
Abstract
The number of intoxications with organophosphorus pesticides (OPs) is estimated at some 3 000 000 per year, and the number of deaths and casualties some 300 000 per year. OPs act primarily by inhibiting acetylcholinesterase (AChE), thereby allowing acetylcholine to accumulate at cholinergic synapses, disturbing transmission at parasympathetic nerve endings, sympathetic ganglia, neuromuscular endplates and certain CNS regions. Atropine is the mainstay of treatment of effects mediated by muscarine sensitive receptors; however, atropine is ineffective at the nicotine sensitive synapses. At both receptor types, reactivation of inhibited AChE may improve the clinical picture.
The value of oximes, however, is still a matter of controversy. Enthusiastic reports of outstanding antidotal effectiveness, substantiated by laboratory findings of reactivated AChE and improved neuromuscular transmission, contrast with many reports of disappointing results. In vitro studies with human erythrocyte AChE, which is derived from the same single gene as synaptic AChE, revealed marked differences in the potency and efficacy of pralidoxime, obidoxime, HI 6 and HLö 7, the latter two oximes being considered particularly effective in nerve agent poisoning. Moreover, remarkable species differences in the susceptibility to oximes were revealed, requiring caution when animal data are extrapolated to humans. These studies impressively demonstrated that any generalisation regarding an effective oxime concentration is inappropriate. Hence, the 4 mg/L concept should be dismissed.
To antagonise the toxic effects of the most frequently used OPs, pralidoxime plasma concentrations of around 80 μmol/L (13.8 mg/L pralidoxime chloride) should be attained while obidoxime plasma concentrations of 10 μmol/L (3.6 mg/L obidoxime chloride) may be sufficient. These concentrations should be maintained as long as circulating poison is expected to be present, which may require oxime therapy for up to 10 days. Various dosage regimens exist to reach this goal. The most appropriate consists of a bolus short infusion followed by a maintenance dosage. For pralidoxime chloride, a 1g bolus over 30 minutes followed by an infusion of 0.5 g/h appears appropriate to maintain the target concentration of about 13 mg/L (70kg person). For obidoxime chloride, the appropriate dosage is a 0.25g bolus followed by an infusion of 0.75 g/24h. These concentrations are well tolerated and keep a good portion of AChE in the active state, thereby retarding the AChE aging rate.
AChE aging is particularly rapid with dimethyl phosphoryl compounds and may thwart the effective reactivation by oximes, particularly in suicidal poisoning with excessive doses. In contrast, patients with diethyl OP poisoning may particularly benefit from oxime therapy, even if no improvement is seen during the first days when the poison load is high. The low propensity to aging with diethyl OP poisoning may allow reactivation after several days, when the poison concentration drops.
Rigorous testing of the benefits of oximes is only possible in randomised controlled trials with clear stratification according to the class of pesticides involved, time elapsed between exposure and treatment and severity of cholinergic symptoms on admission.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1. Treatment of Organophosphate Poisoning: A Global Challenge
Organophosphorus pesticides (OPs) are used increasingly as pest-control agents in the agricultural industry, as organochlorine compounds such as dichlorodiphenyltrichloroethane (DDT) have been mostly banned because of their long-lasting persistence in the environment. Particularly in the hot climate of developing countries, pest control has been considered mandatory to meet food production requirements. If not adhering to the Food and Agriculture Organisation of the United Nations integrated pest management system, extensive use of pesticides has often been adopted to prevent crop reduction by pests.[1]
OPs have two major advantages over DDT and related organochlorine compounds: (i) they are readily degraded; and (ii) they do not accumulate either in humans or in food chains. However, this advantage is paid for dearly, as OPs are far more toxic to mammals. The number of accidental, suicidal and homicidal intoxications with OPs is high. The number of victims worldwide is estimated at having surpassed 300 000 per year.[2]
Most pesticide poisonings recorded in hospital surveys are the result of self-poisoning and result in a high death rate.[3] In Sri Lanka, for example, in 1998 more than 3000 fatalities occurred in 50 000 hospital admissions due to poisoning, including snakebites. While the case fatality ratio was 0.5% with snakebites, it reached 9.6% with pesticides and probably much more with the OPs.[1,4] The high death rate with anticholinesterases, particularly in rural areas of developing countries, is a result of large distances to medical help and poor resources in medical facilities. There are too many patients, too few physicians, too few drugs and ventilators, and not enough good evidence about how to treat patients with life-threatening OP poisoning.
The epidemic proportions of deliberate self-poisoning, particularly in South-East Asian countries, have been related to the lack of strong religious or legal prohibitions against suicide. This could lead people to consider suicide as an acceptable way of relieving their personal misery or of reducing the financial and emotional burden they cause their family.[5] Moreover, the ready availability of potent pesticides in the homes of most residents in rural areas makes OPs the preferred compounds of deliberate self-harm. Thus, dichlorvos and parathion were the most frequently ingested compounds used in committing suicide in China.[5] It has been argued that the lethality of the method used, and the lack of well-trained medical personnel along with poor medical resources may result in the particularly high death rates among people who impulsively ingest pesticides but do not intend to die. Instead, the acts were used to gain attention or to express distress, and to gain revenge causing distress to another person. This situation is particularly tragic in the young, economically active age group.[5,6]
In addition to self-poisoning, accidental exposure to pesticides is also common in tropical areas due to a number of factors. First, it is considered impractical to use safety equipment in a hot and humid climate; in addition, such equipment may not be affordable. Secondly, protecting worker’s health is considered costly and irrelevant to employers. Thirdly, many workers are illiterate, and safety instructions on containers are often written in unfamiliar languages. Lastly, the product instructions may be quite unrealistic: after coming into dermal contact with the concentrate ‘wash off at once’ is difficult to follow when there is no water available.[7]
From the above, it appears mandatory to evaluate medical interventions that may reduce the high death rates in OP poisoning and which can be afforded by a small medical budget, as in developing countries. Proper antidotal treatment of less severely intoxicated patients could save the limited ventilators available for those cases where artificial ventilation is essential.
2. Chemistry of Organophosphorus Pesticides
OPs are normally esters, thiol esters or anhydride derivatives of phosphorus acid. The double-bonded atom at the phosphorus may be oxygen (oxons) or sulphur (phosphorothioates; this nomenclature has superseded the older terms ‘thiophosphate’ or ‘thionophosphate’). The phosphorothioates are usually very weak inhibitors of the serine hydrolases and may be compared with prodrugs that require metabolic activation to the ultimate active compounds. However, some commercial preparations contain a variable percentage of the oxon derivative due to auto-oxidation of the parent compound that can produce an early onset of toxicity.
Two hydroxy groups attached to the phosphorus atom are usually esterified with alcohols or phenols. In some cases one residue comprises a thioalcohol. If one or two residues are directly linked to the phosphorus atom via a C-P bond, a phosphonate or a phosphinate is the result. In phosphoroamidates, a carbon atom is linked via a nitrogen atom to phosphorus. The third hydroxy group on the phosphorus atom is replaced by a labile ‘leaving group’ that influences the reactivity of the OP compound. The rate of phosphylation of a given alcohol group, e.g. nucleophilic displacement of X- by a serine hydroxyl, depends inversely on the strength of the P-X bond and the pKa value of the conjugate acid of the leaving group HX.[8] Figure 1 shows the general structural formula as developed by Schrader in the 1930s.[9]

Schrader formula of an organophosphorus insecticide. The ultimate reactive compounds, the ‘oxons’, bear an oxygen atom at the double bond to phosphorus whilst the thioate prodrugs bear a sulphur atom. In the most commonly used insecticides, R1 and R2 are identical and consist of either O-methyl or O-ethyl groups.
Of the more than 100 OP pesticides used worldwide, the majority are either dimethyl phosphoryl or diethyl phosphoryl compounds, examples of which are listed in table I. These compounds do not have a chiral centre and hence all form the same dimethyl phosphorylated or diethyl phosphorylated enzyme regardless of the leaving group, simplifying therapeutic decision-making. In some cases, however, identification of the OP leaving group may be difficult, e.g. with phosphoroamidates or with phosphorodithioates. Here, modern mass spectroscopic procedures, such as matrix-assisted laser desorption ionisation or surface-enhanced laser desorption ionisation time of flight procedures may provide an answer.[10,11]

3. Targets of Organophosphate Toxicity
Studies of the toxic mechanisms of OP pesticides have historically focused on their interaction with and irreversible inhibition of serine hydrolases, particularly acetylcholinesterase (AChE; EC 3.1.1.7). This enzyme is encoded by a single gene in each animal species, although a wide variety of molecular forms exist.[15] The molecular diversity is believed to reflect post-transcriptional mechanisms and post-translational modifications of the membrane-anchoring tail rather than the catalytic unit. Therefore, it is generally assumed that the active site of mammalian AChE is identical in all tissues at all ages.[16]
Most AChE is localised in the outer basal lamina of the synapse where the enzyme metabolises the neurotransmitter acetylcholine. The neuromuscular endplate is particularly rich in this enzyme, enabling the hydrolysis of acetylcholine in submilliseconds.[17]
While inhibition of this key enzyme explains most of the prominent symptoms of acute OP poisoning, subtle differences among the toxic actions of different OPs remain to be explained. OPs inhibit a variety of other serine hydrolases, such as chymotrypsin and trypsin,[18] carboxylesterases (EC 3.1.1.1), plasma pseudocholinesterase (butyrylcholinesterase [BChE], EC 3.1.1.8) and neuropathy target esterase (NTE),[19,20] by covalent binding to a serine residue at the catalytic site. Variable inhibition of NTE may result in chronic effects elicited by some but not all OPs. Furthermore, processing of other signal peptides, e.g. their formation from pre-proteins or their inactivation by hydrolytic cleavage, may possibly be altered by OPs.
In addition to inhibition of serine hydrolases, recent experimental evidence also suggests OPs directly target acetylcholine receptors. Acetylcholine receptors have been traditionally subclassified into muscarine- and nicotine-sensitive structures. Muscarine-sensitive m-receptors (five subtypes) belong to the metabotropic, G-protein receptor family coupled to cyclic adenosine monophosphate (cAMP) and inositol triphosphate signal transduction processes, and leading either to decreases in cAMP or calcium mobilisation. Nicotine-sensitive n-receptors are pentameric structures with ionotropic functions. Both receptor types are located post- and presynaptically. OP concentrations necessary for direct effects at n-receptor sites are usually much higher than those capable of phosphorylating AChE. Hence, toxicological effects at n-receptor sites appear less important in OP poisoning. In contrast, m-receptors are affected at toxicologically relevant OP concentrations and may thus be involved in modulating toxic OP effects. Like acetylcholine itself, OPs may inhibit transmitter release by a negative feedback loop. However, they may differ from each other and hence elicit different effects even if the extent of their anticho-linesterase action is identical (for review see Pope[21]).
Notwithstanding these considerations, OPs primarily exert their acute toxic actions by interfering with cholinergic transmission due to inhibition of AChE, which almost completely explains all signs and symptoms of acute OP poisoning.[22]
4. Mechanisms of Acetylcholinesterase Inhibition, Spontaneous Reactivation and Aging
The principle reactions of an OP oxon with AChE, the most important target of toxicity, are shown in figure 2. Inhibition of AChE by an OP was first detected by Gross in the late 1930s while testing the insecticides synthesised by Schrader.[9]

Inhibition of a cholinesterase by an organophosphate. Reaction 1 depicts reversible formation of a Michaelis complex with Kd = k-1/k+1. Reaction 2 indicates phosphorylation of a hydroxyl group of the enzyme (E) with liberation of the leaving group (HX). Reaction 3 illustrates the aging process with loss of an alkoxy residue and formation of a negatively charged phosphorus moiety. Reaction 4 shows the spontaneous reactivation of the enzyme with liberation of an inactive, acidic organophosphate.
Phosphorylation was proposed by Burgen[23] and Burgen and Hobbiger[24] as the principle mechanism of irreversible AChE inhibition. Ten years later, Jansz et al.[25] conclusively demonstrated that a serine residue was phosphorylated by diisopropyl fluorophosphate (DFP) in plasma cholinesterase from horse serum. Schaffer et al.[26] in 1953 had already detected phosphorylated serine in chymotrypsin treated with DFP. In 1985, it was unequivocally established that the active site with which DFP reacts is Ser200 in AChE of Torpedo californica.[27] Since then, the three-dimensional structure of AChE from the electric organ of Torpedo californica has been determined by x-ray analysis to 2.8A resolution.[28] More recently, site-directed mutagenesis with replacement of individual amino acid residues in AChE and x-ray analysis of the tailored enzyme have provided a unique tool for understanding the microscopic events occurring during enzyme catalysis and inhibition by organophosphates.[29]
Phosphorylation of the active site serine alcohol group in AChE is facilitated by the features of the catalytic site and the reactivity of the organophosphate. The electrophilic phosphorus atom activated by an appropriate leaving group, such as p-nitrophenyl in paraoxon, is attacked by the nucleophilic alcohol group in serine that is activated by an adjacent imidazole nitrogen of histidine. As a result, the serine residue is phosphorylated. The resulting phosphoryl ester intermediate resembles the intermediate serine-acetate that is formed during normal acetylcholine hydrolysis but has significantly greater stability. The acetyl moiety is released with a half-life (t1/2) of less than 1ms, whereas the phosphoryl esters show half-lives of hours to days.[30]
Inhibition of cholinesterases by oxons (reaction 1 in figure 2) is preceded by reversible formation of a Michaelis complex followed by a unimolecular phosphorylation reaction (2). The affinity constant (k+1/k-1= 1/Kd) of most oxons derived from OPs is usually much higher than the concentrations found in the plasma of intoxicated patients. Hence, the inhibition rate increases proportionally to the increase in oxon concentration.[31,32] The marked species differences observed in the apparent second order rate constants, i.e. ki approaching kp/Kd, are due to differences in both Kd and kp values (for nomenclature of the constants see figure 2).[33,34] Table II compiles various second-order rate constant values of enzyme inhibition (ki), illustrating the wide variation in the phosphorylating potential of the OP oxons.

Rate constants for the inhibition of acetylcholinesterase (AChE) by organophosphorus pesticide oxonsa
Table III presents the half-lives of the spontaneous reactivation (reaction 4 in figure 2) of human blood cholinesterase enzymes under approximate physiological conditions. It is apparent that the spontaneous recovery of the dimethyl phosphorylated enzymes is some 50 times faster than that of the diethyl phosphorylated enzymes and that BChE recovers by an order of magnitude slower than AChE.[24,38–41]

While water is too weak as a nucleophile to allow the cleavage of the phosphoryl-serine bond under physiological conditions, stronger nucleophiles are markedly effective. In 1951, Wilson succeeded in splitting phosphorylated AChE with hydroxylamine.[42] At the concentrations, and thus the dosage, required in vivo, hydroxylamine was too toxic because it produced lethal methaemoglobinaemia. However, when hydroxylamine was allowed to react with JV-methyrpyridinium carbaldehyde the resulting oxime met the requirement of low toxicity and sufficient effectiveness in vivo. In 1955, 2-pyridine aldoxime methiodide) [2-PAM] was introduced by Wilson and Ginsburg[43] and independently described by Childs et al.[44] as the first reactivating antidote against OP poisoning. This compound was able to reactivate AChE in vivo and in vitro.
In the same year, another property of phosphorylated AChE was recognised. Hobbiger[45] reported that enzyme preparations treated with paraoxon or DFP could be almost completely reactivated if an oxime was added soon after inhibition, but only partially if the reactivator was added some hours later. Hobbiger surmised that the phosphorylated enzyme had altered its structure, with other amino acid residues participating.[45] The chemical basis of the ‘aging process’ of DFP-inhibited plasma cholinesterase was elucidated in 1959 by Berends et al.[46] who found that one isopropyl group from the diisopropylphosphorylated serine was released into solution and that the monisopropyl phosphorylated enzyme was no longer able to be reactivated. This ‘aging’ phenomenon (reaction 3 in figure 2) holds true for AChE, BChE and NTE, differs between species and OPs, and is currently understood as an acid-catalysed process. The unimolecular scission of the C-O bond in the alkoxy chain of phosphorylated AChE involves a proton-catalysed carbocationic transition state resulting in the release of a carbonium ion and of a negatively charged phosphate diester. Several residues in the active site gorge facilitate this reaction; the most important residues are H(440), E(199),W(84) and D(72) [numbers refer to numbering of amino acid residues in AChE of Torpedo californica].[41–49] From this, it is conceivable that the aging phenomenon for an OP differs between serine hydrolases and between species.
Table IV summarises the aging half-lives for dimethyl and diethyl phosphorylated human AChE and BChE. The rate of aging depends on the ease with which the alkoxy group can form a carbonium ion. The latter is stabilised in the presence of highly branched side chains such as the pinacolyl group in the nerve agent soman.[8] Here, aging half-lives of a few minutes are observed. Resistance of the aged enzyme to reactivators is understandable since the negatively charged phosphate repels the negatively charged nucleophile, thereby increasing the stability of the phosphorylated enzyme. Furthermore, the aging reaction is not restricted to the scission of the C-O bond but is also observed with P-S and P-N bond cleavage, such as in isomalathion[11] and tabun.[50]

5. Clinical Features of Acute Organophosphate Poisoning
The clinical manifestations of acute OP poisoning reflect the degree of accumulation of the neurotransmitter acetylcholine, causing overstimulation at the cholinergic receptors of various organs. The level of AChE inhibition associated with clinical symptoms varies between tissues. In the brain, 50–80% of AChE must be inactivated before symptoms are noted and 85–90% inhibition is associated with severe toxicity.[22] In rat diaphragm muscles, neuromuscular dysfunction was not observed until 70% of AChE was inhibited. Similar observations have been made with human muscles[51] suggesting AChE activity is usually much higher than necessary for regular cholinergic transmission.
Depending on the receptor structure, initial stimulation by acetylcholine is followed by refractoriness to acetylcholine. However, while the effects at m-receptors are long-lasting, n-receptors rapidly desensitise to acetylcholine leading, for example, to ganglion blockade and muscle paralysis. Hence, it is common to divide clinical manifestations into muscarinic and nicotinic features.[30] The practical significance of this subclassification is that atropine or other cholinolytics only antagonise the muscarinic effects, but not the nicotinic effects.
5.1 Muscarinic Features
Symptoms include increased glandular secretions with bronchorrhoea, salivation, lacrimation and increased sweating, increased tonicity of smooth muscles with bronchoconstriction, miotic pupils, abdominal cramps, involuntary defecation and urination. Cardiac effects include bradycardia and QT prolongation with several types of arrhythmia including torsades de pointes.[52] Most CNS symptoms of OP poisoning are also mediated via m-receptors, although nicotinergic pathways do contribute. The interplay of the cholinergic with other transmitter systems, i.e. gamma aminobutyric acid and glutamate, is very complex and not yet fully understood, particularly in humans. The most prominent CNS symptoms of OP poisoning are headache, dizziness, drowsiness, nausea, confusion, anxiety, slurred speech, ataxia, tremor, psychosis, convulsions, coma and respiratory depression. Most of these symptoms are ameliorated by cholinolytics, although benzodiazepines are superior to cholinolytics for the management of convulsions and seizures. For more in-depth information on the CNS toxicity of OPs the reader is referred to Savolainen.[53]
5.2 Nicotinic Features
The most prominen target structures are the muscle endplate, the sympathetic ganglia and the adrenal medulla. The electrophysiological consequences at the neuromuscular junction are characterised by single electrical stimulus-induced repetitive compound muscle action potentials (CMAPs) and, in response to repetitive nerve stimulation, by a decrement or decrement/increment of CMAP.[51,54,55] The former phenomenon is usually seen at high degrees of AChE inhibition, the latter at comparably lower inhibition. Clinically, twitching of fine muscles, fasciculations and hyperreflexia are observed, which may progressively lead to muscle weakness and flaccid paralysis with reduced tendinous reflexes. This is life-threatening when the diaphragm and respiratory muscles are affected.
At sympathetic ganglia, slightly elevated acetylcholine concentrations first cause enhanced firing with persistent depolarisation as higher concentrations develop. In the first effect, muscarinergic (presynaptic?) receptors are also involved, while in the latter effect postsynaptic n-receptors are mainly thought to be responsible.[17] Plasma epinephrine levels in OP-poisoned patients were enhanced by 10-fold for some hours after OP intoxication, pointing to transient stimulation of n-receptors of chromaffin cells in the adrenal medulla.[56,57] These short-lasting effects of adrenergic/ noradrenergic stimulation may be responsible for the transient tachycardia that was observed in the majority of patients exposed to sarin in the Tokyo subway attack.[58] Similarly, Sidell reported that of 199 patients exposed to mild-to-moderate concentrations of nerve agents only 13 presented with a heart rate below 64 beats/ min while 69 had a heart rate >90 beats/min.[59] In a comprehensive review of OP-poisoning, it was pointed out that heart rate increases in the acute stage of poisoning but may decline later.[60] Hence, heart rate alone may be a misleading indicator.
Lastly, delayed symptoms after acute intoxication including the ‘intermediate syndrome’[61,62] and organophosphate-induced delayed neuropathy,[19] as well as long-term neuropsychopathological effects have been described, and are reviewed by Lotti.[22]
6. Treatment of Acute Organophosphate Poisoning
6.1 General Supportive Therapy
After dermal exposure, washing the poisoned person and removing contaminated clothes appears rational to reduce further dermal absorption, although no systematic trials appear to exist. Care should be taken with the use of gloves, aprons and eye protection to protect healthcare workers from contamination.[63]
After oral ingestion, drinking milk has been occasionally recommended. The rational of this measure is not clear. From a theoretical point of view, milk may disperse highly lipid-soluble OPs, thus facilitating their rate of absorption. Since no benefits are to be expected, milk should be avoided. The same holds true for measures to induce emesis, including ipecacuanha. There is considerable risk of aspiration, which may be particularly hazardous because most OP compounds are dissolved in petroleum distillates that induce pneumonitis and acute respiratory distress syndrome (ARDS) when aspirated.[64,65] Gastric lavage followed by instillation of activated charcoal is common practice, although no systematic controlled trials appear to exist. Adsorption of OPs to activated charcoal has been repetitively described[66] and its effectiveness in animal experiments was conclusive.[67,68] Nonetheless, it remains to be demonstrated that charcoal is effective in human OP overdose poisoning. Gastric emptying, not necessarily lavage, though somewhat delaying administration of activated charcoal, should not be excluded in every case because of a theoretical benefit: the large volumes of solvents and the presence of emulsifiers, penetrants and safeners[69] may reduce the binding capacity of activated charcoal or partly desorb OPs from it. Hence, reduction of the solvent burden by gastric emptying could increase the effectiveness of charcoal. A final decision of the benefits of these measures still awaits a controlled clinical trial as being performed in Sri Lanka.[63] At any rate, careful airway protection is highly recommended to prevent aspiration during these gastrointestinal procedures.
Broncho-pharyngeal secretions require suction and respiration may be assisted by using a respirator with a high fractional inspired oxygen concentration (FiO2). In parallel, antidotal treatment should be instituted.
6.2 Antidotes
6.2.1 Atropine
Atropine represents the cornerstone in the treatment of OP poisoning. This competitive antagonist at m-receptors prevents early life-threatening symptoms such as bronchoconstriction, excessive bronchosecretion and impaired respiratory drive. Atropine is less effective in counteracting convulsions which, however, respond favourably to benzodiazepines,[30,70] with diazepam 10–20mg intravenous (IV) recommended.[71,72]
While there is general consensus for using atropine in OP poisoning, recommendations on the administration regimen in adults vary widely.[73] For the treatment of severe symptoms, Grob[74] preferred 4–6mg atropine followed by further 2mg doses at 3- to 5-minute intervals until bronchial secretions ceased. A total of atropine 24–48mg was considered sufficient for the first day. Another suggested approach starts with atropine 2–4mg and doubles the dose every 5–10 minutes until bronchosecretion is cleared. Thereafter, the dose should be adjusted to maintain the effect.[75] To this end, we found continuous infusion with atropine 1–2 mg/h sufficient in most cases, resulting in plasma concentrations of about 10 nmol/L.[76] Aggressive atropinisation protocols, that obviously exceed the goal of controlling excessive acetylcholine at m-receptors, can also be found in the literature.[77,78] Excessive atropine concentrations may block presynaptic m2-autoceptors that regulate the acetylcholine output at cholinergic nerve endings, and may result in the symptoms becoming refractory to atropine.[79]
Development of ventricular arrhythmia including fibrillation was observed in a patient with cyanosis due to impaired respiration. From this it was recommended that, when practical, the patient should be oxygenated in advance. This, however, should not prevent the quick administration of atropine in settings where oxygen is not available. In addition, paralytic ileus may develop, resulting in retarded absorption of the OP and relapses when peristalsis resumes. Finally, care should be taken when administering atropine in hot climates, particularly with children, because sweating is inhibited.[80]
To conclude, a regimen with careful titration of atropine sufficient to control bronchosecretion and bronchoconstriction, to preserve normal bowel sounds, and to keep the heart-rate at about 80 beats/min seems most appropriate.™ Such a moderate approach is facilitated when oximes are also used to support cholinergic function as specific antidotes.[81]
6.2.2 Oximes
While atropine is highly effective in antagonising acetylcholine at most peripheral m-receptors and is somewhat effective at central m-receptors, it fails at n-receptors. Here, reactivating oximes can act as specific antidotes.
In 1955, Wilson and Ginsburg[43] in the US and Childs et al.[44] in England researched and published independently on the efficacy of the compound 2-PAM iodide (molecular weight 264.1Da) as a reactivator of phosphorylated cholinesterases. Since then, several salts of pralidoxime (2-PAM) have been used: pralidoxime chloride (Protopam chloride®Footnote 1; molecular weight 172.6Da); pralidoxime methanesulfonate (P2S; molecular weight 232.3Da); and pralidoxime methylsulfate (Contrathion®; molecular weight 248.3Da).Footnote 2
In a frequently cited article, Namba and Hiraki wrote in 1958:[82] “Hithertoo, alkylphosphate poisoning has been treated mainly by atropine, but now atropine is replaced by pralidoxime”. This enthusiasm was criticised by others and there are at least as many reports on disappointing results.
Another compound, obidoxime (Toxogonin®), was synthesised by Lüttringhaus and Hagedorn in 1964.[83] The first clinical trials showed that this bispyridinium aldoxime was clearly more potent than pralidoxime in reactivating AChE in OP-poisoned patients;[84] yet, its effectiveness was questioned by others.[85] This debate is ongoing and it appears that two opposing views exist: (i) those who promote oximes as the first-line antidote with atropine being an adjunct;[81] and (ii) those who discount any beneficial action of oximes and reject their administration in any organophosphate poisoning.[86] Obviously, such a dichotomy calls for an in-depth search of the underlying reasons of the failure of oxime therapy.[38,87]
-
There may be several reasons why oximes may be ineffective in OP poisoning:[30,38]
-
1. There may be very slow reactivation rates because of steric or electronic effects. This is particularly known with the phosphoroamidates such as tabun when inhibited AChE is notoriously resistant towards reactivation.
-
2. The rate of re-inhibition may be faster than that of reactivation because the oxime dose is too low or the poison load is too high. The latter is particularly observed in mega-dose poisoning seen in suicide attempts.
-
3. Reactivation produces rather stable phosphoryloximes that inhibit AChE at much higher rates than the parent oxons and are slowly eliminated. Such a situation may be more common with 4-pyridinium aldoximes than with the more labile phosphoryloximes from 2-pyridinium aldoximes.
-
4. The optimal oxime concentration is not maintained for long enough, for it is (mostly erroneously) believed that oxime therapy is ineffective due to aging, making reactivation impossible. In vivo, however, aging is usually slower than observed in vitro because acetylcholine accumulates at the synapse and competes with the OP for the active site of AChE. Moreover, sufficient oxime therapy keeps a percentage of AChE active and protects it from aging.
-
5. Oxime therapy is frequently terminated too early. Highly lipophilic OP compounds may persist in deep compartments much longer than expected, leading to re-inhibition and precipitating a secondary crisis due to acetylcholine overstimulation of the end-plate, such as the Intermediate Syndrome.[62]
Success or failure of oxime therapy critically depends on all of these factors. Thus, similar outcomes achieved with and without oxime therapy should also consider differentiating the underlying poison, the estimated poison load, the time elapsed between OP ingestion and institution of oxime therapy, and the duration and dosage of the oxime therapy. These pertinent issues are discussed in more detail in the sections below.
Oxime-Induced Reactivation
There is ample evidence that the oxime reactivators form an associative complex with a similar orientation to that expected for the phosphylation reaction (this term includes phosphorylation, phosphonylation and phosphinylation). Figure 3 depicts the essential features. While reaction 1 is thought to be fully reversible with Kd = k-1/k+1, nucleophilic attack of the oximate on phosphorus leads to a pentacoordinate transition state of the intermediate phosphyloxime, a reaction that is facilitated by polarisation of the phosphorus oxygen bond in the oxyanion hole of the enzyme.

Proposed scheme of chemical reactions involved in the reactivation of phosphorylated cholinesterase. Reactivation is expected to proceed from a tetrahedral ground state of the phosphoryl moiety via a reversible adduct (reaction 1) to a pentacoordinate transition state having a trigonal bipyramidal geometry that liberates the reactivated enzyme along with a phosphoryl oxime (reaction 2).[88]
Ideally, the nucleophile and the leaving group, i.e. the reactivated enzyme, would occupy apical positions when assuming an in-line Sn2 displacement reaction.[88] However, owing to spatial constraints of the gorge and steric limitations of the bulky oximes, optimal geometry for the interaction is not achieved. Accordingly, reactions with oximes are usually much slower than phosphylation reactions.[89]
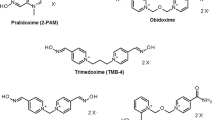
The oximes that shall be addressed here (for formulae see figure 4) are the two marketed compounds, pralidoxime (various salts) and obidoxime chloride, and the experimental oximes HI 6 dichloride (asoxime chloride[90]) and HLö 7 dimethanesulfonate, the proposed reactivators of choice against nerve agents. HLö 7 with its broad antidotal spectrum has very promising properties, high efficacy against nerve agents along with high efficacy against the classical OP pesticides.[38,91–93] However, its synthesis is difficult and its development is much less advanced than that of HI 6.[94–96] HI 6 is available for military use in some countries, has already been manufactured in autoinjectors,[97–99] and was fielded by Canada during the 2nd Gulf War.[100]

Structural formulae of some important pyridinium oximes. 2-PAM = pralidoxime.
Oxime reactivators appear to show distinctive preferred entry routes for attack of the phosphyl moiety, and experiments with site directed mutagenesis have shown that simple side-chain substitutions in AChE may significantly alter the reactivity with oximes.[89] Hence, marked species differences in susceptibility of their phosphylated cholinesterases towards oximes are to be expected and have been proven experimentally with erythrocyte AChE.[93]
The lower part of figure 3 deserves additional comment with regard to reaction 2. This reaction is in fact reversible since the free phosphyloxime (POX) is in itself a powerful phosphylating agent. The identification of these POXs was mostly achieved by synthetic means.[101–105] There is, however, a large body of data showing that POXs are also generated in reactivation media, and a recent study confirmed formation of ethoxy methyl-POX by nuclear magnetic resonance of the naturally abundant non-radioactive 31P isotope (31P-NMR) spectroscopy, arising during reactivation of the respective AChE conjugate with obidoxime and trimedoxime (TMB-4).[106] A high inhibitory potency of the POXs towards AChE has been demonstrated, which may exceed the inhibitory potency of the parent OP by one or two orders of magnitude.[102,107–110] Also, unpublished experiments in the author’s laboratory have shown that POX generated from paraoxon and obidoxime, i.e. diethylphosphoryl-obidoxime, inhibits human erythrocyte AChE 300 times faster than the parent paraoxon (pH 7.4; 37°C).
Despite the high intrinsic inhibitory activity of POXs, their deleterious effects are generally small as concentrations are very low. Given that enzyme recovery only occurs because of oxime-induced reactivation, formation of POX equals formation of free enzyme on a molar basis. In human blood, the AChE concentration is about 3 nmol/L,[111], hence complete reactivation of fully inhibited AChE would produce only 3 nmol/L POX. Nevertheless, such a concentration is roughly equivalent to 1 Limol/L paraoxon, which inhibits half of erythrocyte AChE within about 30 seconds. This phenomenon is usually not detected when in vitro reactivation experiments are performed with dilute enzyme preparations, which makes the bimolecular re-inhibition reactions much slower.[38] With concentrated enzyme solutions, the reactivation curves usually deviate from pseudo-first-order kinetics, and when the rate of re-inhibition by POX equals that of the reactivation by the oxime net reactivation no longer increases.[112,113]
Few data exist on the stability of the various POXs that are formed during reactivation of phosphylated cholinesterases by different oximes. Generally, POXs formed from pyridinium-2-aldoxime derivatives, such as pralidoxime and HI 6, are very unstable and do not accumulate to any appreciable amount.[88,102,105,107,108] In contrast, the stability of diethylphosphoryl-obidoxime (unpublished data) and of ethoxy methylphosphonyl obidoxime is considerable with half-lives of 10–20 minutes under physiological conditions. Thus, it is evident that the effectiveness of oxime reactivators is largely determined by the decay rate of the POX formed in this process.
There has been much concern about destroying POX in vivo to enhance the effectiveness of oxime therapy. Bacterial enzymes have been shown to degrade POX rapidly,[105] and there is evidence that human plasma has an enzyme activity catalysing the degradation of dimethyl- and diethyl-POX formed during the reactivation by obidoxime and TMB–4, respectively.[114], It has been shown in our laboratory that diethylphosphoryl-obidoxime decays non-enzymatically with formation of an intermediate nitrile that eventually liberates a pyridone along with cyanide. Such a reaction has been previously postulated[115], and was underlined by experiments.[116], In contrast to this cyanogenic pathway, plasma-dependent POX degradation results in formation of intact oxime along with dialkoxyphosphoric acid and appears to be a true hydrolysis reaction. The genetic polymorphism that underlies the expression of this enzyme in plasma is discussed in the section below.
Kinetics of Oxime-Induced Reactivation
The kinetic constants of AChE from human erythrocyte membranes inhibited with paraoxon ethyl or paraoxon methyl have been determined. The rate of reactivation was followed under physiological conditions, i.e. at pH 7.4 and 37°C. Details are published elsewhere.[13,38] Table V compiles the kinetic data for the reactivation of dimethyl phosphoryl and diethyl phosphoryl AChE by various oximes. The efficiency (k2 = kr/Kd) of obidoxime was superior for both phosphorylated enzymes compared with the other oximes. HLö 7 was also quite efficient while pralidoxime and particularly HI 6 were clearly inferior to obidoxime. Figure 5 shows the dependence of the reaction t1/2 versus the oxime concentration of the two compounds on the market, pralidoxime and obidoxime, in a double-logarithmic plot to cover the whole range of interest. The curves were calculated for human erythrocyte AChE inhibited by either paraoxon ethyl or paraoxon methyl. It is obvious that the reactivation t1/2 increase proportionally when the oxime concentration falls below 10-5 mol/L, but little is gained at oxime concentrations above 2 x 10-4 mol/L. Thus, a therapeutic plasma concentration of about 10-4 mol/L would be optimal for both oximes, but the efficacy of obidoxime is much higher than that of pralidoxime for both dialkoxy phosphoryl enzymes.

Kinetic data for the reactivation by oximes of dimethyl phosphoryl (diMe) and diethyl phosphoryl (diEt) acetylcholinesterase of human erythrocytesa

Double-logarithmic plot showing the relationship between reactivation t1/2 (min) and oxime concentration. Using the equation: t1/2 = In 2 x (Kd + [Ox])/(kr x [Ox]), the t1/2 was calculated for the reactivation of erythrocyte acetylcholinesterase inhibited by paraoxon ethyl (bold lines) or paraoxon methyl (hair lines) and reactivated with obidoxime (solid) and pralidoxime (broken), respectively. The kinetic parameters are listed in table V. Kd = dissociation constant of the Michaelis complex; kr = reactivation rate constant; Ox = oxime concentration; dt1/2 = half-life; 2-PAM = pralidoxime.
The concentrations of oximes necessary to achieve 20% net reactivation within 5 minutes are shown in table VI. A residual AChE activity in this range is usually considered sufficient for normal cholinergic transmission.[30] From table VI it becomes clear that the statement “a therapeutic oxime plasma concentration of 4 mg/L” irrespective of the organophosphate involved or the oxime used is insufficient as a dosage guideline.

Oxime concentrations necessary to arrive at 20% reactivation of dimethyl and diethyl phosphoryl acetylcholinesterase (AChE) within 5 min
Kinetics of Oxime-Induced Reactivation in the Presence of Organophosphorus Pesticide
With the kinetic constants of inhibition and reactivation at hand, the fraction of active AChE at equilibrium in the presence of oximes and organophosphates can be calculated. Figure 6 shows the calculated curves for paraoxon and the marketed oximes; these data have been verified by experimental data from human erythrocytes (pH 7.4, 37°C).[13,38] It is obvious that with pralidoxime significant enzyme activity at paraoxon concentrations above 10-7 mol/L can only be maintained at pralidoxime concentrations above 100 μmol/L (requiring continuous infusion at 12 g/day, see figure 7 and the Proposed Oxime Dosage Regimen section) while the same effect is achieved with 10 μmol/L obidoxime (attained with 0.75 g/day). The curves in figure 6 can be shifted to the right with higher oxime concentrations but approach an asymptotic value when the concentration markedly surpasses the oxime Kd (see table V).

Calculated acetylcholinesterase (AChE) activity in the presence of various concentrations of paraoxon and obidoxime (accumulation of phosphoryloxime not considered). The inhibition rate constants for human erythrocyte AChE are given in table II, the reactivation constants in table V, and the formula used for calculation is given in figure 12 (equation 11). 2-PAM = pralidoxime.

Calculated plasma concentration of pralidoxime following two regimes. Regime A uses intermittent bolus doses of pralidoxime chloride 1g qid (injection time 2 minutes). Regime B consists of a short-time infusion of pralidoxime chloride 1g over 30 minutes, followed by a continuous infusion at a rate of 0.5 g/h. The time course was calculated for a 70kg person, using the kinetic data and methods of calculation as given by Sidell et al.[119], qid = four times daily; 2-PAM = 2-pyridine aldoxime methochloride.

Calculation of acetylcholinesterase inhibition in the presence of organophosphate (P) and oxime (Ox).
To establish a proper oxime dosage regimen the pharmacokinetic data of the therapeutic oximes of interest should be revisited. In the next section, the data for pralidoxime, obidoxime and HI 6 dichloride are presented. Where necessary, animal data are also included.
Pharmacokinetics of Therapeutic Oximes
It should be noted that pharmacokinetic data are predominantly from volunteer studies and do not necessarily reflect oxime kinetics in severely poisoned patients.
Distribution
Following intravenous injection, pralidoxime distributes in most body fluids and is not significantly bound to plasma proteins or haemoglobin. Pralidoxime penetrates the red cell membrane in vivo and in vitro in a concentration-independent manner, presumably by simple diffusion. The transfer follows first-order kinetics with a t1/2 of about 4.5 hours.[117], Behaviour of the 3-hydroxy derivative of pralidoxime was similar. However, the penetration t1/2 under physiological conditions was much shorter and approached 6 minutes with this derivative.[118], In contrast, bispyridinium oximes such as obidoxime, TMB-4 and HLö 7 did not penetrate the red cell membrane to any appreciable extent.[117,118], Data for the apparent volume of distribution at steady state (Vdss) is in agreement with these findings: Vdss for pralidoxime chloride 5 mg/kg IV was 0.7 ± 0.1 L/kg in human volunteers.[119,120], A contrasting value was found after 12.5 mg/kg intramuscularly (IM) with wide variations 2.7 ± 2.1 L/kg.[121], This figure was similar in OP-poisoned patients, 2.8 ± 2.2 L/kg.[121], Willems and coworkers, after administering a loading dose of pralidoxime methylsulfate 4.4 mg/kg IV followed by continuous infusion at 2.1 mg/kg/h, found a volume of distribution (Vd) in the ß-phase of 2.77 ± 1.45 L/kg in OP-poisoned patients.[122], Hence, it appears that the volume of distribution of pralidoxime in OP-poisoned patients is markedly higher than in healthy persons.
Obidoxime chloride exhibited a much smaller Vdss in healthy volunteers, 0.17 ± 0.02 L/kg.[119], Similarly, Vd was 0.25 ± 0.04 L/ kg for another bispyridinium compound, HI 6 dichloride, administered IM in combination with atropine sulfate 2mg to healthy young men.[123], In OP-poisoned patients treated with a continuous infusion of obidoxime chloride at 31 mg/h (n = 27) we found a two-compartment distribution at steady-state with V1 = 0.32 ± 0.11 L/kg and V2 = 0.28 ± 0.12 L/kg (unpublished data). An even larger Vd of obidoxime, 0.85 L/kg, was reported for a methamidophos-poisoned patient who also had renal failure.[124],
The deep compartment for such a polar compound with quite a high molecular weight (359Da for obidoxime) was surprising. Autoradiographic studies in rats showed radioactive material from pralidoxime, obidoxime, HI 6 and other pyridinium oximes was concentrated in the kidney and mucopolysaccharide-containing tissue such as cartilage and intervertebral disc.[125–129] Some radioactive material also appeared to be concentrated in the liver,[129], and was found in stomach, bile and intestine.[125], The enrichment of cationic substances in chondroitin sulfate-rich tissue is not unusual and is most probably based on ionic interactions.[130–132] These data indicate that proteoglycan-containing tissues may represent a deep compartment for pyridinium aldoximes from which the compounds are slowly released.
A more intriguing question is the accessibility of the CNS to pyridinium oximes. There are numerous contradictory reports of animal experiments showing occasionally detectable oxime levels in brain and cerebrospinal fluid (CSF). Mainly, oxime doses far beyond those administered to humans were used and the positive assays in brain tissue may reflect damage to the blood-brain barrier rather than natural permeability to oximes. In a recent report, pralidoxime was detected by microdialysis technique in the striatal extracellular fluid after pralidoxime iodide 10, 50 and 100 mg/kg IV to rats, the ratio of brain and blood concentration being approximately 1 : 10.[133], Pralidoxime was undetectable in the spinal fluid of a 36-year-old epileptic man infused with pralidoxime iodide for 1 hour (total dose 44 mg/kg).[134], A central action of pralidoxime chloride (500mg IV given over 15 minutes) was assumed in the case of a 3.5-year-old child poisoned with parathion. The physicians could not relate the dramatic improvement of the EEG pattern and the simultaneous restoration of consciousness to any improvement in circulatory or respiratory function.[135]
The bispyridinium oxime HI 6 was detected in the brain of anaesthetised rats when given in a dosage of 50 mg/kg IV, the maximum content amounting to about 10 nmol/g tissue.[136], HI 6 kinetics were also followed in the CSF of beagle dogs HI 6 18.5 mg/kg IV. HI 6 did not produce behavioural changes or toxic effects on the cardiovascular system.[137], The oxime concentration slowly increased in CSF, peaking at 1.6 μmol/L after 90 minutes, when plasma concentration had already fallen from its maximum level (140 μmol/L after 15 minutes) to 65 μmol/L.[137], Obidoxime 4 μmol/L in CSF was also detected in a parathion-poisoned patient on day 6 upon continuous obidoxime infusion when the plasma concentration was 17 μmol/L. CSF puncture in this patient was necessary because of some meningism which, however, was not to be associated with inflammatory processes. The same CSF sample still contained some 5 nmol/L paraoxon while the concentration in plasma was 29 nmol/L (unpublished data). Obidoxime also was detected in the frontal cortex tissue of a deceased patient following parathion poisoning. Four days after discontinuation, obidoxime in brain tissue amounted to 0.85 nmol/g wet weight while plasma contained 0.5 nmol/g. All these data indicate that the blood-brain barrier is not entirely impermeable to quaternary pyridinium compounds and may become even more permeable in stress situations, such as in severe intoxication.[138,139]
Metabolism and Excretion
When 1-[14C]methyl-pyridinium-2-aldoxime iodide (40 or 100 mg/kg) was administered to rats intraperitoneally, about 90% of the radioactivity was excreted in the urine within 1 day, 80–90% of the radioactivity being associated with unchanged pralidoxime while 5% was associated with 1-methyl-2-pyridone and about the same amount was associated with 1-methyl-2-cyanopyridinium ion. Less than 0.1% of the radioactivity was exhaled.[140], In a more recent study, pralidoxime iodide 20 mg/kg IM ([14C]-labelled in the oxime group; radiochemical purity >98%) was administered to male rats. Within 72 hours, 90.8 ± 2.3% of the radioactivity was excreted in the urine and 5.8 ± 1.8% in the faeces. Exhalation of [14]CO2 was not detected. About 90% of the urinary radioactivity was associated with intact pralidoxime.[129], These data indicate that after parenteral administration of pralidoxime to rats more than 80% is excreted unchanged.
In humans, 1-methyl-2-cyanopyridinium ion was detected in the urine of four male volunteers who received oral pralidoxime methanesulfonate 3g, suggesting some cyanogenic metabolism. However, urinary thiocyanate did not increase significantly.[141,142], Since in human volunteers the total urinary recovery of pralidoxime chloride 5 mg/kg IV was 89.8 ± 2.6% of the dose, cyanide formation is probably not a toxicological problem.
Obidoxime is chemically more stable than pralidoxime and no metabolites have been reported. In dogs (n = 4), following 10mg administration of obidoxime chloride 28 μmol/kg IV, 77% of the dose was recovered unchanged in urine in the first 24 hours.[118], In human volunteers (n = 5) who received 5 mg/kg obidoxime chloride IV, 68 ± 8% of the dose was eliminated unchanged in urine in the first 24 hours.[119], The metabolism and excretion of the remaining 30% of the obidoxime dose has not been reported. Although we screened the urine chromatograms we were not able to detect material indicative of a pyridinium compound.[118], Hence, some elimination by the biliary/faecal route cannot be ruled out.
The bispyridinium oxime HI 6 dichloride is chemically less stable than obidoxime in solution. When 50 mg/kg IV was administered to rats only 57 ± 2% of HI 6 was recovered unchanged in the urine in the first 24 hours. Two metabolites, with most likely a pyridone structure, were identified comprising some 20% of the administered dose.[143], In dogs receiving HI 6 dichloride 38 mg/kg IV, 79.8 ± 1% (n = 3) of HI 6 was recovered unchanged in the urine in the first 6 hours. About 4% of HI 6 equivalents were found in cyanide that had been trapped in methaemoglobin-containing erythrocytes and was calculated to stem exclusively from abiotic degradation of HI 6 in the body and not from biotransformation.[144], In a subsequent dog study with [14C]aminocarbonyl-HI 6 38 mg/kg IV, 94% of the dose was excreted in the urine within 5 days, 83% of it being associated with intact HI 6. The remaining radioactivity was found in the corresponding pyridone (5%) and in the deaminated carboxylic acid derivative of HI 6 (2%). Free isonicotinamide (3%) and isonicotinic acid (1%) indicated cleavage of the aminal-acetal bridge.[145], These data suggest that in the dog probably 10% of HI 6 is cleaved at the aminal-acetal bridge and about 5% channelled through the cyanogenic pathway yielding equivalent amounts of pyridone and cyanide. Most of this ‘metabolism’ appears to stem from abiotic transformation of HI 6[146], in vivo. Hence, similar results can be expected in humans.
Elimination
Disappearance of pralidoxime chloride administered in doses of 2.5–10 mg/kg IV to male human volunteers could be described by a two-compartment open model with a distribution half-life (t1/2α) of 8.8 ± 2.0 minutes and a terminal elimination half-life (t1/2ß) of 73.7 ± 15.2 minutes.[147], In another volunteer study, pralidoxime chloride or pralidoxime methanesulfonate 5 mg/kg IV showed a t1/2ß of 78.6 ± 7.8 and 84.6 ± 14.4 minutes, respectively.[119], The t1/2ß of pralidoxime chloride after IM administration of 2.5–10 mg/kg was also in this range, i.e. 83 minutes after 10 mg/kg. In another study, pralidoxime plasma concentrations declined more slowly, when approximately 12.5 mg/kg was administered IM to healthy servicemen. Upon fitting the data to a one-compartment open kinetic model, a t1/2ß of 148.9 ± 65.7 minutes was calculated.[121], The difference in t1/2ß between the two studies has not been addressed.[121], However, recalculation of the latter data points to a plateau at infinite time indicating a possible artefact in determination. If such a ‘blank’ is accepted, the mean t1/2ß is shortened to 96 minutes
In OP-poisoned patients, the pralidoxime t1/2ß was considerably prolonged in two studies: 174.4 ± 70.9 minutes (n = 6)[121], and 206.4 ± 54 minutes (n = 9).[122], In the former study, approximately 13 mg/kg pralidoxime was injected IM; in the latter, elimination was measured after intravenous infusion of pralidoxime methyl-sulfate (Contrathion®) 2.14 mg/kg/h. It has been concluded that as the mean clearance of pralidoxime was similar in patients and volunteers, the much higher volume of distribution in patients (see ‘Distribution’ section) may have caused the delayed elimination.[122]
Obidoxime chloride 0.5–1.0 mg/kg IV administered to volunteers was eliminated with a t1/2 of 72 ± 9.6 minutes (n = 5).[119], After 2.5–10 mg/kg IM a mean t1/2 of 83 minutes was found.[148], After administration of 250mg IM, adults eliminated obidoxime with a mean t1/2 of 120 minutes.[149]
In our recent clinical study of OP-poisoned patients (n = 27) who received obidoxime chloride 250mg IV bolus followed by continuous infusion at 750 mg/24h, obidoxime was eliminated biphasically with half-lives of 2.15 ± 1.29 hours and 13.9 ± 6.7 hours.[150], The latter phase is indicative of the deep compartment as already mentioned in the ‘Distribution’ section. The transfer rate constant into and from this deep compartment are slow and in the order of 0.135 per hour (unpublished data). Accordingly, no indication of a deep compartment was found following a single IV bolus of obidoxime chloride to an OP-poisoned patient who exhibited a t1/Jp of 2.1 hours.
In human studies, HI 6 dichloride 250 or 500mg IM administered to male healthy volunteers was eliminated with half-lives of 84.9 and 79.9 minutes, respectively. Some 60% of the dose was eliminated unchanged in the urine.[94] In another study, ascending doses of HI 6 dichloride 60, 125, 250 and 500mg IM administered to healthy young men were eliminated with half-lives of some 80 minutes. Urinary recovery of unchanged HI 6 averaged around 67% at all four dose levels. When HI 6 dichloride 500mg IM was administered to OP-poisoned patients t1/2ß of about 2 hours were observed.[151], The t1/2ß of HI 6 in dogs was shorter, about 50 minutes,[137], suggesting a higher proportion of HI 6 may undergo abiotic degradation in humans. Even if one assumes 10% cyanogenic decomposition of the 500mg HI 6 dichloride (1.33 mmol), only 3.6mg hydrocyanic acid will be produced, an amount that is detoxified within 1 hour (1 μg/kg/min).[152], Thus, cyanide intoxication is not to be expected following a single HI 6 500mg dose.
It has already been suggested that pathophysiological changes following anticholinesterase poisoning may markedly alter the rate of distribution of therapeutic agents. This effect is most likely a consequence of changes in blood flow to various organs.[153], Thus, pharmacokinetic data for pyridinium oximes in healthy humans probably cannot be extrapolated to severely poisoned patients.
Safety and Tolerability of the Therapeutic Oximes The therapeutic pyridinium oximes have been selected for use as antidotes in OP poisoning because of their high affinity for phosphylated AChE. This property also implies interactions with substrate hydrolysis of the enzyme. Table VII shows the inhibition constants of the four oximes as obtained with electric eel and human erythrocyte AChE under physiological conditions. It can be seen that the Ki value describing the dissociation constant of the substrate-free enzyme is around 300 μmol/L, while the Kii value describing the dissociation constant of the oxime from the enzyme-substrate complex is 5- to 10-times higher. That means 10 μmol/L oxime is virtually without any effect while 100 μmol/L is expected to inhibit acetylcholine hydrolysis to an appreciable extent.

Inhibition constants [mmol/L] of oximes towards acetylcholinesterases (AChE)[154],a
When young volunteers (n = 18) received pralidoxime iodide 15–30 mg/kg IV over 2–4 minutes, the following symptoms were found: dizziness (n = 8), blurred vision (n = 13), diplopia (n = 9), headache (n = 4), impaired accommodation (n = 4) and nausea (n = 3).[155], Blurred vision, diplopia and dizziness were also caused by pralidoxime methanesulfonate, lasting a few minutes following administration of 30 mg/kg IV.[156], Administration of pralidoxime chloride 30 mg/kg IM resulted in ECG changes with T-wave elevation and increased blood pressure.[157], In another study, IM injection of pralidoxime chloride up to 600mg caused no changes in heart rate or blood pressure and only mild pain at the site of injection.[158], Burning and stinging at the injection site following 0.5–1.0g pralidoxime chloride IM also was reported by others.[159], Volunteers experienced dizziness and blurred vision when plasma pralidoxime levels approached 80 μmol/L, corresponding to 14 mg/L pralidoxime chloride.[160]
A serious adverse effect was related to pralidoxime iodide infusion in a coumaphos-intoxicated patient. Following infusion of 0.4g over 2 minutes the patient experienced a cardiac arrest in asystole. He responded rapidly to intravenous sodium bicarbonate and adrenaline. When the pralidoxime infusion was recommenced, the patient had a further cardiac arrest within 2 minutes (0.4g). It was unclear whether the small amounts of neuroleptics or the hydrocarbon solvents co-ingested were causally related to this serious adverse event.[161]
Vomiting following rapid IV injection of pralidoxime chloride 1g was frequently seen in OP-poisoned patients (Eddleston M, personal communication). Besides vomiting, transient impairment of respiration (slow and shallow breathing, probably a peripheral effect at the neuromuscular junction) was also reported following rapid injection of pralidoxime.[159], Rapid injection was also associated with tachycardia, laryngeal spasm, muscle rigidity and transient neuromuscular blockade.[162], Hence it appears wise to avoid excess peak plasma concentrations of pralidoxime that may elicit toxic symptoms.
Obidoxime chloride 250mg IM had a tolerability profile different to that observed after injection of pralidoxime, which included a hot and tight feeling in the skin around the mouth and inhibition of the face muscles, developing within 15 minutes of injection.[148,149,163], In addition, a cold sensation in the rhino-pharyngeal space during inspiration, similar to the effect of menthol, was reported.[148,149], At a higher dose, 10 mg/kg IM, dry mouth and a hot feeling in the throat was a unanimous symptom in all subjects who also experienced a variety of paraesthesias. All subjects showed a mild-to-moderate transient increase in systolic and diastolic blood pressure reaching peak values at about 30 minutes, along with an increase in heart rate (30 beats/min after 10 mg/kg IM).[148], In another study, eleven volunteers of both sexes were given obidoxime chloride 250mg IM to screen for possible liver toxicity. In the follow-up period of 1 month, no abnormal liver data were found. The volunteers experienced adverse events similar to those ascribed above to obidoxime.[164], Following high obidoxime doses (several grams per day) in severely OP-poisoned patients, hepatotoxic effects were occasionally observed, including increased serum transaminases, jaundice and cholestasis.[165–167]
From clinical data[168], and from a discussion statement[169], it appears that the hepatotoxicity of obidoxime is a transient phenomenon and occurs in about 10% of patients with severe poisoning.[30] Obidoxime-treated patients in general had normal liver function on discharge. In our recent Munich study, all patients received an obidoxime 250mg bolus followed by a continuous IV infusion of 750 mg/24h. No patient with dimethoate poisoning (n = 6) showed pathological liver laboratory findings. Five patients with oxydemeton-methyl poisoning (n = 12) had transient pathological liver findings, one of whom died. Eight patients with parathion poisoning (n = 13) had transient pathological liver findings, five of whom died. Hence, it cannot be ruled out that the type of poison may have some influence on liver function. Cardiac arrhythmias have occasionally been associated with obidoxime therapy (one 2nd grade atrioventricular block; two atrial fibrillation, n = 17).[159], We also observed some transient arrhythmias in the Munich study; however, these were considered not related to obidoxime.
When HI 6 dichloride 10% in water was injected IM into the gluteal muscle of healthy male servicemen (62.5 to 500mg) there were no significant changes found in blood pressure, heart rate, ECG, blood chemistry or urine analysis, except for the elevated potassium levels found in those receiving the 500mg dose (n = 8).[94]
Ascending doses of HI 6 dichloride 62.5 to 500mg in conjunction with atropine sulfate 2mg IM were well tolerated by healthy male volunteers (double-blind, placebo-controlled) and no serious clinical complaints were reported. No clinically significant changes in heart rate, ECG, blood pressure, respiration or mental acuity were noted. Blood chemistry, urine and semen analysis over the 24-hour observation period were normal. Creatine phosphokinase and aspartate aminotransferase were elevated, particularly in the high dose group (n = 6), probably as a result of the hypertonic solution injected in the rectus femoris muscle.[96] These data indicate that HI 6 dichloride 500mg is better tolerated than obidoxime 250mg.
-
From the theoretical considerations discussed in the sections above, the pharmacokinetic data of the oximes and the tolerability concerns, the following two conclusions may be drawn:
-
The recommendation of ‘a therapeutic oxime plasma concentration’, generally about 4 mg/L[94,151,170], should be dismissed. Such a concentration was originally proposed by Sundwall in 1961 for P2S as the minimal effective concentration that reversed neuromuscular block in cats intoxicated with a quaternary sarin-analogue.[171], However, this concentration has been promulgated by others, irrespective of the underlying poison or oxime and the species involved, including humans.
-
Considering the low propensity of diethyl phosphoryl AChE for aging, one must query the usually recommended duration of oxime administration. For example, the product information for Toxogonin® (obidoxime chloride) advises that obidoxime should be given as soon as possible after intoxication, and that a second dose can be administered after 2–4 hours, while later administration is to be avoided. This recommendation probably dates back to the opinion of Erdmann in 1968.[172], Such a scheme, however, largely ignores the long residence time of lipophilic OPs that will re-inhibit the reactivated enzyme if the reactivator is no longer present. In addition, keeping the enzyme in the active state will reduce the amount of enzyme prone to the stochastic event of aging.[173], Even with the notoriously fast aging dimethyl phosphoryl AChE, aging will be retarded in the presence of oximes, which should therefore be given for longer periods.[30] Since a high concentration of active poison may overcome the reactivation power of an oxime, net reactivation will usually not be observed with a heavy poison load, as occasionally found in suicide attempts. It would be a clinical tragedy to discontinue oxime administration at this point and to surmise that AChE has already aged.[173], Rather, oxime administration should be continued over much longer periods to become effective when the inhibitor falls below the critical concentration (see figure 6). Hence, prolonged maintenance of an appropriate oxime concentration appears desirable [60,81,174–180]
Proposed Oxime Dosage Regimen
Various dosage regimens have been recommended from intermittent oxime administration to continuous infusion following a loading dose. For pralidoxime chloride, intermittent doses of 1g every 6 hours have been used[162,174], or alternatively a 1g short infusion followed by continuous infusion at 0.5 g/h.[174,180], The latter approach appears more appropriate.[177], Model simulations[174], of these two regimens are illustrated in figure 7 for a healthy 70kg person. It is obvious that the deep troughs following the peak levels are less appropriate. On the other hand, peak values approaching 50 mg/L may produce appreciable adverse effects, e.g. when a 1g bolus is injected within 2 minutes. From the data presented by Medicis et al. short-term infusion of 1g pralidoxime chloride within 30 minutes to healthy volunteers (about 70kg) resulted in plasma levels, corresponding to 30 mg/L pralidoxime chloride, that elicited a highly significant increase in diastolic blood pressure of 20mm Hg.[160], The computer simulation in figure 7 indicates a similar peak value after the short-term infusion (1g per 30 minutes) and a concentration of 13 mg/L at steady-state which is below the 14 mg/L threshold that can produce dizziness and blurred vision.[160], A plasma concentration of13 mg/L is reached 8 minutes after starting the short-term infusion. Inspection of figure 5 clearly points to the need for plasma concentrations between 50 and 100 μmol/L for reactivation t1/2 <10 minutes, necessary to cope with higher poison concentrations (see figure 6). Hence it appears that such a regimen should be both effective and safe.
There had been some concern whether the calculations based on data from young healthy volunteers[119,160], also apply to poisoned patients. There are some data showing that the targeted plasma concentration of 4 mg/L, calculated from the data from volunteers, was approximately obtained in OP-poisoned patients (between 2.1 and 9; n = 9) with pralidoxime methylsulfate.[122], The variation (also intraindividually) was explained by changes in the clinical condition of the patients. Interestingly, the total clearance in patients was very similar to the values found in healthy volunteers.[119], The significantly longer t1/2ß was balanced by an essentially larger Vd. With pralidoxime methanesulfonate (P2S; MW 232) given either by bolus 30 mg/kg IV every 4 hours or by intravenous infusion at 10 mg/kg/h, concentrations ranging between 25 and 840 μmol/L (peak and trough) or 130 and 210 μmol/ L were obtained, respectively.[178], The concentrations found during steady-state are significantly higher than the 80 μmol/L expected (see figure 7; the dosage corresponds to 0.5 g/h pralidoxime chloride in a 70kg person). Hence, diminished clearance in the intoxicated patient may have caused some accumulation. A large variation in the steady-state plasma concentrations (22 ± 12 mg/L, mean ± standard deviation, range 6.9–47, n = 11) was also found in OP-poisoned children receiving pralidoxime chloride 14 mg/kg/ h.[181], Nonetheless, the mean value of 22 mg/L compares well with 25.8 mg/L as calculated with the pharmacokinetic data from healthy volunteers.[119],
For obidoxime, concentrations around 10 μmol/L were adopted. Calculated plasma concentrations in a 70kg person, again using the kinetic data from healthy volunteers from Sidell et al.[119], are shown in figure 8 (for convenience, the amount of one ampoule Toxogonin®, 250mg, was used as the loading dose followed by 3 ampoules in 24 hours). The anticipated concentration was 11.4 μmol/L. OP-poisoned patients, treated with this regime without adjustment for their bodyweight, exhibited plasma concentrations between 14.8 and 7.4 μmol/L (n = 28). Linear regression analysis of the data, figure 9, gave an intercept at 70kg bodyweight of 10.6 μmol/L instead of the calculated 11.4 μmol/L. For this analysis, only patients with a serum creatinine of <1.5 mg/100mL were included. Hence, pharmacokinetic data from healthy volunteers also reasonably apply to intoxicated patients, at least during the first day of intoxication (unpublished data).

Calculated plasma concentration of obidoxime, resulting from a bolus dose of 0.25g obidoxime chloride (injection time 2 minutes) followed by a continuous infusion at a rate of 0.75 g/24h. The time course was calculated for a 70kg person, using the kinetic data and methods of calculation as given by Sidell et al.[119]

Obidoxime plasma concentrations found in organophosphorus pesticide-poisoned patients at steady-state following the standard regimen (250mg intravenous bolus followed by 750 mg/24h). Plasma concentrations at steady-state (between 10 and 20 hours after start) were considered in patients (n = 28) with serum creatinine values below 1.5 mg/100mL (<130 μmol/L). The enlarged dot symbol indicates the calculated value for a 70kg person, using the kinetic data obtained in healthy human volunteers.[119], Linear regression analysis gave y = -0.033 x +12.9, the slope not significantly different from zero.
Data on a suitable HI 6 dosage scheme in OP-poisoned patients are rare. In one major study, HI 6 dichloride 0.5g IM every 6 hours was judged to be at least as effective as pralidoxime chloride 1g IM every 6 hours in quinalphos-poisoned patients. Peak concentrations of about HI 6 50 μmol/L were found after 30 minutes and HI 6 disappeared from plasma with a t1/2 of about 110 minutes.[151], This figure in patients was not much different from the values found in human volunteers (maximum plasma drug concentration [Cmax] 42 μmol/L, time to reach maximum plasma concentration [tmax] 40 minutes t1/2, 80 minutes).[94] In this study, plasma concentrations of pralidoxime were not given. Since plasma pralidoxime peaked at 30 μmol/L after pralidoxime chloride 0.5g IM in human volunteers,[158], Cmax in the patients was probably in the range of 60 μmol/L.
Comparison of the Efficacy of the Various Oxime Dosage Regimens
Using the reactivation parameters found for human erythrocyte AChE (table VI) the t1/2 of reactivation can be calculated with t1/2 = ln 2 x (Kd + [oxime])/kr x [oxime].[182], The following consideration deals with diethyl phosphoryl erythrocyte AChE.
In the pralidoxime regimen with 1g pralidoxime chloride given as short infusion followed by continuous infusion at 0.5 g/h, a steady-state concentration of some 80 μmol/L is attained. This results in a reactivation t1/2 of 12 minutes. In contrast, the fluctuating regimen, 1g four times daily, leads to troughs where reactivation is negligible. For example, 5 hours after the bolus injection, the reactivation t1/2 is about 3 hours. Hence, the proportion of re-inhibited enzyme will be high and prone to aging.
In our favoured obidoxime regime with a loading dose of 250mg obidoxime chloride followed by 750 mg/24h,[116], the steady-state concentration is 10 μmol/L with a reactivation t1/2 of 4.3 minutes.
In the HI 6 dichloride regimen with IM injections of 500mg every 6 hours, the peak plasma concentration is 50 μmol/L, leading to a reactivation t1/2 of 35 minutes. After 4 hours, however, at a plasma concentration of 4 mg/L, i.e. 10.6 μmol/L HI 6, the reactivation t1/2 is around 2.5 hours. Again, such a regimen promotes aging of the enzyme. Curiously, the quinalphos-poisoned patients treated with this HI 6 regimen showed faster reactivation of erythrocyte AChE than the patients treated with pralidoxime[151], while the reactivation t1/2 would have predicted the opposite, i.e. 35 versus 15 minutes. Possibly, the groups were not comparable because of different plasma concentrations of circulating oxon.
On the basis of the three regimens outlined above, obidoxime might be preferred because of its higher efficacy which allows it to counteract higher organophosphate concentrations than pralidoxime chloride or HI 6 dichloride. Concerning tolerability and safety, an exact judgement is not possible as no study directly compares these agents at equieffective oxime dosages. Finally, economic aspects have to be considered when favouring a certain oxime. At present, the daily costs for obidoxime (750mg) are €17 in Germany (about $US20) [2004 values]. The daily costs for pralidoxime (12g) vary widely between different countries and amount, e.g. in Canada, to some $US200 per day. However, one major question remains to be answered that is addressed in the next section: how much does the toxicity of the phosphoryloximes formed by reactivation contribute to the clinical syndrome?
As already outlined, POXs generated from pyridinium oximes with the oxime position ortho to the quaternary nitrogen are highly unstable and prone to rapid breakdown. Hence, the POX problem hardly concerns pralidoxime and HI 6. The O,O-diethyl phosphoryl oxime and possibly also the O,O-dimethyl derivative from obidoxime, however, are more stable and can easily re-inhibit the reactivated enzyme, as the inhibitory activity of O,O-diethyl phosphoryl obidoxime is approximately two orders of magnitude higher than that of paraoxon.[112,114]
There is good evidence that a plasma enzyme, most probably identical to human paraoxonase 1 (PON1) is able to rapidly destroy the POXs. However, PON1 exhibits a substrate-dependent activity polymorphism determined by an Arg/Gln (R/Q) substitution at amino acid residue 192.[183], It appears that the homozygous RR192 form has only weak activity to hydrolyse the POXs, while the heterozygous QR192 and particularly the homozygous QQ192 isoform are active enough to destroy POX effectively.[184], While the studies undertaken in our laboratory relate to in vitro experiments, it was of utmost interest to elucidate whether such a POX effect also occurs in patients. One approach to answer this question was based on a comparison of the erythrocyte AChE activity we actually found with that calculated on the basis of the reaction rate constants and the concentrations of paraoxon and obidoxime in plasma. As deduced in figure 12, such a calculation can tell us whether the paraoxon concentration found can explain the degree of inhibition or whether inhibition is much higher, due to POX interaction.
Figure 10 shows two examples. In case BR 3/97 (a) the measured inhibition of erythrocyte AChE was not different from the calculated inhibition at the given concentrations of obidoxime and paraoxon, indicating this patient had sufficient PON1 with high POX hydrolase activity. We subsequently found only one patient (n = 20), case GJ 2/97 (b), with parathion poisoning who showed appreciably higher erythrocyte AChE inhibition than calculated. This patient probably belonged to the RR 192 phenotype (not analysed). The frequency of the PON1 genotype expressing RR 192 in a population of 376 white individuals was 6%.[185], This is in accordance with our findings of only one suspected person in 20 parathion poisoned patients. It should be noted that people with the RR 192 variant nevertheless would benefit from obidoxime therapy, since a good part of the inhibited non-aged enzyme will always be reactivated as seen in case (b) of figure 10. Finally, one should keep in mind that the POX molecule formed by reactivation of the inhibited enzyme can only inhibit one molecule of reactivated enzyme. Hence POX formation will not cause more harm than already present without giving the oxime. We, therefore, think that obidoxime is the favourite oxime in organophosphorus insecticide poisoning. With regard to nerve agent poisoning the picture changes and asymmetrical bispyridinium oximes, such as HI 6 and HLö 7, are considerably more effective.[93,186–188]

Erythrocyte acetylcholinesterase (EryAChE) activity referred to the haemoglobin content in the blood samples of two poisoned patients in vivo, case BR 3/97 (a) and GJ 2/97 (b), and after reactivation in diluted blood (in vitro) vs time elapsed post-incorporation (p.i.) of parathion. The solid squares refer to the measured activities in vivo. The solid circles are calculated values based on the plasma obidoxime concentrations and the plasma concentrations of unbound paraoxon, assuming 55% protein binding. The open squares represent AChE activities determined in diluted blood samples after reactivation in vitro (see Eyer et al.[65]). For calculation, see figure 12 (equation 11); pharmacokinetic data for human EryAChE are shown in tables II and table V.
The Current Status of Oximes in the Treatment of Organophosphorus Pesticide Poisoning
Until now, no RCTs appear to exist that clearly stratify patients according to the class of pesticide taken (O,O-diethyl or O,O-dimethyl phosphoryl compounds or other OPs), time of presentation and severity of cholinergic crisis on admission. Only those patients that present with clear signs and symptoms of anticholin-esterase poisoning should be included. Patients with definite carbamate poisoning should be excluded from RCTs as there is ongoing concern that oximes may aggravate poisoning by carbaryl.[189], This phenomenon does not necessarily hold true for all other carbamates. In fact, obidoxime therapy in children poisoned with anticholinesterases did not influence the clinical course in those cases where the poison was later identified as a carbamate, specifically methomyl and aldicarb. The authors concluded that obidoxime therapy is not indicated in carbamate poisoning since it does not offer any marked benefit. On the other hand, obidoxime did not aggravate the condition of the patients. In case of doubt on the identity of the causative anticholinesterase, the use of oximes was recommended.[190]
RCTs should adhere to a strict protocol of atropine dosage and of determining when the patient will be discontinued from oxime treatment and weaned from the ventilator. The existing trials as published until 2002 lack one or more of the above prerequisites. For details the reader is referred to Eddleston et al.[6]
From existing data it appears that oximes, if administered in doses that produce sufficiently high plasma concentrations to allow reactivation t1/2 of 15 minutes or less, are effective in moderate poisoning as long as the aging process is not too advanced. If the rate of re-inhibition is clearly higher than the rate of reactivation, oximes may initially disappoint, but can become effective in the later course of intoxication if the aging process is slow. This is particularly true with the O,O-diethyl phosphoryl compounds. Unfortunately, this class of compounds (see table I) is generally highly lipophilic (logP octanol/water > 3), distributes in the fat tissue and is re-distributed for days and even weeks. Therefore oxime treatment should be for long enough to prevent relapses and to keep as much as possible of the enzyme active to retard aging.
These considerations lead to the obvious question of how to decide when oxime treatment can be discontinued. It has been proposed to use serial plasma cholinesterase determinations. If the activity is steadily increasing, circulating poison is probably no longer present and the oxime can be withdrawn. It is wise to control the enzyme activity even further by recommencing oxime therapy if enzyme activity drops again.[191], This indicator is hardly altered by therapeutic oxime concentrations because phosphorylated plasma cholinesterase is notoriously resistant to reactivation. Another approach is to incubate the patient’s plasma with an appropriate enzyme source to look for possible inhibition. This can be done with plasma cholinesterase[192], or with erythrocyte AChE.[193]
While this approach tells us whether oxime therapy should be continued because of remaining poison, it does not indicate whether further oxime therapy may be effective. Oximes are of little value when all the AChE is completely in the aged state. This can be easily checked by incubating the inhibited erythrocyte AChE with a high concentration of oxime in vitro. To this end, the blood sample is diluted, e.g. 1 : 100 with water, and incubated with 0.1 mmol/L of the oxime for 1 hour. Since any inhibitor present is also diluted, reactivation takes place as long as some portion of the enzyme has not already aged. By testing the so-called reactivatability one can easily decide whether further oxime administration is rational. We have used this technique in many cases and also determined whether an O,O-diethyl phosphoryl compound (aging t1/2 >30 hours) was the causative poisoning agent or possibly an O,O-dimethyl phosphoryl derivative (aging t1/2 mostly <6 hours).[193–198] If these tests could be performed in the intensive care unit, unnecessary oxime therapy could be avoided.
From the above, it becomes clear that oximes will be particularly effective in moderately severe poisonings caused by O,O-diethyl phosphoryl compounds. In fact, in an ongoing RCT of activated charcoal in OP-poisoned patients treated with the traditional pralidoxime scheme, 1g four times daily, the case fatality ratio (CFR) in patients with chlorpyrifos intoxication (n = 200) was significantly less (7.5%) than in dimethoate poisoning (n = 110; fatality rate 20.9%) [unpublished data]. Curiously, in the completed Munich study of OP-poisoned patients treated with obidoxime, 250mg bolus IV, followed by continuous infusion at 750 mg/24h, the CFR was much higher with the O,O-diethyl phosphoryl compound parathion (5 deaths, 8 survivors) than with the O,O-dimethyl phosphoryl compound dimethoate (no deaths, 6 survivors) or with oxydemeton-methyl (1 death, 11 survivors). Interestingly, only two parathion-poisoned patients could not be sufficiently reactivated. This was because of an extremely high poison load. The other patients died because of life-threatening aspiration before admission to the hospital, which resulted in acute respiratory distress syndrome, or because of late complications (e.g. lung embolism on the day of discharge; stress ulcer with overt peritonitis).[200]
The huge differences in precipitation of life-threatening symptoms amongst patients following OP-poisoning deserve some additional comment. Figure 11 illustrates the inhibition kinetics of erythrocyte AChE activity at concentrations of paraoxon or oxydemeton-methyl inhibiting the enzyme at similar rate. In the absence of any oxime, spontaneous reactivation prevents complete inhibition by oxydemeton-methyl, even in the presence of a constant concentration of the poison. Thus, a steady-state at about 20% of residual enzyme activity is reached with oxydemeton-methyl (hatched curve) while enzyme activity would be nil in the case of paraoxon. The slow second phase of inhibition in the case of oxydemeton-methyl results from gradual aging of the inhibited part of the enzyme and also leads finally to complete inhibition. This picture may explain why a patient with parathion poisoning is in a life-threatening situation; after 1 hour there is less than 5% active enzyme. In contrast, the oxydemeton-methyl intoxicated patient has some 20% of active enzyme at this time and keeps this amount for some hours. Hence, patients intoxicated with O,O-dimethyl-phosphoryl compounds are usually less severely intoxicated when arriving at the hospital than those intoxicated with parathion. If the patient does not present with life-threatening complications on admission, those intoxicated with O,O-diethyl phosphoryl compounds will have a better chance of reactivation than patients intoxicated by dimethyl phosphoryl compounds. These basic differences stress once more that unequivocal stratification of patients is a prerequisite for proper judgement of oxime effectiveness.

Calculated acetylcholinesterase (AChE) activity in the presence of oxydemeton-methyl (2.5 μmol/L) or paraoxon (0.025 μmol/L). The dotted line shows the calculated curve for oxydemeton-methyl when ignoring aging. The squares are experimental values obtained with 2.5 μmol/L oxydemeton-methyl in a human erythrocyte preparation (pH 7.4, 37°C). The following values were used for calculation (see figure 13): oxydemeton-methyl: ki = 1.32 x 106L x mol-1 x h-1; ks = 1.01 h-1; ka = 0.186 h-1;[13] paraoxon: ki = 1.32 x 108L x mol-1 x h-1; ka = 0.022 h-1; ks = 0.022 h-1.[38]

7. Conclusions
The above considerations clearly indicate that oximes may have an important role in the management of OP poisoning. Their potential, however, is only exploited if the oximes at sufficient dosage and duration are given in a regimen that avoids troughs where aging occurs. Patients with poisoning by O,O-diethyl phosphoryl OPs will probably derive most benefit from oximes, while intoxication with O,O-dimethyl phosphoryl OPs will only benefit if treated shortly after intoxication (usually less than 12 hours) using high oxime concentrations to keep the portion of inhibited enzyme (that undergoes aging) small. These expectations, however, have to be rigorously tested in RCTs with appropriate stratification, such as planned in a forthcoming trial in Sri Lanka.[6] It is hoped that objective parameters regarding the cholinesterase status of patients are gathered for quantitative assessment of oxime effectiveness. Determination of erythrocyte AChE near the ward would be ideal. This may be possible in the near future when reliable and cheap instruments become available.
Notes
The use of trade names is for product identification purposes only and does not imply endorsement.
In the following sections, doses of oxime salts are given in mass units while concentrations in biological fluids or buffer solutions are presented in mol/L. The molecular weights of the various pralidoxime salts as indicated in the sections above, enables easy conversion into the desired dimension.
References
Roberts DM, Karunarathna A, Buckley N, et al. Influence of pesticide regulation on acute poisoning deaths in Sri Lanka. Bull World Health Organ 2003; 81: 789–98
Jeyaratnam J. Acute pesticide poisoning: a major global health problem. World Health Stat Q 1990; 43(3): 139–44
Eddieston M. Patterns and problems of deliberate self-poisoning in the developing world. Q J Med 2000; 93: 715–31
Gunnell D, Eddieston M. Suicide by intentional ingestion of pesticides: a continuing tragedy in developing countries. Int J Epidemiol 2003; 32: 902–9
Phillips MR, Li X, Zhang Y. Suicide rates in China, 1995–1999. Lancet 2002; 359: 835–40
Eddleston M, Szinicz L, Eyer P, et al. Oximes in acute organophosphate poisoning: a systematic review of clinical trials. Q J Med 2002; 95: 1–9
Eddleston M, Karalliedde L, Buckley N, et al. Pesticide poisoning in the developing world: a minimum pesticide list. Lancet 2002; 360: 1163–7
Black RM, Harrison JM, editors. The chemistry of organophosphorus chemical warfare agents. Chichester: John Wiley, 1996
Schrader G. Organische Phosphor-Verbindungen als neuartige Insektizide. Angew Chemie 1950; 62: 471–3
Jennings LL, Malecki M, Komives EA, et al. Direct analysis of the kinetic profiles of organophosphate-acetylcholinesterase adducts by MALDI-TOF mass spectrometry. Biochemistry 2003; 42: 11083–91
Doom JA, Thompson CM, Christner RB, et al. Stereoselective inactivation of Torpedo californica acetylcholinesterase by isomalathion: inhibitory reactions with (1R)- and (1-S)-isomers proceed by different mechanisms. Chem Res Toxicol 2003; 16: 958–65
Worek F, Kirchner T, Bäcker M, et al. Reactivation by various oximes of human erythrocyte acetylcholinesterase inhibited by different organophosphorus compounds. Arch Toxicol 1996; 70: 497–503
Worek F, Diepold C, Eyer P. Dimethylphosphoryl-inhibited human cholinesterases: inhibition, reactivation, and aging kinetics. Arch Toxicol 1999; 73: 7–14
Worthing CA, editor. The pesticide manual: a world compendium. 7th ed. Lavenham: The British Crop Protection Council, 1983
Taylor P, Radic Z. The cholinesterases: from genes to proteins. Annu Rev Pharmacol Toxicol 1994; 34: 281–320
Mortensen SR, Brimijoin S, Hooper MJ, et al. Comparison of the in vitro sensitivity of rat acetylcholinesterase to chlorpyrifos-oxon: what do tissue IC50 values represent? Toxicol Appl Pharmacol 1998; 148: 46–9
Taylor P. Anticholinesterase agents. In: Hardman JG, Limbird LE, editors. Goodman and Gilman’s the pharmacological basis of therapeutics. 9th ed. New York: McGraw-Hill, 1996: 161–76
Schaffer NK, Engle RR, Simet L, et al. Phosphopeptides from chymotrypsin and trypsin after inactivation with 32P-labelled DFP and sarin. J Biochem 1958; 230: 185–92
Johnson MK. Organophosphorus esters causing delayed neurotoxic effects: mechanism of action and structure/activity studies. Arch Toxicol 1975; 34: 259–88
Williams DJ, Johnson MK. Gel-electrophoretic identification of hen brain neurotoxic esterase, labelled with tritiated di-isopropyl phosphofluoridate. Biochem J 1981; 199: 323–33
Pope CN. Organophosphorus pesticides: do they all have the same mechanism of toxicity? J Toxicol Environ Health B Crit Rev 1999; 2(2): 161–81
Lotti M. Clinical toxicology of anticholinesterase agents in humans. In: Krieger RI, editor. Handbook of pesticide toxicology. 2nd ed. San Diego (CA): Academic Press, 2001: 1043–85
Burgen ASV. The mechanism of action of anticholinesterase drugs. Br J Pharmacol 1949; 4: 219–28
Burgen ASV, Hobbiger F. The inhibition of cholinesterases by alkylphosphates and alkylphenolphosphates. Br J Pharmacol 1951; 6: 593–605
Jansz HS, Brons D, Warringa MGPJ. Chemical nature of the DFP-binding site of pseudocholinesterase. Biochim Biophys Acta 1959; 34: 573–5
Schaffer NK, May SC, Summerson WH. Serine phosphoric acid from di-isopropylphosphoryl chymotrypsin. J Biol Chem 1953; 202: 67–76
MacPhee-Quingley K, Taylor P, Taylor S. Primary structures of the catalytic subunits from two molecular forms of acetylcholinesterase: a comparison of NH2-terminal and active center sequences. J Biol Chem 1985; 260: 12185–9
Sussman JL, Harel M, Frolow F, et al. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 1991; 253: 872–9
Kovarik Z, Radic Z, Berman HA, et al. Acetylcholinesterase active centre and gorge conformations analysed by combinatorial mutations and enantiomeric phosphonates. Biochem J 2003; 373: 33–40
Johnson MK, Jacobsen D, Meredith TJ, et al. Evaluation of antidotes for poisoning by organophosphorus pesticides. Emerg Med 2000; 12: 22–37
Aldridge WN, Reiner E. Enzyme inhibitors as substrates: interactions of esterases with esters of organophosphorus and carbamic acids. Amsterdam: North-Holland Publishing Co., 1972
Eyer F, Eyer P. Enzyme-based assay for quantification of paraoxon in blood of parathion poisoned patients. Hum Exp Toxicol 1998; 17: 645–51
Cohen SD, Williams RA, Killinger JM, et al. Comparative sensitivity of bovine and rodent acetylcholinesterase to in vitro inhibition by organophosphate insecticides. Toxicol Appl Pharmacol 1985; 81: 452–9
Johnson JA, Wallac KB. Species-related differences in the inhibition of brain acetylcholinesterase by paraoxon and malaoxon. Toxicol Appl Pharmacol 1987; 88: 234–41
Wilson IB, Meislich EK. Reactivation of acetylcholinesterase inhibited by alkylphosphates. J Am Chem Soc 1953; 75: 4628–9
Radic Z, Taylor P. The influence of peripheral site ligands on the reaction of symmetric and chiral organophosphates with wildtype and mutant acetylcholinesterases. Chem Biol Interact 1999; 119: 111–7
Amitai G, Moorad D, Adani R, et al. Inhibition of acetylcholinesterase and butyrylcholinesterase by chlorpyrifos-oxon. Biochem Pharmacol 1998; 56: 293–9
Worek F, Bäcker M, Thiermann H, et al. Reappraisal of indications and limitations of oxime therapy in organophosphate poisoning. Hum Exp Toxicol 1997; 16: 466–72
Skrinjaric-Spoljar M, Simeon V, Reiner E. Spontaneous reactivation and aging of dimethylphosphorylated acetylcholinesterase and cholinesterase. Biochim Biophys Acta 1973; 315: 363–9
Mason HJ, Waine E, Stevenson A, et al. Aging and spontaneous reactivation of human plasma cholinesterase activity after inhibition by organophosphorus pesticides. Hum Exp Toxicol 1993; 12: 497–503
Hobbiger F. Inhibition of cholinesterases by irreversible inhibitors in vitro and in vivo. Br J Pharmacol 1951; 6: 21–30
Wilson IB. Acetylcholinesterase XI: reversibility of tetraethylpyrophosphate inhibition. J Biol Chem 1951; 190: 111–7
Wilson IB, Ginsburg S. A powerful reactivator of alkylphosphate-inhibited acetylcholinesterase. Biochim Biophys Acta 1955; 18: 168–70
Childs AF, Davies DR, Green AL, et al. The reactivation by oximes and hydroxamic acids of cholinesterase inhibited by organo-phosphorus compounds. Br J Pharmacol 1955; 10: 462–5
Hobbiger F. Effect of nicotinhydroxamic acid methiodide on human plasma cholinesterase inhibited by organophosphates containing a dialkylphosphato group. Br J Pharmacol 1955; 10: 356–62
Berends F, Posthumus CH, Sluys IVD, et al. The chemical basis of the ‘ageing process’ of DFP-inhibited pseudocholinesterase. Biochim Biophys Acta 1959; 34: 576–9
Saxena A, Doctor BP, Maxwell DM, et al. The role of glutamate-199 in the aging of cholinesterase. Biochem Biophys Res Commun 1993; 197: 343–9
Shafferman A, Ordentlich A, Barak D, et al. Aging of phosphylated human acetylcholinesterase: catalytic processes mediated by aromatic and polar residues of the active centre. Biochem J 1996; 318: 833–40
Masson P, Fortier P-L, Albaret C, et al. Structural and hydration changes in the active site gorge of phosphorylated butyrylcholinesterase accompanying the ageing process. Chem-Biol Interact 1999; 119-120: 17–27
Barak D, Ordentlich A, Kaplan D, et al. Evidence for P-N bond scission in phosphoroamidate nerve agent adducts of human acetylcholinesterase. Biochemistry 2000; 39: 1156–61
Besser R, Weilemann LS, Schollmeyer U, et al. Synaptic transmisson during pesticide poisoning: the neuromuscular block. In: Szinicz L, Eyer P, Klimmek R, editors. Role of oximes in the treatment of anticholinesterase agent poisoning. Heidelberg: Spektrum, Akademischer Verlag, 1996: 19–31
Ludomirsky A, Klein HO, Sarelli P, et al. Q-T prolongation and polymorphous (‘torsade de pointes’) ventricular arrhythmias associated with organophosphorus insecticide poisoning. Am J Cardiol 1982; 49: 1654–8
Savolainen K. Understanding the toxic actions of organophosphates. In: Krieger RI, editor. Handbook of pesticide toxicology. 2nd ed. San Diego: Academic Press, 2001: 1013–41
Besser R, Gutman L, Weilemann LS. Inactivation of end-plate acetylcholinesterase during the course of organophosphate intoxications. Arch Toxicol 1989; 63: 412–5
Besser R, Gutmann L, Dillmann U, et al. End-plate dysfunction in acute organophosphate intoxication. Neurology 1989; 39: 561–7
Kauert G, Schoppek B, von Clarmann M, et al. Plasma-Katecholamin-Verlauf bei Alkylphosphat-Intoxikationen und deren Therapie. Klin Wochenschr 1989; 67: 456–62
Kauert G, Schoppek B, von Clarmann M, et al. Plasma- und Urin-Katecholamine bei einer 7 Tage überlebten Parathion-Intoxikation. Klin Wochenschr 1990; 68: 96–100
Suzuki T, Morita H, Ono K, et al. Sarin poisoning in Tokyo subway. Lancet 1995; 345: 980
Sidell F. Nerve agents. In: Sidell F, Takafuji ET, Franz DR, editors. Medical aspects of chemical and biological warfare. Washington, DC: Walter Reed Army Medical Center, 1997: 130–79
Namba T, Nolte CT, Jackrel J, et al. Poisoning due to organophosphate insecticides: acute and chronic manifestations. Am J Med 1971; 50: 475–92
Wadia RS, Sadagopan C, Amin RB, et al. Neurological manifestations of organophosphorous insecticide poisoning. J Neurol Neurosurg Psychiatry 1974; 37: 841–7
Senanayake N, Karalliedde L. Neurotoxic effects of organophosphorus insecticides: an intermediate syndrome. N Engl J Med 1987; 316: 761–3
Eddleston M, Singh S, Buckley N. Acute organophosphate poisoning. Clin Evid 2002; 8: 1436–46
Zwiener RJ, Ginsburg CM. Organophosphate and carbamate poisoning in infants and children. Pediatrics 1988; 81: 121–6
Eyer F, Meischner V, Kiderlen D, et al. Human parathion poisoning: a toxicokinetic analysis. Toxicol Rev 2003; 22(3): 143–63
Guven H, Tuncok Y, Gidener S, et al. In vitro adsorption of dichlorvos and parathion by activated charcoal. J Toxicol Clin Toxicol 1994; 32: 157–63
Tuncok Y, Gelai A, Apaydin S, et al. Prevention of oral dichlorvos toxicity by different activated charcoal products in mice. Ann Emerg Med 1995; 25: 353–5
Tomimaru A, Arimori K, Inotsume N, et al. Effect of activated charcoal and atropine on absorption and/or exsorption of organophosphorus compounds in rats. J Pharm Pharmacol 1996; 48: 351–6
Simpson Jr WM, Schuman SH. Recognition and management of acute pesticide poisoning. Am Fam Physician 2002; 65: 1599–604
Marrs TC. Organophosphate poisoning. Pharmacol Ther 1993; 58: 51–66
Vale JA, Scott GW. Organophosphorus poisoning. Guys Hosp Rep 1974; 123: 13–25
Minton NA, Murray VSG. A review of organophosphate poisoning. Med Toxicol 1988; 3: 350–75
Eddleston M, Singh S, Buckley N. Acute organophosphorus poisoning. Clin Evid 2003; 9: 1542–53
Grob D. The manifestations and treatment of poisoning due to nerve gas and other organic phosphate anticholinesterase compounds. Arch Intern Med 1956; 98: 221–39
Aaron CK, Howland MA. Insecticides: organophosphates and carbamates. In: Goldfrank TR, Flomenbaum NE, Lewin NA, et al., editors. Toxicologic emergencies. Norwalk (CT): Appleton & Lange, 1994: 1111
Thiermann H, Worek F, Szinicz L, et al. On the atropine demand in organophosphate poisoned patients. J Toxicol Clin Toxicol 2003; 41: 457
Golsousidis H, Kokkas V. Use of 19590mg atropine during 24 days of treatment after a case of unusually severe parathion poisoning. Hum Toxicol 1985; 4: 339–40
LeBlanc FN, Benson BE, Gilg AD. A severe organophosphate poisoning requiring the use of an atropine drip. J Toxicol Clin Toxicol 1986; 24: 69–76
Helm U. Nervenkampfstoffvergiftung (Alkylphosphatvergiftung). In: Rebentisch E, editor. Wehrmedizin. München: Urban & Schwarzenberg, 1980
Heath AJW, Meredith T. Atropine in the management of anticholinesterase poisoning. In: Ballantyne B, Marrs TC, editors. Clinical and experimental toxicology of organophosphates and carbamates. Oxford: Butterworth-Heinemann Ltd, 1992: 543–54
De Kort WLAM, Kiestra SH, Sangster B. The use of atropine and oximes in organosphosphate intoxications: a modified approach. J Toxicol Clin Toxicol 1988; 26: 199–208
Namba T, Hiraki K. PAM (pyridine-2-aldoxime methiodide) therapy for alkylphosphate poisoning. JAMA 1958; 166: 1834–9
Lüttringhaus A, Hagedorn I. Quartäre Hydroxyiminomethyl-pyridiniumsalze: das dichlorid des bis-[4-hydroxyiminomethyl-pyridinium-(1)-methyl]-äthers (‘LüH6’), ein neuer Reaktivator der durch organische Phosphorsäureester gehemmten Acetylcholin-Esterase. Arzneimittel Forschung 1964; 14: 1–5
Erdmann WD, von Clarmann M. Ein neuer Esterase-Reaktivator für die Behandlung von Vergiftungen mit Alkylphosphaten. Dtsch Med Wochenschr 1963; 88: 2201–6
Oettel H. Erfahrungen einer Giftinformationsstelle. Dtsch Ärztebl 1967; 64: 2787–91
De Silva HJ, Wijewickrema R, Senanayake N. Does pralidoxime affect outcome of management in acute organophosphate poisoning? Lancet 1992; 339: 1136–8
Johnson MK, Vale JA, Marrs TC, et al. Pralidoxime for organophosphorus poisoning. Lancet 1992; 340: 64
Ashani Y, Radic Z, Tsigelny I, et al. Amino acid residues controlling reactivation of organophosphonyl conjugates of acetylcholinesterase by mono- and bis-quaternary oximes. J Biol Chem 1995; 270: 6370–80
Taylor P, Wong L, Radic Z, et al. Analysis of cholinesterase inactivation and reactivation by systematic structural modification and enantiomeric selectivity. Chem Biol Interact 1999; 119-120: 3–15
Parfitt K, editor. Martindale. The complete drug reference. 32nd ed. London: The Pharmaceutical Press, 1999
Eyer P, Hagedorn I, Klimmek R, et al. HLö 7 dimethanesulfonate, a potent bispyridinium-dioxime against anticholinesterases. Arch Toxicol 1992; 66: 603–21
Clement JG, Hansen AS, Boulet CA. Efficacy of HLö-7 and pyrimidoxime as antidotes of nerve agent poisoning in mice. Arch Toxicol 1992; 66: 216–9
Worek F, Reiter G, Eyer P, et al. Reactivation kinetics of acetylcholinesterase from different species inhibited by highly toxic organophosphates. Arch Toxicol 2002; 76: 523–9
Kusic R, Boskovic B, Vojvodic V, et al. HI-6 in man: blood levels, urinary excretion, and tolerance after intramuscular administration of the oxime to healthy volunteers. Fundam Appl Toxicol 1985; 5: S89–97
Rousseaux CG, Dua AK. Pharmacology of HI-6, an H-series oxime. Can J Physiol Pharmacol 1989; 67: 1183–9
Clement JG, Madill HD, Bailey D, et al. Clinical study of a new therapy for nerve agent poisoning: ascending dose tolerance study of HI 6 and atropine. Suffield: Defense Research Establishment, 1994. Report no. 597
Schlager JW, Dolzine TW, Stewart JR, et al. Operational evaluation of three commercial configurations of atropine/HI-6 wet/dry autoinjectors. Pharm Res 1991; 8: 1191–4
Thiermann H, Spöhrer U, Klimmek R, et al. Operational evaluation of wet/dry autoinjectors containing atropine in solution and powdered HI 6 or HLö 7. Int J Pharm 1994; 109: 35–43
Göransson-Nyberg A, Cassel G, Jeneskog T, et al. Treatment of organophosphate poisoning in pigs: antidote administration by a new binary autoinjector. Arch Toxicol 1995; 70: 20–7
Clement JG. Toxicity of the combined nerve agents GB/GF in mice: efficacy of atropine and various oximes as antidotes. Arch Toxicol 1994; 68: 64–6
Green AL, Saville B. The reaction of oximes with isopropyl methylphosphonofluoridate (sarin). J Chem Soc 1956; 3: 3887–92
Hackley Jr BE, Steinberg GM, Lamb JC. Formation of potent inhibitors of AChE by reaction of pyridinaldoximes with isopropyl methylphosphonofluoridate (GB). Arch Biochem Biophys 1959; 80: 211–4
Portmann R, Niederhauser A, Hofmann W, et al. 32. Synthesis of 4-(([(isopropyloxy)methylphosphoryloxy]imino)methyl)-1-methylpyridinium iodide and its characterisation. Helv Chim Acta 1991; 74: 331–5
Becker G, Kawan A, Szinicz L. Direct reaction of oximes with sarin, soman, or tabun in vitro. Arch Toxicol 1997; 71: 714–8
Leader H, Vincze A, Manisterski B, et al. Characterization of O,O-diethylphosphoryl oximes as inhibitors of cholinesterases and substrates of phosphotriesterases. Biochem Pharmacol 1999; 58: 503–15
Luo C, Saxena A, Smith M, et al. Phosphoryl oxime inhibition of acetylcholinesterase during oxime reactivation is prevented by edrophonium. Biochemistry 1999; 38: 9937–47
Lamb JC, Steinberg GM, Hackley Jr BE. Isopropyl methylphosphonylated bis-quaternary oximes; powerful inhibitors of cholinesterase. Biochim Biophys Acta 1964; 89: 174–6
Nenner M. Phosphonylierte aldoxime: hemmwirkung auf acetylcholinesterase und hydrolytischer abbau. Biochem Pharmacol 1974; 23: 1255–62
De Jong LPA, Ceulen DI. Anticholinesterase activity and rate of decomposition of some phosphylated oximes. Biochem Pharmacol 1978; 27: 857–63
Harvey B, Scott RP, Sellers DJ, et al. In vitro studies on the reactivation by oximes of phosphylated acetylcholinesterase, I: on the reactions of P2S with various organophosphates and the properties of the resultant phosphylated oximes. Biochem Pharmacol 1986; 35: 737–44
Lockridge O, Masson P. Pesticides and susceptible populations: people with butyrylcholinesterase genetic variants may be at risk. Neurotoxicology 2000; 21: 113–26
Kiderlen D, Worek F, Klimmek R, et al. The phosphoryl oxime-destroying activity of human plasma. Arch Toxicol 2000; 74: 27–32
Worek F, Eyer P, Kiderlen D, et al. Effect of human plasma on the reactivation of sarin-inhibited human erythrocyte acetylcholinesterase. Arch Toxicol 2000; 74: 21–6
Kiderlen D, Meischner V, Worek F, et al. Phosphoryl oxime-hydrolase in human serum influences oxime effectiveness in organophosphate poisoning. Drug Metab Rev 2001; 33Suppl. 1: 110
Hagedorn I, Gündel WH, Schoene K. Reaktivierung phosphorylierter acetylcholinesterase mit oximen: beitrag zum Studium des reaktionsablaufes. Arzneimittel Forschung 1969; 19: 603–6
Eyer P. Optimal oxime dosage regimen, a pharmacokinetic approach. In: Szinicz L, Eyer P, Klimmek R, editors. Role of oximes in the treatment of anticholinesterase agent poisoning. Heidelberg: Spektrum, Akademischer Verlag, 1996: 33–51
Ellin RI, Groff WA, Sidell FR. Passage of pyridinium oximes into human red cells. Biochem Pharmacol 1974; 23: 2663–70
Spöhrer U. HPLC-analytische Untersuchungen zur Pharmakokinitk von Pyridiniumaldoximen [dissertation]. Munich: Ludwig-Maximilians-University, 1994
Sidell FR, Groff WA, Kaminskis A. Toxogonin and pralidoxime: kinetic comparison after intravenous administration to man. J Pharm Sci 1972; 61: 1765–9
Josselson J, Sidell FR. Effect of intravenous thiamine on pralidoxime kinetics. Clin Pharmacol Ther 1978; 24: 95–100
Jovanovic D. Pharmacokinetics of pralidoxime chloride: a comparative study in healthy volunteers and in organophosphorus poisoning. Arch Toxicol 1989; 63: 416–8
Willems JL, Langenberg JP, Verstraete AG, et al. Plasma concentrations of pralidoxime methylsulphate in organophosphorus poisoned patients. Arch Toxicol 1992; 66: 260–6
Clement JG, Bailey DG, Madill HD, et al. The acetylcholinesterase oxime reactivator HI-6 in man: pharmacokinetics and tolerability in combination with atropine. Biopharm Drug Dispos 1995; 16: 415–25
Bentur Y, Nutenko I, Tsipiniuk A, et al. Pharmacokinetics of obidoxime in organophosphate poisoning associated with renal failure. Clin Toxicol 1993; 31: 315–22
Waser PG, Sammett R, Schönenberger E, et al. Pharmacokinetics of [14C]-sarin and its changes by obidoxime and pralidoxime. In: Hanin I, editor. Dynamics of cholinergic function. New York: Plenum Press, 1986: 743–55
Waser PG, Streichenberg C. Metabolism, kinetics and interaction of 14C-sarin and 14C-obidoxime. Toxicol Environ Chem 1988; 18: 1–10
Ligtenstein DA, Moes GWH, Kossen SP. In vivo distribution of organophosphate antidotes: autoradiography of [14C]HI-6 in the rat. Toxicol Appl Pharmacol 1988; 92: 324–9
Garrigue H, Maurizis JC, Nicolas C, et al. Disposition and metabolism of two acetylcholinesterase reactivators, pyrimidoxime and HI6, in rats submitted to organophosphate poisoning. Xenobiotica 1990; 20: 699–709
Garrigue H, Maurizis JC, Madelmont JC, et al. Disposition and metabolism of acetylcholinesterase reactivators 2PAM-I, TMB4 and R665 in rats submitted to organophosphate poisoning. Xenobiotica 1991; 21: 583–95
Boyd ES, Neuman WF. The surface chemistry of bone: V. the ion-binding properties of cartilage. J Biol Chem 1951; 193: 243–51
Dunstone JR. Some cation-binding properties of cartilage. Biochem J 1959; 72: 465–73
Maurizis JC, Ollier M, Nicolas C, et al. In vitro binding of oxime acetylcholinesterase reactivators to proteoglycans synthesized by cultured chondrocytes and fibroblasts. Biochem Pharmacol 1992; 44: 1927–33
Sakurada K, Matsubara K, Shimizu K, et al. Pralidoxime iodide (2-PAM) penetrates across the blood-brain barrier. Neurochem Res 2003; 28: 1401–7
Jager BV, Stagg GN, Green N, et al. Studies on distribution and disappearance of pyridine-2-aldoxime methiodide (PAM) and of diacetylmonoxime (DAM) in man and in experimental animals. Bull Johns Hopkins Hosp 1958; 102: 225–34
Lotti M, Becker CE. Treatment of acute organophosphate poisoning: evidence of a direct effect on central nervous system by 2-PAM (pyridine-2-aldoxime methyl chloride). Clin Toxicol 1982; 19: 121–7
Ligtenstein DA, Kossen SP. Kinetic profile in blood and brain of the cholinesterase reactivating oxime HI-6 after intravenous administration to the rat. Toxicol Appl Pharmacol 1983; 71: 177–83
Klimmek R, Eyer P. Pharmacokinetics and pharmacodynamics of the oxime HI 6 in dogs. Arch Toxicol 1986; 59: 272–8
Tochino Y, Schanker LS. Active transport of quaternary ammonium compounds by the choroid plexus in vitro. Am J Physiol 1965; 208: 666–73
Grange-Messent V, Bouchaud C, Jamme M, et al. Seizure-related opening of the blood-brain barrier produced by the anticholinesterase compound, soman: new ultrastructural observations. Cell Mol Biol 1999; 45: 1–14
Enander I, Sundwall A, Sörbo B. Metabolic studies on N-methylpyridinium-2-aldoxime, III: experiments with the 14C-labelled compound. Biochem Pharmacol 1962; 11: 377–82
Enander I, Sundwall A, Sörbo B. Metabolic studies on N-methylpyridinium-2-aldoxime, I: the conversion to thiocyanate. Biochem Pharmacol 1961; 7: 226–31
Enander I, Sundwall A, Sörbo B. Metabolic studies on N-methylpyridinium-2-aldoxime, II: the conversion to N-methylpyridinium-2-nitrile. Biochem Pharmacol 1961; 7: 232–6
Ligtenstein DA, Wils ERJ, Kossen SP, et al. Identification of two metabolites of the cholinesterase reactivator HI-6 isolated from rat urine. J Pharm Pharmacol 1987; 39: 17–23
Eyer P, Kawan A, Ladstetter B. Formation of cyanide after i.v. administration of the oxime HI 6 to dogs. Arch Toxicol 1987; 61: 63–9
Ladstetter B. Stabilität und metabolisches Schicksal neuer Antidote gegen Organophosphate [dissertation]. Munich: Ludwig-Maximilians-University, 1990
Eyer P, Hell W, Kawan A, et al. Studies on the decomposition of the oxime HI 6 in aqueous solution. Arch Toxicol 1986; 59: 266–71
Creasey HN, Green AC. 2-Hydroxyiminomethyl-N-methyl-pyridiniummethansulfonate (P2S), an antidote to organophosphorus poisoning: its preparation, estimation, and stability. J Pharm Pharmacol 1959; 11: 485–90
Sidell FR, Groff WA. Toxogonin: blood levels and side effects after intramuscular administration in man. J Pharm Sci 1970; 59: 793–7
Erdmann WD, Bosse I, Franke P. Zur Resorption und Ausscheidung von Toxogonin nach intramuskulärer Injektion am Menschen. Dtsch Med Wochenschr 1965; 90: 1436–8
Thiermann H, Worek F, Szinicz L, et al. Obidoxime plasma levels in organophosphate poisoned patients. J Toxicol Clin Toxicol 2002; 40: 318–9
Kusic R, Jovanovic D, Randjelovic S, et al. HI-6 in man: efficacy of the oxime in poisoning by organophosphorus insecticides. Hum Exp Toxicol 1991; 10: 113–8
Schulz V, Gross R, Pasch T, et al. Cyanide toxicity of sodium nitroprusside in therapeutic use with and without sodium thiosulfate. Klin Wochenschr 1982; 60: 1393–400
Green MD, Jones DE, Hilmas DE. Sarin intoxication elevates plasma pralidoxime. Toxicol Lett 1985; 28: 17–21
Mast U. Reaktivierung der Erythrozyten-Acetylcholinesterase durch Oxime: ermittlung enzymkinetischer Konstanten und ihre Bedeutung für die Therapie einer Organophosphat-Vergiftung, [dissertation]. Munich: Ludwig-Maximilians-University, 1997
Jager BV, Stagg GN. Toxicity of diacetylmonoxime and of pyridine-2-aldoxime methiodide in man 15–30 mg/kg 2-PAM: no effects on EEG in a healthy man. Bull Johns Hopkins Hosp 1958; 102: 203–11
Sundwall A. Plasma concentration curves of N-methylpyridinium-2-aldoxime methane sulphonate (P2S) after intravenous, intramuscular and oral administration in man. Biochem Pharmacol 1960; 5: 225–30
Calesnick B, Christensen JA, Richter M. Human toxicity of various oximes: 2-pyridine aldoxime methyl chloride, its methane sulfonate salt, and 1,1′-trimethylenebis-(4-formylpyridinium chloride). Arch Environ Health 1967; 15: 599–608
Sidell FR, Groff WA. Intramuscular and intravenous administration of small doses of 2-pyridinium aldoxime methochloride to man. J Pharm Sci 1971; 60: 1224–8
Xue SZ, Ding XJ, Ding Y. Clinical observation and comparison of the effectiveness of several oxime cholinesterase reactivators. Scand J Work Environ Health 1985; 11: 46–8
Medicis JJ, Stork CM, Howland MA, et al. Pharmacokinetics following a loading plus a continuous infusion of pralidoxime compared with the traditional short infusion regimen in human volunteers. Clin Toxicol 1996; 34: 289–95
Scott RJ. Repeated asystole following PAM in organophosphate self-poisoning. Anaesth Intensive Care 1986; 14: 458–60
Ellenhorn MJ, Barceloux DG. Medical toxicology: diagnosis and treatment of human poisoning. New York: Elsevier, 1988
Simon GA, Tirosh MS, Edery H. Administration of obidoxime tablets to man: plasma levels and side reactions. Arch Toxicol 1976; 36: 83–8
Boelcke G, Creutzfeldt W, Erdmann WD, et al. Untersuchungen zur Frage der Lebertoxizität von Obidoxim (Toxogonin®) am Menschen. Dtsch Med Wochenschr 1970; 95: 1175–8
von Gaisberg U, Dieterle K. Organ-Parenchymschäden nach E-605-Vergiftung bzw. hochdosierter Toxogoninbehandlung. Dtsch Arztebl 1967; 64: 1791–6
Prinz HJ. Therapie akuter Alkylphosphat-Vergiftungen. Dtsch Arztebl 1967; 36: 1845–9
Boelcke G, Gaaz JW. Zur Frage der Lebertoxizität von Nitrostigmin (E605 forte®) und Obidoxim (Toxogonin®) an Hunden. Arch Toxicol 1970; 26: 93–101
Finkelstein Y, Kushnir A, Raikhlin-Eisenkraft B, et al. Antidotal therapy of severe acute organophosphate poisoning: a multihospital study. Neurotoxicol Teratol 1989; 11: 593–6
Taitelman U. Round table discussion. In: Szinicz L, Eyer P, Klimmek R, editors. Role of oximes in the treatment of anticholinesterase agent poisoning. Heidelberg: Spektrum, Akademischer Verlag, 1996: 78
Zech R, Erdmann WD, Engelhard H. Grenzen der Therapie mit Oximen bei Vergiftungen mit Insektiziden Alkylphosphaten. Arzneimittel Forschung 1967; 17: 1196–202
Sundwall A. Minimum concentrations of N-methylpyridinium-2-aldoxime methane sulphonate (P2S) which reverse neuromuscular block. Biochem Pharmacol 1961; 8: 413–7
Erdmann WD. Antidotbehandlung bei Alkylphosphatvergiftungen. Arch Toxicol 1968; 24: 30–40
Vale JA. Rationale for oxime therapy: pralidoxime as an antidote in OP insecticide poisoning [abstract]. Hum Exp Toxicol 1996; 15: 77
Thompson DF, Thompson GD, Greenwood RB, et al. Therapeutic dosing of pralidoxime chloride. Drug Intell Clin Pharm 1987; 21: 590–3
Farrar HC, Wells TG, Kearns GL. Use of continuous infusion of pralidoxime for treatment of organophosphate poisoning in children. J Pediatr 1990; 116: 658–61
Willems JL, Belpaire FM. Anticholinesterase poisoning: an overview of pharmacotherapy. In: Ballantyne B, Marrs T, editors. Clinical and experimental toxicology of organophosphates and carbamates. Oxford: Butterworth-Heinemann Ltd, 1992: 536–42
Willems JL, De Bisschop HC, Verstraete AG, et al. Cholinesterase reactivation in organophosphorus poisoned patients depends on the plasma concentrations of the oxime pralidoxime methylsulphate and of the organophosphate. Arch Toxicol 1993; 67: 79–84
Casey PB, Gosden E, Blakely L, et al. Plasma pralidoxime concentrations following bolus injection and continuous infusion [abstract]. Przegl Lek 1995; 52: 203
Tush GM, Anstead MI. Pralidoxime continuous infusion in the treatment of organophosphate poisoning. Ann Pharmacother 1997; 31: 441–3
Singh S, Chaudhry D, Behera D, et al. Aggressive atropinisation and continuous pralidoxime (2-PAM) infusion in patients with severe organophosphate poisoning: experience of a northwest Indian hospital. Hum Exp Toxicol 2001; 20: 15–8
Schexnayder S, James LP, Kearns GL, et al. The pharmacokinetics of continuous infusion pralidoxime in children with organophosphate poisoning. Clin Toxicol 1998; 36: 549–55
Green AL, Smith HJ. The reactivation of cholinesterase inhibited with organophosphorus compounds: 1. reactivation by 2-oxoaldoximes. Biochem J 1958; 68: 28–31
Furlong CE, Li W-F, Shih DM, et al. Genetic factors in susceptibiliy: serum PON1 variation between individuals and species. Hum Ecol Risk Assess 2002; 8: 31–43
Kiderlen D. On the phosphoryl oxime hydrolase of plasma, an enzyme that markedly increases the efficacy of oximes in organophosphate poisoning [dissertation]. Munich: Ludwig-Maximilians-University, 2003
Brophy VH, Jampsa RL, Clendenning JB, et al. Effects of 5′ regulatory-region polymorphism on paraoxonase-gene (PON1) expression. Am J Genet 2001; 68: 1428–36
Dawson RM. Review of oximes available for treatment of nerve agent poisoning. J Appl Toxicol 1994; 14: 317–31
Kassa J. Review of oximes in the antidotal treatment of poisoning by organophosphorus nerve agents. J Toxicol Clin Toxicol 2002; 40: 803–16
Marrs TC, Rice P, Vale JA. The role of oximes in the treatment of nerve agent poisoning in civilian casualties. J Toxicol Clin Toxicol 2003; 41: 453–4
Lieske CN, Clark JH, Maxwell DM, et al. Studies of the amplification of carbaryl toxicity by various oximes. Toxicol Lett 1992; 62: 127–37
Lifshitz M, Rotenberg M, Sofer S, et al. Carbamate poisoning and oxime treatment in children: a clinical and laboratory study. Pediatrics 1994; 93: 652–5
Dawson A, Buckley N, Whyte J. What target pralidoxime concentration? J Toxicol Clin Toxicol 1997; 35: 227–8
Mahieu P. Severe and prolonged poisoning by fenthion: significance of the determination of the anticholinesterase capacity of plasma. J Toxicol ClinToxicol 1982; 19: 425–32
Eyer P. Pharmacokinetic aspects for the improvement of oxime therapy [abstract]. Przegl Lek 1995; 52: 202
Eyer P, Worek F, Thiermann H. Easy laboratory tests to follow the acetylcholinesterase status during oxime therapy in organophosphate poisoning. In: XIX International Congress of the European Association of Poison Centres and Clinical Toxicologists; 1999 Jun 22–25; Dublin: 110
Thiermann H, Mast U, Klimmek R, et al. Cholinesterase status, pharmacokinetics and laboratory findings during obidoxime therapy in organophosphate poisoned patients. Hum Exp Toxicol 1997; 16: 473–80
Thiermann H, Szinicz L, Eyer F, et al. Modern strategies in therapy of organophosphate poisoning. Toxicol Lett 1999; 107: 232–9
Worek F, Mast U, Kiderlen D, et al. Improved determination of acetylcholinesterase activity in human whole blood. Clin Chim Acta 1999; 288: 73–90
Zilker T, Felgenhauer N, Hibler A, et al. Factors influencing the efficacy of obidoxime in organophosphate pesticides poisoning. Przeg Lek 1997; 54: 662–4
Riggs DS. The mathematical approach to physiological problems. Baltimore (MD): The William & Wilkins Company, 1963
Eyer P, Kiderlen D, Meischner V, et al. The current status of oximes in the treatment of OP poisoning: comparing two regimes. J Toxicol Clin Toxicol 2003; 41: 441–3
Acknowledgements
The author is grateful to Prof. B. Fichtl for help with the mathematics in figure 13 and to Prof. A. Dawson, Dr M. Eddleston and Prof. E. Reiner for their helpful comments. No sources of funding were used to assist in the preparation of the review. The author has no conflicts of interest that are directly relevant to the content of this review.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Eyer, P. The Role of Oximes in the Management of Organophosphorus Pesticide Poisoning. Toxicol Rev 22, 165–190 (2003). https://doi.org/10.2165/00139709-200322030-00004
Published:
Issue Date:
DOI: https://doi.org/10.2165/00139709-200322030-00004