

Abstract
Background: The second-generation antipsychotic drug olanzapine is an effective pharmacological treatment for psychosis. However, use of the drug is commonly associated with a range of metabolic side effects, including glucose intolerance and insulin resistance. These symptoms have been accurately modelled in rodents.
Methods: We compared the effects of 3 distinct classes of antidiabetic drugs, metformin (100 and 500 mg/kg, oral), rosiglitazone (6 and 30 mg/kg, oral) and glyburide (2 and 10 mg/kg, oral), on olanzapine-induced metabolic dysregulation. After acutely treating female rats with lower (7.5 mg/kg) or higher (15 mg/kg) doses of olanzapine, we assessed glucose intolerance using the glucose tolerance test and measured insulin resistance using the homeostatic model assessment of insulin resistance equation.
Results: Both doses of olanzapine caused pronounced glucose dysregulation and insulin resistance, which were significantly reduced by treatment with metformin and rosiglitazone; however, glucose tolerance did not fully return to control levels. In contrast, glyburide failed to reverse the glucose intolerance caused by olanzapine despite increasing insulin levels.
Limitations: We evaluated a single antipsychotic drug, and it is unknown whether other antipsychotic drugs are similarly affected by antidiabetic treatments.
Conclusion: The present study indicates that oral hypoglycemic drugs that influence hepatic glucose metabolism, such as metformin and rosiglitazone, are more effective in regulating olanzapine-induced glucose dysregulation than drugs primarily affecting insulin release, such as glyburide. The current model may be used to better understand the biological basis of glucose dysregulation caused by olanzapine and how it can be reversed.
Introduction
Second-generation antipsychotics (SGAs; also known as atypical antipsychotics) are effective pharmacological treatments for psychotic conditions, including schizophrenia and bipolar disorder.1 On- and off-label use of SGAs has increased in recent years to include additional indications, such as mood and anxiety disorders.2 The widespread use of these drugs has been ascribed to their lower propensity to induce neurologic side effects, such as extrapyramidal symptoms, compared with first-generation antipsychotics.3 Importantly though, the past decade of clinical research has reported that most SGAs can cause serious metabolic side effects, resulting in a metabolic syndrome that substantially increases the risk for cardiometabolic disorders, such as type II diabetes mellitus and cardiovascular disease.4–6 The identifying characteristics of metabolic syndrome are weight gain, hypertension, hyperlipidemia, hyperglycemia, glucose intolerance and insulin resistance.7
Despite the similarity of SGA-induced metabolic syndrome to other forms of prediabetes, the paucity of knowledge about the underlying physiology of the condition has hindered the development of optimal treatment strategies for controlling metabolic dysregulation. Nevertheless, health care providers have recognized the serious nature of SGA-induced metabolic side effects and have sought to ameliorate them through various interventions.8 Consistent with the literature on type II diabetes mellitus, some success has been obtained through lifestyle changes, including exercise and dietary modifications.9 However, these changes may be more challenging in the psychiatric population,10 therefore the mainstay of treatment remains the use of antidiabetic drugs. A number of different antidiabetic drugs are currently used to treat metabolic syndrome11 and type II diabetes mellitus, but unlike antipsychotic drugs that all work primarily through a similar mechanism in regards to clinical efficacy (blockade of dopamine D2 receptors12), the antidiabetic drugs operate through diverse physiological pathways. For instance, the efficacy of metformin (a biguanide) is mediated in part by an AMP-dependent kinase (AMPK) signalling pathway, which does not directly stimulate insulin secretion.13,14 The main mode of action for rosiglitazone (a thiazolidinedione) involves the activation of the peroxisome proliferator-activated receptor γ (PPARγ), a nuclear transcriptional protein that belongs to the family of PPARs, which regulate genes involved in lipid and glucose metabolism.15 Rosiglitazone-induced acute effects are also independent of direct insulin release.16 In contrast to both metformin and rosiglitazone, glyburide (a sulfonylurea) directly increases insulin secretion in the pancreas by inhibiting the ATP-sensitive potassium channel in β cells.17 It is therefore important to determine whether specific classes of antidiabetic drugs are more efficacious in treating SGA-induced metabolic syndrome, as this form of metabolic dysregulation may be more or less sensitive to individual antidiabetic drugs.
The symptoms of metabolic syndrome can be modelled in rodents, and preclinical paradigms have reliably reproduced many of the metabolic symptoms of SGAs observed in humans.18–20 We and others have previously shown that 2 of the key symptoms of SGA-induced metabolic dysregulation (i.e., glucose intolerance and insulin resistance) are faithfully reproduced in rats following both acute and chronic treatment with SGAs.21–25 Importantly, these changes in glucose metabolism occur rapidly and have been demonstrated repeatedly to be independent of changes in body weight, both in the clinical setting and in rodent models.24,26,27 To date, the effects of most of the main classes of antidiabetic drugs on SGA-induced metabolic dysregulation remain undetermined in preclinical models. It is important to perform such studies, as findings may not only provide knowledge about the biological pathways that are affected, but also offer insights into optimal treatment approaches in the clinic.
We therefore conducted the present study to determine the effects of 3 of the most commonly used classes of oral hypoglycemic drugs (i.e., biguanides, thiazolidinediones and sulfonylureas) on the metabolic dysregulation caused by the SGA olanzapine. Olanzapine is a widely used SGA with a low propensity for neurological side effects that has proven to be superior in controlling psychosis and preventing rehospitalization to other SGAs in a major head-to-head trial.28 However, enthusiasm for the use of olanzapine is tempered by evidence that it causes serious metabolic side effects that may be second only in severity to those associated with clozapine.29,30 We therefore tested the effects of metformin, rosiglitazone and glyburide on glucose intolerance and insulin resistance caused by acute treatment with olanzapine in a rat model that we have used previously.
Methods
Animals
Adult female Sprague-Dawley rats (Charles River) initially weighing 250–275 g were pair-housed and maintained on a 12-hour light–dark cycle (lights on at 07:00h) in a temperature-controlled colony (mean 22ºC ± 1ºC). Rats were allowed to habituate to the University of British Columbia (UBC) colony for 1 week before experimental testing. Food and water were freely available. Animals were treated in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The Animal Care and Use Committee at UBC approved our study protocol.
Drugs
The doses of olanzapine (7.5 and 15 mg/kg, intraperitoneal, hereafter referred to as “lower” and “higher” doses, respectively), which we purchased from Toronto Research Chemicals Inc., were carefully chosen to represent the middle-to-upper range of physiologically relevant levels in vivo and were based on doses used in previous behavioural studies.20,31,32 The vehicle solution for olanzapine consisted of 50% polyethylene glycol 400, 40% distilled water and 10% ethanol (PEG solution). Olanzapine was administered intraperitoneally in a volume of 1 mL/kg as a single injection 60 minutes before the glucose challenge (refer to section on Acute antidiabetic treatment). The doses of metformin (100 and 500 mg/kg, oral) and rosiglitazone (6 and 30 mg/kg, oral), which we purchased from Toronto Research Chemicals Inc., and of glyburide (2 and 10 mg/kg, oral), which we purchased from Sigma-Aldrich Inc., were based on doses used in previous preclinical studies33–35 and represented a 5-fold range from low to high doses in the acute setting of various antidiabetic animal models. The vehicle solutions for metformin and rosiglitazone consisted of heated 0.9% saline (which was allowed to cool before administration), whereas the vehicle for glyburide consisted of PEG solution. All hypoglycemic drugs were administered orally (gastric gavage) once daily for 2 consecutive days (refer to section on Acute antidiabetic treatment). The duration of oral hypoglycemic drug treatment was set to 2 consecutive days to ensure that baseline fasting metabolic parameters (measured both before and after olanzapine administration) and postprandial measures could be examined under antidiabetic drug exposure. All solutions were compounded fresh daily, and the use of all other chemical compounds were commercially available and of reagent grade.
Baseline Intraperitoneal Glucose Tolerance Test
See Appendix 1, Figure S1, available at cma.ca/jpn, for a representation of the sequence of events. Prior to the administration of the first antidiabetic trial (metformin), all rats were subjected to a baseline glucose tolerance test (day 1). Briefly, animals were wrapped in a towel to minimize stress, and a small drop of saphenous venous blood was procured through the use of a 25-gauge needle for the baseline blood glucose measurement at t = 0 minutes. Subsequently, all animals received a glucose challenge (1 g/kg/mL, intraperitoneal) followed by repeated sampling of blood glucose readings at t = 15, 45, 75 and 105 minutes. All blood glucose measurements were determined by a hand-held glucometer (One Touch Ultra). Rats were left untreated from days 2–7 before the first antidiabetic drug administration (day 8) and the subsequent intraperitoneal glucose tolerance test (IGTT; day 9). As the present longitudinal study exposed the rats consecutively to 3 different antidiabetic drugs that could theoretically have residual carryover effects, a similar “washout” procedure was performed 1 week after each drug treatment (rats were left untreated during the week after each olanzapine/antidiabetic drug trial, days 10–14). Any putative carryover effects would be detected as a change in IGTT results in the absence of drug challenge.
Acute antidiabetic treatment
See Appendix 1, Figure S1 for a representation of the sequence of events. Rats (n = 8–10 per group) were rank-ordered based on the baseline IGTT and the initial total body weight, and they were then randomized into 1 of 9 treatment groups: higher dose olanzapine (15 mg/kg) and higher dose metformin (500 mg/kg), higher dose olanzapine and lower dose metformin (100 mg/kg), higher dose olanzapine and no metformin (0.9% saline vehicle), lower dose olanzapine (7.5 mg/kg) and higher dose metformin, lower dose olanzapine and lower dose metformin, lower dose olanzapine and 0.9% saline, no olanzapine (PEG vehicle solution) and higher dose metformin, no olanzapine and lower dose metformin, and no olanzapine and no metformin (0.9% saline vehicle).
Each rat received a single gavage administration of either metformin or 0.9% saline on day 8 (at 11:00h). On day 9, rats that were fasted overnight (mean 16 [SD 2] hr) had their baseline blood glucose levels measured and then received a single intraperitoneal injection of either olanzapine (7.5 or 15 mg/kg) or PEG vehicle (t = 0 minutes). After a 60-minute delay, animals were subjected to a 100 μL saphenous blood draw, whereby plasma was centrifuged (10 000 revolutions per minute for 10 minutes at 4°C) and stored at –80°C for the analysis of insulin levels. The animals then received the second dose of metformin or vehicle by gavage (60 min postolanzapine administration) followed by an intraperitoneal challenge injection of glucose (1 g/mL/kg). Glucose levels were then measured every 15 minutes for a duration of 120 minutes. An identical protocol was repeated for the 2 additional antidiabetic drugs, rosiglitazone (6 or 30 mg/kg, oral) and glyburide (2 or 10 mg/kg, oral). For the entirety of the study, each animal handler was blinded to drug treatment group.
Insulin measurement by enzyme-linked immunosorbent assay
Individual plasma samples extracted during day 2 from each of the 3 antidiabetic IGTTs were analyzed for insulin content using ultra sensitive rat insulin enzyme-linked immunosorbent assay (ELISA) kits (Crystal Chem Inc.), as previously performed.21,36 Briefly, 5 μL plasma samples were added and analyzed, in duplicate, on each 96-well plate according to the specific time points studied (t = 60 and t = 90 minutes). Samples were incubated at 4°C for 2 hours followed by repeated washes. Substrate was added for 40 minutes, and absorbance was measured at 450–630 nm. Calibrators provided with the ELISA kit were used to generate a curve to interpolate sample insulin values. In addition, a reference (nonfasted) animal’s plasma added to all plates served as a reference standard; this confirmed a high intraplate reliability, with a mean run-to-run correlation of 0.996 (range 0.994–0.999).
Insulin resistance
To determine acute insulin resistance in drug-treated rats, we calculated the homeostatic model assessment of insulin resistance (HOMA-IR). This equation takes into account the product of both fasting levels of glucose (expressed as mmol/L) and insulin (μU/mL) at 60 minutes postolanzapine treatment and divides by a constant of 22.5 ([I0 × G0]/22.5), where I0 and G0 are fasting insulinemia and glycemia. A larger calculated HOMA-IR value denotes greater insulin resistance.
Statistical analysis
We performed a 2-factor analysis of variance (ANOVA), with antipsychotic drug (2 doses of olanzapine and vehicle) and antidiabetic drug (2 doses and vehicle) as the between-subject factors, with an α of p < 0.05. Individual glucose measurements at the 8 time points during the IGTT were integrated to generate a single area under the curve (AUC) value. The variables analyzed included fasting levels of glucose before and 60 minutes after the antipsychotic drug challenge, the AUC for the glucose tolerance test, fasting postdrug insulin and HOMA-IR values. When appropriate, we conducted least significant difference post hoc tests. Data were analyzed with SPSS software version 16.
Results
Olanzapine and metformin
Fasting levels of glucose in the rats before olanzapine administration did not differ between the groups (Table 1). However, fasting levels of glucose measured 60 minutes after treatment with olanzapine but before the administration of the second metformin dose and the glucose load showed a highly significant effect of antipsychotic drug treatment (F2,63 = 19.18, p < 0.001) but no interaction with antidiabetic drug treatment (there were no significant interactions between these 2 factors on any variable for any of the 3 antidiabetic drugs). Post hoc analysis indicated that all olanzapine-treated groups had higher fasting glucose levels than the vehicle-treated groups (p < 0.001; Table 1). Interestingly, the higher dose olanzapine-treated rats that were not given metformin had higher fasting glucose levels than all other groups (p = 0.011), including the 2 other higher dose olanzapine-treated groups that received metformin the day before. This suggests that the first day of treatment may have had a residual effect on glucose levels after challenge with the antipsychotic drug.
Mean concentration of fasting glucose, insulin and HOMA-IR scores in rats treated with oral hypoglycemic drugs*
Analysis of insulin levels postolanzapine administration but before the metformin and glucose load indicated a significant main effect of antipsychotic drug treatment (F2,63 = 29.71, p < 0.001), whereby insulin levels were significantly increased in all groups treated with olanzapine (Table 1). Insulin resistance was calculated using the HOMA-IR equation. The ANOVA indicated a significant main effect of olanzapine treatment on HOMA-IR values (F2,63 = 24.36, p < 0.001), whereby they were significantly higher in all groups treated with olanzapine than in those treated with vehicle; HOMA-IR values were also significantly higher in the 15 mg/kg dose olanzapine groups than the 7.5 mg/kg dose groups (p = 0.013), indicating a dose-dependent effect of olanzapine on insulin resistance. The effects of metformin on olanzapine-induced glucose dysregulation were directly assessed with the IGTT (Fig. 1A and Appendix 1, Figure S2A). The ANOVA indicated significant main effects of both olanzapine (F2,63 = 24.796, p < 0.001) and metformin (F2,63 = 5.146, p = 0.009) treatment in the IGTT. Post hoc analysis revealed that olanzapine produced a dose-dependent increase in the glucose values during the IGTT, with the 15 mg/kg dose causing the greatest degree of glucose intolerance (p = 0.008). Both doses of metformin caused a significant reduction in olanzapine-induced glucose intolerance (p = 0.002); however, this effect did not differ between the 2 doses of metformin. Glucose levels were still higher in groups that received olanzapine and metformin than in those that did not receive olanzapine (p = 0.046), reflecting a partial rather than full reversal of glucose intolerance.
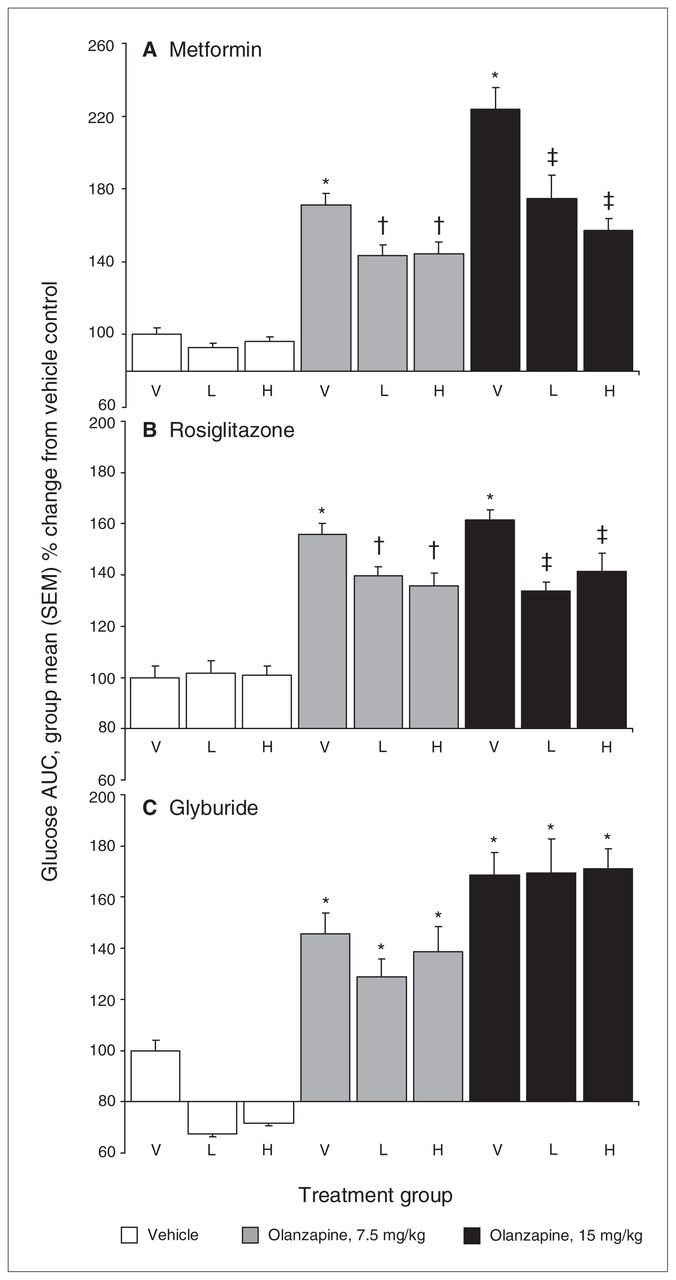
Animals (n = 8–10 per group) received 2 daily gavages of either (A) metformin (100 and 500 mg/kg, oral), (B) rosiglitazone (6 and 30 mg/kg, oral) or (C) glyburide (2 and 10 mg/kg, oral) immediately after olanzapine treatment (7.5 and 15 mg/kg, intraperitoneal) and overnight fasting. Subsequently, all rats were subjected to a glucose tolerance test, receiving an intraperitoneal challenge injection of 1 g/mL/kg of glucose, and blood glucose levels were measured every 15 minutes for the next 2 hours. Total cumulative glucose levels for each treatment group are summed as areas under the curve (AUC) and are presented as percent change from vehicle control. H = high dose; L = low dose; SEM = standard error of the mean; V = vehicle. *Significantly greater than vehicle-treated rats (p = 0.010). †Significantly greater than vehicle-only treated rats (p = 0.009) but lower than rats treated with 7.5 mg/kg olanzapine and no antidiabetic drug (p = 0.045). ‡Significantly greater than vehicle-only treated rats (p = 0.007) but lower than rats treated with 15 mg/kg olanzapine and no antidiabetic drug (p = 0.045).
Olanzapine and rosiglitazone
Comparison of glucose levels in the baseline IGTT and the washout IGTT after metformin treatment (i.e., 1 week before and 1 week after metformin treatment) indicated no carryover effect of drug treatment, so animals were rerandomized to 2 days of treatment with rosiglitazone the following week.
Fasting levels of glucose in the rats on the second day of rosiglitazone treatment before olanzapine administration did not differ between the groups. Olanzapine increased fasting levels of glucose measured 60 minutes after antipsychotic drug treatment (F2,63 = 23.29, p < 0.001; Table 1). This reflected increased glucose levels for the olanzapine-treated groups compared with groups not treated with olanzapine (p < 0.001). Fasting insulin levels were similarly increased in all olanzapine-treated groups compared with vehicle-treated groups (F2,63 = 17.31, p < 0.001; Table 1). Analysis of HOMA-IR values revealed a significant effect of olanzapine (F2,63 = 17.29, p < 0.001), whereby HOMA-IR values were significantly higher in all groups treated with olanzapine. Whereas HOMA-IR values were lower in all groups that had received rosiglitazone on the previous day, this effect did not approach significance, unlike with metformin.
Analysis of the data from the IGTT indicated that there was both an effect of treatment with olanzapine (F2,63 = 27.95, p < 0.001) and an effect of treatment with rosiglitazone (F2,63 = 5.43, p = 0.007). Similar to the effects of metformin, both doses of rosiglitazone caused a significant reduction in glucose intolerance in olanzapine-treated rats (p = 0.010; Fig. 1B and Appendix 1, Figure S2B) but did not completely reverse glucose intolerance, as glucose levels still remained significantly higher than those in rats not treated with olanzapine (p = 0.014).
Olanzapine and glyburide
Comparison of glucose levels in the washout IGTTs before and after treatment with rosiglitazone indicated no difference in glucose tolerance, therefore the rats were rerandomized to treatment with glyburide the following week.
Analysis of fasting glucose levels on the second day of glyburide treatment revealed a highly significant main effect of antidiabetic drug treatment (F2,63 = 32.79, p < 0.001), unlike with metformin and rosiglitazone. This was due to a large reduction of about 50% in fasting glucose levels in animals treated with glyburide, demonstrating that glyburide has hypoglycemic actions even 24 hours after administration (Table 1). Sixty minutes following treatment with olanzapine there was a main effect of treatment with both olanzapine (F2,63 = 29.11, p < 0.001) and glyburide (F2,63 = 4.66, p = 0.013) on fasting glucose levels. Further analysis revealed that olanzapine had a dose-dependent effect on glucose levels, with both doses of olanzapine causing increases compared with vehicle-treated rats (p < 0.001) and a greater effect of the 15 mg/kg dose compared with the 7.5 mg/kg dose (p = 0.032). The effect of glyburide, representing residual effects from the first day of treatment, was evident, as decreased fasting glucose levels compared with rats not treated with the antidiabetic drug: while all glyburide-treated groups showed decreases, this was only significant in the rats not treated with olanzapine. Fasting insulin levels revealed main effects of both olanzapine treatment (F2,63 = 23.59, p < 0.001) and glyburide treatment (F2,63 = 3.15, p = 0.049) 24 hours previously (Table 1). Olanzapine, relative to vehicle, caused an increase in insulin levels (p < 0.001). Glyburide treatment 24 hours previously increased insulin levels, but only in the higher dose groups (10 mg/kg; p = 0.017). Insulin resistance, measured by HOMA-IR, exhibited a main effect of olanzapine treatment (F2,63 = 26.65, p < 0.001) but no effect of glyburide, as olanzapine increased HOMA-IR values. Glucose intolerance during the IGTT following the second dose of glyburide also revealed a main effect of olanzapine treatment (F2,63 = 39.35, p < 0.001) but no effect of glyburide treatment (Fig. 1C and Appendix 1, Fig. S2C). As mentioned previously, olanzapine caused a dose-dependent increase in glucose intolerance, regardless of glyburide treatment group, with both doses of olanzapine increasing glucose intolerance significantly (p < 0.001), and a greater effect of the 15 mg/kg olanzapine dose compared with the 7.5 mg/kg dose (p = 0.002). Whereas glyburide decreased glucose levels in the animals not treated with olanzapine, this effect did not quite achieve significance and had no effect in olanzapine-treated animals, unlike with metformin and rosiglitazone.
Interestingly, the magnitude of the response to olanzapine during the IGTT showed a slight reduction with time across the entire study, as AUC glucose levels modestly (but non-significantly) declined with both doses of olanzapine between the first exposure to olanzapine and the second exposure (with rosiglitazone), although there was no further drop between the second and third olanzapine exposures.
Discussion
In the present study, we tested the effects of 3 distinct classes of oral hypoglycemic drugs on glucose dysregulation and insulin resistance in adult female rats treated with lower and higher doses (7.5 mg/kg and 15 mg/kg) of the SGA olanzapine. The hypoglycemic drugs were administered once daily for 2 consecutive days, and included a biguanide (metformin), thiazolidinedione (rosiglitazone) and sulfonylurea (glyburide).
A major conclusion from the present results is that olanzapine-induced glucose dysregulation can be alleviated, in part, by antidiabetic drug mechanisms that are independent of direct insulin release. Under current experimental conditions, improvement of glucose intolerance and hyperglycemia was demonstrated by both metformin and rosiglitazone, but not glyburide treatment. It is unlikely that the 2 doses of glyburide used were too low to have an effect, as these are doses commonly used efficaciously in other rat models of metabolic dysregulation and type 2 diabetes.33 Furthermore, our 5-fold dose range of glyburide reduced fasting glucose levels by almost 50% and nearly doubled plasma insulin levels in control animals, consistent with glyburide’s known insulin-secreting action. It appears that increasing insulin levels alone is insufficient to decrease the glucose dysregulation induced by olanzapine. Working through mechanisms independent of direct insulin release, metformin and rosiglitazone were able to cause a respective 39%–54% and 29%–50% decrease in glucose intolerance in the IGTT. The effects of metformin and rosiglitazone were not dose-dependent, as the higher dose of each drug did not have a greater effect, so doses might have to be substantially higher to produce additional effects on glucose dysregulation. It is also unlikely that more extended dosing could produce a greater effect, as our pilot studies found no further benefit to extending hypoglycemic drug treatment beyond 1 week (data not shown). It is possible that the inability of these drugs to completely reverse olanzapine-induced glucose dysregulation reflects the complex physiologic effects of the antipsychotics through multiple pathways.
Consistent with previous studies, olanzapine caused significant metabolic dysregulation,22,24,31,37–43 evident as elevated fasting glucose levels, insulin resistance (greater HOMA-IR values) and glucose intolerance in the IGTT. To our knowledge, we assessed the effects of metformin, rosiglitazone and glyburide on these metabolic side effects in rats for the first time. Metformin showed an effect on glucose dysregulation after the first day of treatment: fasting glucose levels were decreased after treatment with the higher dose of the antipsychotic. Importantly, after the second dose, metformin significantly reduced glucose intolerance in the IGTT, although values still remained above those of controls. Rosiglitazone did not exhibit effects after the first day of treatment, but the second dose resulted in a reduction of glucose intolerance in the IGTT similar to metformin, causing a significant reduction of glucose intolerance but, again, not a complete return to control values. In contrast, glyburide had a strong hypoglycemic effect on fasting glucose levels in rats not treated with olanzapine. However, the drug did not decrease fasting glucose levels after olanzapine treatment, and unlike the other 2 antidiabetic drugs, glyburide had no effect on glucose intolerance in the IGTT. Previously, we have reported that intermittent treatment with olanzapine can sensitize glucose intolerance.31 This was not observed in the present study, likely owing to factors, including the duration of treatment, rerandomization of animals after each antidiabetic drug, injection regimen and potential influence of exposure to antidiabetic drugs.
The selective effects of metformin and rosiglitazone versus glyburide on glucose homeostasis are consistent with the known effects of olanzapine on glucose dysregulation. Evidence suggests that the pathogenesis of SGA-induced glucose dysregulation stems mainly from inadequate hepatic glucose control,24,44,45 reflecting hepatic insulin insensitivity. Hyperinsulinemic-euglycemic clamp studies have demonstrated that olanzapine significantly decreases hepatic insulin sensitivity and increases hepatic glucose output (HGO) in rodent models.22,24,44 For both metformin and rosiglitazone, in vitro evidence indicates that suppression of liver HGO is mediated independently of the effects of insulin.14,46 In comparison, sulfonylureas, such as glyburide, produce their therapeutic effects by directly stimulating insulin secretion from the pancreas, giving rise to sustained levels of circulating insulin.17 In theory, the increased levels of insulin caused by treatment with glyburide should stimulate type 1 processes that lower glucose levels in response to a hyperglycemic state, such as hepatic glucose uptake, peripheral glucose disposal and inhibition of glucogenic responses. However, the clear failure of glyburide to affect olanzapine-induced hyperglycemia strongly suggests that the therapeutic effects of metformin and rosiglitazone occur via their insulin-independent mechanisms. As metformin’s pharmacological action involves suppressing HGO by curtailing gluconeogenesis in addition to enhancing peripheral glucose utilization,47,48 there are shared physiologic pathways between both antipsychotic and antidiabetic drugs. The “cellular energy sensing” AMPK-signalling pathway has been proposed as a mechanism of antidiabetic action. Both liver and muscle AMPK activity is increased by metformin, facilitating inhibition of lipogenesis, gluconeogenesis and increased glucose uptake.49,50 Metformin also blocks hypothalamic AMPK activity, resulting in anorexigenic effects.51,52 Several recent studies have documented elevated levels of phosphorylated hypothalamic AMPK after chronic olanzapine treatment,45,53 which were associated with weight gain and increased food intake.54 Evidence also suggests that metformin modulates the incretin axis via an AMPK-independent mechanism. Enhanced plasma levels of the insulinotropic hormone glucagon-like peptide 1 (GLP-1) have been reported after metformin treatment in humans and in preclinical models.55–57 Among other beneficial antidiabetic effects, GLP-1 suppresses the hyperglycemic action of glucagon, causing decreased HGO and lower circulating glucose levels. Recent studies by Smith and colleagues41,58 demonstrated that olanzapine-, clozapine- and quetiapine-induced glucose dysregulation was associated with decreased GLP-1 production and enhanced glucagon secretion, leading to stimulated HGO. These studies, together with our present findings, suggest common targets for both metformin and antipsychotic drug action. The opposing effects of SGAs and metformin on glucagon, GLP-1 and AMPK may explain why hypoglycemic drug treatment has been only partially successful in relieving SGA-induced metabolic side effects in the clinic.59 Rosiglitazone, via activation of PPARγ receptors, causes reduced expression of genes required for hepatic gluconeogenesis, such as pyruvate carboxylase and glucose-6-phosphatase, enhancing suppression of HGO and increasing peripheral glucose disposal,60 similar to metformin.
To our knowledge, 4 other studies have determined the effects of antidiabetic agents on SGA-induced glucose intolerance. In the study by Lykkegaard and colleagues,61 treatment of female rats with liraglutide, a GLP-1 analogue, alleviated metabolic indices, including olanzapine-induced glucose intolerance. There was no effect on fasting plasma insulin levels, but importantly, only a single dose of both olanzapine and liraglutide were tested. In a separate study, treatment with the GLP-1 receptor agonist exendin-4 decreased glucose levels in the GTT after treatment with an acute 10 mg/kg dose of clozapine.58 Arulmozhi and colleagues62 assessed the effects of 3 different PPARγ modulators (glimepiride, rosiglitazone and fenofibrate) on ziprasidone-, clozapine- and chlorpromazine-induced hyperglycemia and hyperinsulinemia in mice. Rosiglitazone and glimepiride reduced hyperglycemia in chlorpromazine-treated animals, whereas all 3 antidiabetics reduced clozapine-induced hyperglycemia, with the greatest effect attributed to rosiglitazone. Adeneye and colleagues63 examined the chronic effects of both metformin (20 mg/kg) and glyburide (0.1 mg/kg) pretreatment on risperidone-induced weight gain, hyperglycemia, insulin resistance and dyslipidemia in male rats. After 60 days of pretreatment, metformin reduced weight gain, fasting hyperglycemia, hyperinsulinemia and dyslipidemia, whereas glyburide had no effect. Our results are therefore consistent with those reported in the 2 latter studies, and also mostly consistent with the clinical literature. Human studies have confirmed that metformin alleviates some of the metabolic effects of olanzapine. A recent meta-analysis concluded that metformin had modest effects on olanzapine-induced weight gain,64 whereas another meta-analysis that included multiple SGAs determined that metformin reduced but did not fully reverse drug-induced insulin resistance.59 There is less evidence regarding the clinical efficacy of rosiglitazone, owing in part to ongoing concern about the cardiovascular side effects of the drug.65 However, a clinical trial noted that rosiglitazone significantly improved glycemic control in patients treated with olanzapine.66 To our knowledge, there has been no reported evaluation of glyburide on the metabolic sequelae of olanzapine or other SGAs, but given our current findings, we would not expect this agent to be efficacious.
Limitations
The principle limitation of the present study is the evaluation of only a single SGA, despite testing multiple doses of both antipsychotic and antidiabetic treatments. Whereas most SGAs cause metabolic dysregulation, the extent to which all such drugs produce effects through shared pathways remains unknown. It will therefore be necessary in future studies to determine whether the current findings with olanzapine generalize across the entire class of SGAs, including newer drugs.67
Conclusion
The present study shows that both metformin and rosiglitazone, but not glyburide, can mitigate glucose intolerance caused by olanzapine in female rats. These findings are consistent with those reported in preclinical and clinical studies. Our findings indicate that drugs that influence hepatic glucose metabolism are most effective. Further studies using representative drugs from other classes of antidiabetic drugs and different models of SGA-induced metabolic abnormality are needed to elucidate the biological basis of SGA-induced metabolic sequelae and how antidiabetic drugs reverse these side effects. Future research should also examine multidrug antidiabetic combinations, as routinely occurs in the clinical setting,68 to identify optimal treatment strategies that may guide future clinical studies.
Acknowledgements
The current research was supported by grants from the British Columbia Provincial Health Services Authority and National Sciences and Engineering Research Council of Canada (NSERC) grant 356069-09 to A.M. Barr, and NSERC grant 355912-11 to C.C.Y. Pang. A.M. Barr is a Canadian Institutes of Health Research (CIHR) New Investigator, and H.N. Boyda is a CIHR Banting scholar.
Footnotes
Competing interests: None declared for H.N. Boyda, L. Tse, E. Hawkes and C.C.Y. Pang. R.M. Procyshyn declares having consulted for AstraZeneca, Bristol-Myers Squibb, Janssen, Sunovion and Pfizer; received lecture fees from AstraZeneca, Bristol-Myers Squibb, Otsuka and Pfizer; and developed educational presentations for Bristol-Myers Squibb and Pfizer. C.H. Jin declares having received a student award from the NSERC to fund a summer research project. W.G. Honer declares advisory board membership with Roche Canada and In Silico Biosciences; having received consultant fees from Novartis and the Canadian Agency for Drugs and Technology in Health; receiving royalties from antibody manufacturers for licenses held by his university; and having received travel support from multiple academic and health authorities for presentations. A.M. Barr declares advisory board membership with Roche Canada; having acted as legal consultant for Eli Lilly Canada; and having a grant pending with BMS Canada through his institution.
Contributors: H.N. Boyda, R.M. Procyshyn, C.C.Y. Pang and A.M. Barr designed the study. H.N. Boyda, L. Tse, E. Hawkes, C.H. Jin and A.M. Barr acquired the data, which H.N. Boyda, W.G. Honer and A.M. Barr analyzed. H.N. Boyda, R.M. Procyshyn, C.C.Y. Pang and A.M. Barr wrote the article, which H.N. Boyda, L. Tse, E. Hawkes, C.H. Jin, W.G. Honer and A.M. Barr reviewed. All authors approved the article’s publication.
- Received September 30, 2011.
- Revision received February 2, 2012.
- Revision received March 12, 2012.
- Revision received March 20, 2012.
- Accepted March 22, 2012.
References

{kind=link}
Article tools





