (16).
Patients with cirrhosis were excluded from the trials that comprised our population pharmacokinetic analysis. However, fibrosis score was collected and included as a covariate. Fibrosis did not show any significant association with clearance or volume parameters. Notably, the degree of fibrosis among these patients was generally classified as mild or moderate in severity. A recent pharmacokinetic study specifically evaluating the 3D regimen in patients with hepatic impairment found that exposure changes for paritaprevir, ombitasvir, dasabuvir, and ritonavir were minimal and did not necessitate dose adjustment for patients with mild or moderate hepatic impairment (20). The 3D regimen is currently not recommended for patients with moderate hepatic impairment (16). Patients with severe hepatic impairment, however, did experience higher paritaprevir and dasabuvir exposures, which would preclude use of the 3D regimen in this population.
The only drug or inhibitor/inducer class with a significant influence on clearance was CYP2C8 inhibitors, which reduced CL/F relative to patients who did not receive any inhibitor or inducer of metabolic enzymes or transporters. The co-medication analysis is subject to several limitations based on the design of the studies and the need to best utilize the available data. Perhaps, the most salient of these relates to the timing and duration of concomitant medication use. A 6-week treatment criterion was utilized as it represents half of the treatment period for 3D regimen therapy. It is possible that concomitant medications used over a shorter duration could have influenced pharmacokinetic findings. It is also likely that patients who did not continue a concomitant medication regimen throughout the study would have had some pharmacokinetic samples taken during co-medication use and one or more without. However, we believe that the analysis as performed is a valid approach to a large and complex dataset. More specific information on the influence of concomitant medications can be gained from formal drug-drug interaction studies, a number of which have been conducted with agents representing different enzyme or transporter substrates and inhibitor/inducer classes. Descriptions of these study results have recently been published (19,21–23) and can be used as a guide for dosing of concomitant medications with the 3D regimen.
Ribavirin may be added to 3D or 2D therapy to augment therapeutic efficacy. It is needed for patients with genotype 1a HCV and patients with genotype 4 HCV (16, 24, 25). The population pharmacokinetic modeling indicates that ribavirin co-administration does not affect the pharmacokinetics of paritaprevir, ombitasvir, dasabuvir, or ritonavir; use of ribavirin was not identified as a covariate in any of the analyses. This is consistent with the fact that ribavirin is primarily eliminated via renal pathway and does not share common disposition pathways with paritaprevir, ombitasvir, dasabuvir, or ritonavir and is, therefore, not expected to contribute to drug interactions. Hence, no dose adjustment is necessary when ribavirin is given with the 3D regimen. The estimated CL/F and calculated apparent steady-state volume of distribution and t1/2,z are in close agreement with the values reported in the literature (26,27). Other estimated population parameters for ribavirin from our assessment were also in good agreement with published results in patients with chronic HCV genotype 1 infection (27). A notable difference between the previous population pharmacokinetic model and our findings was a 50% greater Vp/F for black versus white patients in the previous study (27). No such relationship was observed in our dataset. In our evaluation, sex and BWT were covariates for volume parameters, and sex and baseline CrCL for CL/F. Demographic distribution may account for some of the differences between studies; for example, the analyses by Jin and colleagues (27) included only 39 females (27% of their study population), whereas the present evaluation included 287 female patients. Jin and colleagues (27) did not assess CrCL because 85% of the participants had normal renal function, and decrements were generally mild in those outside the reference range. Our finding of an influence of CrCL is not unexpected, given the known effect of renal impairment on the pharmacokinetics of ribavirin, a renally cleared compound (28–30).
Calculation of steady-state exposure (C max and AUC) values for patient groups identified as significant covariates for CL/F or apparent volume parameters demonstrated generally modest variance from the overall population estimates. The resulting changes in exposure were within 0.5- to 2.0-fold of exposures from approved doses. A change in the exposures within a window of 0.5- to 2.0-fold from the population mean exposures for all three DAAs (paritaprevir, ombitasvir, dasabuvir) are not anticipated to alter the efficacy or safety profile to an extent that requires dose adjustments. Doses of ombitasvir up to 200 mg once daily, paritaprevir/r up to 250/100 mg once daily, and dasabuvir up to 800 mg twice daily, which are associated with substantially higher exposures than doses used for HCV treatment, produce comparable efficacy and safety profiles to that of the studied regimen (ombitasvir/paritaprevir/r, 25/150/100 once daily, and dasabuvir, 400 mg twice daily) (10–12). Moreover, no differences in virologic response rates for the 3D regimen have been observed based on age, sex, race, ethnic group, fibrosis score, or baseline viral load in phase 3 clinical trials (2–6). Interleukin 28B genotype, which is known to influence antiviral efficacy in patients with HCV (31), was associated with response rate in one phase 3 study (3), but was not significant in the other phase 3 studies (2,4–6). A pharmacokinetic study of ombitasvir and paritaprevir/r with or without dasabuvir in patients with mild, moderate, or severe renal impairment detected no clinically significant influence of decreased CrCL on DAA exposures (32). Together, these data support standard dosing of the 3D regimen across patient groups. Dosing of ribavirin, however, may require adjustment (17,18). Ribavirin dose modification is recommended for patients who experience adverse reactions or laboratory abnormalities during treatment with ribavirin. A priori ribavirin dose reduction is also advised for patients with renal impairment.
The strength of these analyses lies in the breadth of pharmacokinetic data available for assessment. However, although the dataset represents a wide range of dose combinations, not every possible permutation of the five drugs involved could be tested clinically. Also, the 2D regimen is used in the treatment of HCV genotype 4 infection, but this population pharmacokinetic model was developed based on observations in patients with HCV genotype 1 infection.
CONCLUSION
Using data from the robust clinical development program for the 3D regimen, population pharmacokinetic models were developed for paritaprevir, ombitasvir, dasabuvir, ritonavir, and ribavirin. The models for each drug adequately described the respective plasma concentration-time data with precise and reliable model parameter estimates and with good predictive performance. Based on the model-predicted, steady-state drug exposures at doses used in the treatment of HCV genotype 1 infection, no patient-related or clinical parameters were identified that would necessitate dose adjustment for the 3D regimen.
References
Thompson JR. Emerging therapeutic options for the management of hepatitis C infection. World J Gastroenterol. 2014;20(23):7079–88. doi:10.3748/wjg.v20.i23.7079.
Andreone P, Colombo MG, Enejosa JV, Koksal I, Ferenci P, Maieron A, et al. ABT-450, ritonavir, ombitasvir, and dasabuvir achieves 97% and 100% sustained virologic response with or without ribavirin in treatment-experienced patients with HCV genotype 1b infection. Gastroenterology. 2014;147(2):359–65. doi:10.1053/j.gastro.2014.04.045. e1.
Ferenci P, Bernstein D, Lalezari J, Cohen D, Luo Y, Cooper C, et al. ABT-450/r-ombitasvir and dasabuvir with or without ribavirin for HCV. N Engl J Med. 2014;370(21):1983–92. doi:10.1056/NEJMoa1402338.
Poordad F, Hezode C, Trinh R, Kowdley KV, Zeuzem S, Agarwal K, et al. ABT-450/r-ombitasvir and dasabuvir with ribavirin for hepatitis C with cirrhosis. N Engl J Med. 2014;370(21):1973–82. doi:10.1056/NEJMoa1402869.
Feld JJ, Kowdley KV, Coakley E, Sigal S, Nelson DR, Crawford D, et al. Treatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 2014;370(17):1594–603. doi:10.1056/NEJMoa1315722.
Zeuzem S, Jacobson IM, Baykal T, Marinho RT, Poordad F, Bourliere M, et al. Retreatment of HCV with ABT-450/r-ombitasvir and dasabuvir with ribavirin. N Engl J Med. 2014;370(17):1604–14. doi:10.1056/NEJMoa1401561.
Kwo PY, Mantry PS, Coakley E, Te HS, Vargas HE, Brown Jr R, et al. An interferon-free antiviral regimen for HCV after liver transplantation. N Engl J Med. 2014;371(25):2375–82. doi:10.1056/NEJMoa1408921.
Eron JJ, Lalezari J, Slim J, Gathe J, Ruane PJ, Wang C, et al. Safety and efficacy of ombitasvir - 450/r and dasabuvir and ribavirin in HCV/HIV-1 co-infected patients receiving atazanavir or raltegravir ART regimens. J Int AIDS Soc. 2014;17(4 Suppl 3):19500. doi:10.7448/IAS.17.4.19500.
Hezode C, Asselah T, Reddy KR, Hassanein T, Berenguer M, Fleischer-Stepniewska K, et al. Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment-naive and treatment-experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL-I): a randomised, open-label trial. Lancet. 2015. doi:10.1016/S0140-6736(15)60159-3.
Sullivan GJ, Rodrigues-Torres M, Lawitz E, Poordad F, Kapoor M, Campbell A, et al. ABT-267 combined with pegylated interferon alpha-2a/ribavirin in genotype 1 (GT1) HCV-infected treatment-naive subjects: 12 week antiviral and safety analysis. J Hepatol. 2012;56 Suppl 2:S480.
Poordad F, Lawitz E, Kowdley KV, Cohen DE, Podsadecki T, Siggelkow S, et al. Exploratory study of oral combination antiviral therapy for hepatitis C. N Engl J Med. 2013;368(1):45–53. doi:10.1056/NEJMoa1208809.
Kowdley KV, Lawitz E, Poordad F, Cohen DE, Nelson DR, Zeuzem S, et al. Phase 2b trial of interferon-free therapy for hepatitis C virus genotype 1. N Engl J Med. 2014;370(3):222–32. doi:10.1056/NEJMoa1306227.
Krishnan P, Beyer J, Mistry N, Koev G, Reisch T, DeGoey D, et al. In vitro and in vivo antiviral activity and resistance profile of ombitasvir, an inhibitor of hepatitis C virus NS5A. Antimicrob Agents Chemother. 2015;59(2):979–87. doi:10.1128/AAC.04226-14.
Polepally A, Mensing S, Khatri A, Liu W, Awni W, Menon R, et al. Dose- and formulation-dependent non-linear pharmacokinetic model of ABT-450, a protease inhibitor for the treatment of HCV: combined analyses from 12 phase 1 studies. J Pharmacokinet Pharmacodyn. 2014;41(Suppl):S7–S101.
Farmer KC. Methods for measuring and monitoring medication regimen adherence in clinical trials and clinical practice. Clin Ther. 1999;21(6):1074–90.
Viekira Pak (ombitasvir, paritaprevir, and ritonavir tablets; dasabuvir tablets) [prescribing information]. North Chicago, IL: AbbVie Inc.; 2015.
Copegus (ribavirin) tablets [prescribing information]. South San Francisco, CA: Genentech USA, Inc.; 2013.
Rebetol (ribavirin USP) capsules [prescribing information]. Whitehouse Station, NJ: Merck Sharp & Dohme Corp; 2013.
Menon RM, Badri PS, Wang T, Polepally AR, Zha J, Khatri A, et al. Drug-drug interaction profile of the all-oral anti-hepatitis C virus regimen of paritaprevir/ritonavir, ombitasvir, and dasabuvir. J Hepatol. 2015;63(1):20–9. doi:10.1016/j.jhep.2015.01.026.
Khatri A, Menon RM, Marbury TC, Lawitz EJ, Podsadecki TJ, Mullally VM, et al. Pharmacokinetics and safety of co-administered paritaprevir plus ritonavir, ombitasvir, and dasabuvir in hepatic impairment. J Hepatol. 2015;63(4):805–12. doi:10.1016/j.jhep.2015.05.029.
Badri PS, King JR, Polepally AR, McGovern BH, Dutta S, Menon RM. Dosing recommendations for concomitant medications during 3D anti-HCV therapy. Clin Pharmacokinet. 2015. doi:10.1007/s40262-015-0317-8.
Badri PS, Dutta S, Wang H, Podsadecki TJ, Polepally AR, Khatri A, et al. Drug interactions with the direct-acting antiviral combination of ombitasvir and paritaprevir/ritonavir (2D regimen). Antimicrob Agents Chemother. 2015. doi:10.1128/AAC.01778-1 .
Polepally AR, King JR, Ding B, Shuster DL, Dumas EO, Khatri A, et al. Drug-drug interactions of commonly used medications with direct acting antiviral HCV combination therapy of paritaprevir/r, ombitasvir and dasabuvir. Presented at the 16th International Workshop on Clinical Pharmacology of HIV and Hepatitis Therapy, Washington, DC, 26–28 May 2015.
Viekirax 12.5 mg/75 mg/50 mg film-coated tablets [Summary of product characteristics]. Berkshire, United Kingdom: AbbVie Limited; 2015.
Feld JJ, Moreno C, Trinh R, Tam E, Bourgeois S, Horsmans Y, et al. Sustained virologic response of 100% in HCV genotype 1b patients with cirrhosis receiving ombitasvir/paritaprevir/r and dasabuvir for 12 weeks. J Hepatol. 2015. doi:10.1016/j.jhep.2015.10.005.
Wade JR, Snoeck E, Duff F, Lamb M, Jorga K. Pharmacokinetics of ribavirin in patients with hepatitis C virus. Br J Clin Pharmacol. 2006;62(6):710–4. doi:10.1111/j.1365-2125.2006.02704.x.
Jin R, Fossler MJ, McHutchison JG, Howell CD, Dowling TC. Population pharmacokinetics and pharmacodynamics of ribavirin in patients with chronic hepatitis C genotype 1 infection. AAPS J. 2012;14(3):571–80. doi:10.1208/s12248-012-9368-z.
Gupta SK, Kantesaria B, Glue P. Pharmacokinetics and safety of single-dose ribavirin in patients with chronic renal impairment. Drug Discov Ther. 2013;7(4):158–63.
Brennan BJ, Wang K, Blotner S, Magnusson MO, Wilkins JJ, Martin P, et al. Safety, tolerability, and pharmacokinetics of ribavirin in hepatitis C virus-infected patients with various degrees of renal impairment. Antimicrob Agents Chemother. 2013;57(12):6097–105. doi:10.1128/AAC.00608-13.
Gupta SK, Kantesaria B, Glue P. Exploring the influence of renal dysfunction on the pharmacokinetics of ribavirin after oral and intravenous dosing. Drug Discov Ther. 2014;8(2):89–95.
Thompson AJ, Muir AJ, Sulkowski MS, Ge D, Fellay J, Shianna KV, et al. Interleukin-28B polymorphism improves viral kinetics and is the strongest pretreatment predictor of sustained virologic response in genotype 1 hepatitis C virus. Gastroenterology. 2010;139(1):120–9. doi:10.1053/j.gastro.2010.04.013. e18.
Khatri A, Dutta S, Marbury T, Preston RA, Rodrigues L, Wang H, et al. The pharmacokinetics and safety of the direct acting antiviral regimen of ABT-450/r, ombitasvir with/without dasabuvir in subjects with mild, moderate and severe renal impairment compared to subjects with normal renal function. Boston: 65th Annual Meeting of the American Association for the Study of Liver Diseases; 2014.
Acknowledgments
This work was supported by AbbVie Inc. AbbVie contributed to the study design, research, and interpretation of data and the writing, reviewing, and approving of the publication. The authors thank Crystal Murcia, PhD, of The JB Ashtin Group, Inc., for the assistance in preparing this manuscript for publication.
Author Contributions
SM and ARP equally contributed to this work. SM, ARP, DK, AK, WL, RMM, and SD contributed to the data analyses. All authors contributed to the study designs, interpretation of results, and writing and reviewing of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
All authors are AbbVie employees and may hold AbbVie stocks or options.
Additional information
Sven Mensing and Akshanth R. Polepally contributed equally to this work.
Rights and permissions
About this article

Cite this article
Mensing, S., Polepally, A.R., König, D. et al. Population Pharmacokinetics of Paritaprevir, Ombitasvir, Dasabuvir, Ritonavir, and Ribavirin in Patients with Hepatitis C Virus Genotype 1 Infection: Combined Analysis from 9 Phase 1b/2 Studies. AAPS J 18, 270–280 (2016). https://doi.org/10.1208/s12248-015-9846-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1208/s12248-015-9846-1