Genomic Profiling of KRAS/NRAS/BRAF/PIK3CA Wild-Type Metastatic Colorectal Cancer Patients Reveals Novel Mutations in Genes Potentially Associated with Resistance to Anti-EGFR Agents

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
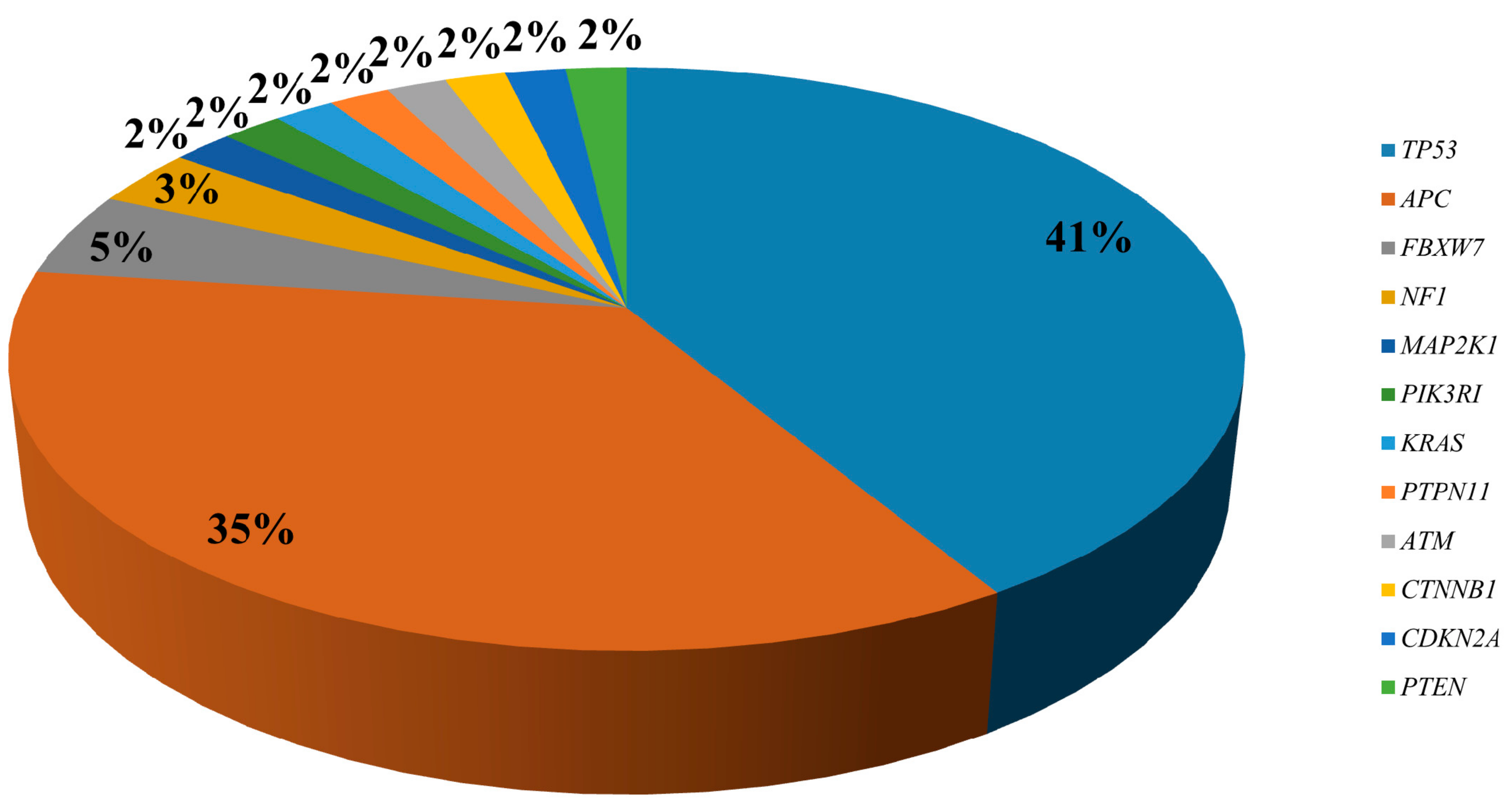
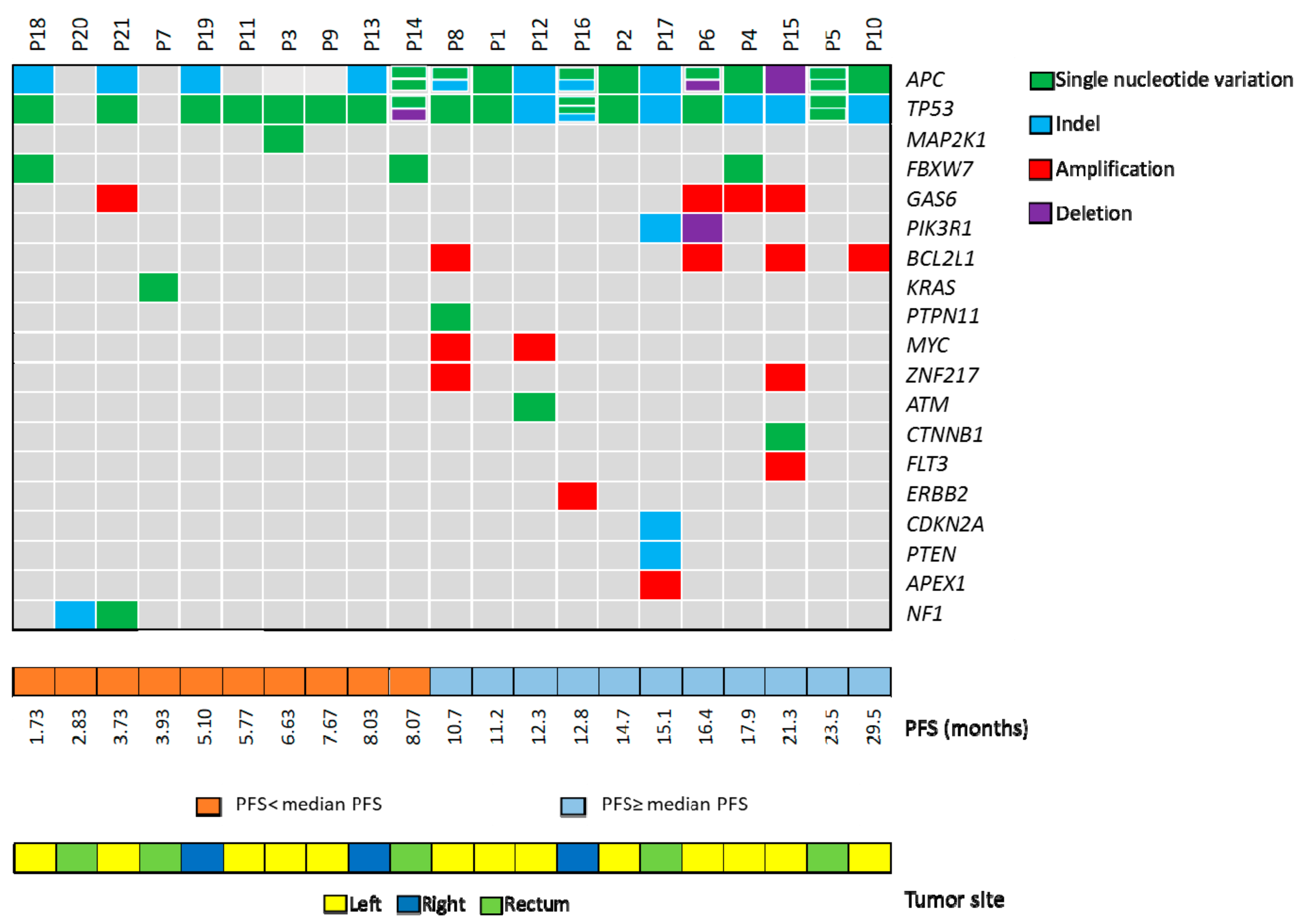
2.1. Targeted Sequencing of KRAS/NRAS/BRAF/PIK3CA Wt mCRC Samples
2.2. Correlation of Genetic Landascape with Patients’ Outcome
2.3. Frequency of Identified Genetic Alterations in CRC
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Targeted Sequencing
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cremolini, C.; Schirripa, M.; Antoniotti, C.; Moretto, R.; Salvatore, L.; Masi, G.; Falcone, A.; Loupakis, F. First-line chemotherapy for mCRC-a review and evidence-based algorithm. Nat. Rev. Clin. Oncol. 2015, 12, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Douillard, J.Y.; Oliner, K.S.; Siena, S.; Tabernero, J.; Burkes, R.; Barugel, M.; Humblet, Y.; Bodoky, G.; Cunningham, D.; Jassem, J.; et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N. Engl. J. Med. 2013, 369, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Lenz, H.J.; Kohne, C.H.; Heinemann, V.; Tejpar, S.; Melezinek, I.; Beier, F.; Stroh, C.; Rougier, P.; van Krieken, J.H.; et al. Fluorouracil, leucovorin, and irinotecan plus cetuximab treatment and RAS mutations in colorectal cancer. J. Clin. Oncol. 2015, 33, 692–700. [Google Scholar] [CrossRef] [PubMed]
- Cremolini, C.; Pietrantonio, F. How the lab is changing our view of colorectal cancer. Tumori 2016, 102, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Tejpar, S.; Morgillo, F.; De Luca, A.; Van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol. 2009, 6, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Arnold, D.; Lueza, B.; Douillard, J.Y.; Peeters, M.; Lenz, H.J.; Venook, A.; Heinemann, V.; Van Cutsem, E.; Pignon, J.P.; Tabernero, J.; et al. Prognostic and predictive value of primary tumour side in patients with RAS wild-type metastatic colorectal cancer treated with chemotherapy and EGFR directed antibodies in six randomized trials. Ann. Oncol. 2017, 28, 1713–1729. [Google Scholar] [CrossRef]
- Bertotti, A.; Migliardi, G.; Galimi, F.; Sassi, F.; Torti, D.; Isella, C.; Cora, D.; Di Nicolantonio, F.; Buscarino, M.; Petti, C.; et al. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 2011, 1, 508–523. [Google Scholar] [CrossRef]
- Cremolini, C.; Morano, F.; Moretto, R.; Berenato, R.; Tamborini, E.; Perrone, F.; Rossini, D.; Gloghini, A.; Busico, A.; Zucchelli, G.; et al. Negative hyper-selection of metastatic colorectal cancer patients for anti-EGFR monoclonal antibodies: The PRESSING case-control study. Ann. Oncol. 2017, 28, 3009–3014. [Google Scholar] [CrossRef]
- De Roock, W.; De Vriendt, V.; Normanno, N.; Ciardiello, F.; Tejpar, S. KRAS, BRAF, PIK3CA, and PTEN mutations: Implications for targeted therapies in metastatic colorectal cancer. Lancet Oncol. 2011, 12, 594–603. [Google Scholar] [CrossRef]
- Pietrantonio, F.; Di Nicolantonio, F.; Schrock, A.B.; Lee, J.; Tejpar, S.; Sartore-Bianchi, A.; Hechtman, J.F.; Christiansen, J.; Novara, L.; Tebbutt, N.; et al. ALK, ROS1, and NTRK Rearrangements in Metastatic Colorectal Cancer. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.; Siravegna, G.; Blaszkowsky, L.S.; Corti, G.; Crisafulli, G.; Ahronian, L.G.; Mussolin, B.; Kwak, E.L.; Buscarino, M.; Lazzari, L.; et al. Tumor Heterogeneity and Lesion-Specific Response to Targeted Therapy in Colorectal Cancer. Cancer Discov. 2016, 6, 147–153. [Google Scholar] [CrossRef]
- Ciardiello, F.; Normanno, N.; Maiello, E.; Martinelli, E.; Troiani, T.; Pisconti, S.; Giuliani, F.; Barone, C.; Carteni, G.; Rachiglio, A.M.; et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: Findings from the CAPRI-GOIM trial. Ann. Oncol. 2014, 25, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Esposito Abate, R.; Lambiase, M.; Forgione, L.; Cardone, C.; Iannaccone, A.; Sacco, A.; Rachiglio, A.M.; Martinelli, E.; Rizzi, D.; et al. RAS testing of liquid biopsy correlates with the outcome of metastatic colorectal cancer patients treated with first-line FOLFIRI plus cetuximab in the CAPRI-GOIM trial. Ann. Oncol. 2018, 29, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Bertotti, A.; Papp, E.; Jones, S.; Adleff, V.; Anagnostou, V.; Lupo, B.; Sausen, M.; Phallen, J.; Hruban, C.A.; Tokheim, C.; et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature 2015, 526, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Marks, J.L.; Gong, Y.; Chitale, D.; Golas, B.; McLellan, M.D.; Kasai, Y.; Ding, L.; Mardis, E.R.; Wilson, R.K.; Solit, D.; et al. Novel MEK1 mutation identified by mutational analysis of epidermal growth factor receptor signaling pathway genes in lung adenocarcinoma. Cancer Res. 2008, 68, 5524–5528. [Google Scholar] [CrossRef]
- Yeh, C.H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Cayre, A.; Manceau, G.; Buc, E.; Bachet, J.B.; Lecomte, T.; Rougier, P.; Lievre, A.; Landi, B.; Boige, V.; et al. Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J. Clin. Oncol. 2009, 27, 5924–5930. [Google Scholar] [CrossRef] [PubMed]
- Loupakis, F.; Pollina, L.; Stasi, I.; Ruzzo, A.; Scartozzi, M.; Santini, D.; Masi, G.; Graziano, F.; Cremolini, C.; Rulli, E.; et al. PTEN expression and KRAS mutations on primary tumors and metastases in the prediction of benefit from cetuximab plus irinotecan for patients with metastatic colorectal cancer. J. Clin. Oncol. 2009, 27, 2622–2629. [Google Scholar] [CrossRef] [PubMed]
- Sartore-Bianchi, A.; Martini, M.; Molinari, F.; Veronese, S.; Nichelatti, M.; Artale, S.; Di Nicolantonio, F.; Saletti, P.; De Dosso, S.; Mazzucchelli, L.; et al. PIK3CA mutations in colorectal cancer are associated with clinical resistance to EGFR-targeted monoclonal antibodies. Cancer Res. 2009, 69, 1851–1857. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra254. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; Rachiglio, A.M.; Lambiase, M.; Martinelli, E.; Fenizia, F.; Esposito, C.; Roma, C.; Troiani, T.; Rizzi, D.; Tatangelo, F.; et al. Heterogeneity of KRAS, NRAS, BRAF and PIK3CA mutations in metastatic colorectal cancer and potential effects on therapy in the CAPRI GOIM trial. Ann. Oncol. 2015, 26, 1710–1714. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Puig, P.; Pekin, D.; Normand, C.; Kotsopoulos, S.K.; Nizard, P.; Perez-Toralla, K.; Rowell, R.; Olson, J.; Srinivasan, P.; Le Corre, D.; et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin. Cancer Res. 2015, 21, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.H.; Cunningham, D.; Werner, B.; Vlachogiannis, G.; Spiteri, I.; Heide, T.; Mateos, J.F.; Vatsiou, A.; Lampis, A.; Damavandi, M.D.; et al. Longitudinal Liquid Biopsy and Mathematical Modeling of Clonal Evolution Forecast Time to Treatment Failure in the PROSPECT-C Phase II Colorectal Cancer Clinical Trial. Cancer Discov. 2018, 8, 1270–1285. [Google Scholar] [CrossRef] [Green Version]
- Normanno, N.; Cervantes, A.; Ciardiello, F.; De Luca, A.; Pinto, C. The liquid biopsy in the management of colorectal cancer patients: Current applications and future scenarios. Cancer Treat. Rev. 2018, 70, 1–8. [Google Scholar] [CrossRef]
- Van Cutsem, E.; Huijberts, S.; Grothey, A.; Yaeger, R.; Cuyle, P.J.; Elez, E.; Fakih, M.; Montagut, C.; Peeters, M.; Yoshino, T.; et al. Binimetinib, Encorafenib, and Cetuximab Triplet Therapy for Patients With BRAF V600E-Mutant Metastatic Colorectal Cancer: Safety Lead-In Results From the Phase III BEACON Colorectal Cancer Study. J. Clin. Oncol. 2019. [Google Scholar] [CrossRef]
- Mao, J.H.; Kim, I.J.; Wu, D.; Climent, J.; Kang, H.C.; DelRosario, R.; Balmain, A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science 2008, 321, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Adua, D.; Di Fabio, F.; Ercolani, G.; Fiorentino, M.; Gruppioni, E.; Altimari, A.; Rojas Limpe, F.L.; Normanno, N.; Pinna, A.D.; Pinto, C. Heterogeneity in the colorectal primary tumor and the synchronous resected liver metastases prior to and after treatment with an anti-EGFR monoclonal antibody. Mol. Clin. Oncol. 2017, 7, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Korphaisarn, K.; Morris, V.K.; Overman, M.J.; Fogelman, D.R.; Kee, B.K.; Raghav, K.P.S.; Manuel, S.; Shureiqi, I.; Wolff, R.A.; Eng, C.; et al. FBXW7 missense mutation: A novel negative prognostic factor in metastatic colorectal adenocarcinoma. Oncotarget 2017, 8, 39268–39279. [Google Scholar] [CrossRef]
- Guinney, J.; Ferte, C.; Dry, J.; McEwen, R.; Manceau, G.; Kao, K.J.; Chang, K.M.; Bendtsen, C.; Hudson, K.; Huang, E.; et al. Modeling RAS phenotype in colorectal cancer uncovers novel molecular traits of RAS dependency and improves prediction of response to targeted agents in patients. Clin. Cancer Res. 2014, 20, 265–272. [Google Scholar] [CrossRef]
- Lupini, L.; Bassi, C.; Mlcochova, J.; Musa, G.; Russo, M.; Vychytilova-Faltejskova, P.; Svoboda, M.; Sabbioni, S.; Nemecek, R.; Slaby, O.; et al. Prediction of response to anti-EGFR antibody-based therapies by multigene sequencing in colorectal cancer patients. BMC Cancer 2015, 15, 808. [Google Scholar] [CrossRef]
- Akitake-Kawano, R.; Seno, H.; Nakatsuji, M.; Kimura, Y.; Nakanishi, Y.; Yoshioka, T.; Kanda, K.; Kawada, M.; Kawada, K.; Sakai, Y.; et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis 2013, 34, 1567–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, Z.; Shao, Y.W.; Lin, P.; Cai, X.; Wang, B.; Ding, Y.; Ma, X.; Wu, X.; Xia, Y.; Zhu, D.; et al. SMAD4 and NF1 mutations as potential biomarkers for poor prognosis to cetuximab-based therapy in Chinese metastatic colorectal cancer patients. BMC Cancer 2018, 18, 479. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Raghav, K.; Loree, J.M.; Morris, J.S.; Overman, M.J.; Yu, R.; Meric-Bernstam, F.; Menter, D.; Korphaisarn, K.; Kee, B.; Muranyi, A.; et al. Validation of HER2 Amplification as a Predictive Biomarker for Anti–Epidermal Growth Factor Receptor Antibody Therapy in Metastatic Colorectal Cancer. JCO Precis. Oncol.ogy. 2019, 1–13. [Google Scholar] [CrossRef]
- Sartore-Bianchi, A.; Amatu, A.; Porcu, L.; Ghezzi, S.; Lonardi, S.; Leone, F.; Bergamo, F.; Fenocchio, E.; Martinelli, E.; Borelli, B.; et al. HER2 Positivity Predicts Unresponsiveness to EGFR-Targeted Treatment in Metastatic Colorectal Cancer. Oncologist 2019. [Google Scholar] [CrossRef]
- Martin, V.; Landi, L.; Molinari, F.; Fountzilas, G.; Geva, R.; Riva, A.; Saletti, P.; De Dosso, S.; Spitale, A.; Tejpar, S.; et al. HER2 gene copy number status may influence clinical efficacy to anti-EGFR monoclonal antibodies in metastatic colorectal cancer patients. Br. J. Cancer 2013, 108, 668–675. [Google Scholar] [CrossRef] [Green Version]
- Bregni, G.; Sciallero, S.; Sobrero, A. HER2 Amplification and Anti-EGFR Sensitivity in Advanced Colorectal Cancer. JAMA Oncol. 2019, 7229. [Google Scholar] [CrossRef]
- Dienstmann, R.; Vermeulen, L.; Guinney, J.; Kopetz, S.; Tejpar, S.; Tabernero, J. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat. Rev. Cancer 2017, 17, 268. [Google Scholar] [CrossRef]
- Sveen, A.; Bruun, J.; Eide, P.W.; Eilertsen, I.A.; Ramirez, L.; Murumagi, A.; Arjama, M.; Danielsen, S.A.; Kryeziu, K.; Elez, E.; et al. Colorectal Cancer Consensus Molecular Subtypes Translated to Preclinical Models Uncover Potentially Targetable Cancer Cell Dependencies. Clin. Cancer Res. 2018, 24, 794–806. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Unselected CRC | KRAS/NRAS/BRAF Wild-Type CRC | |||||
|---|---|---|---|---|---|---|
| Genes | All | Left-Sided | Right-Sided | All | Left-Sided | Right-Sided |
| MAP2K1 | 1.7% (61/3473) | 1.1% (10/878) | 3.1% (13/416) | 2.7% (27/1011) | 1.6% (8/497) | 6% (7/117) |
| NF1 | 4.9% (169/3473) | 3.9% (34/878) | 7.2% (30/416) | 5% (51/1011) | 4.2% (21/497) | 7.7% (9/117) |
| FBXW7 | 12.5% (433/3473) | 13.3% (117/878) | 15.6% (65/416) | 8.5% (86/1011) | 10.3% (51/497) | 10.3% (12/117) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rachiglio, A.M.; Lambiase, M.; Fenizia, F.; Roma, C.; Cardone, C.; Iannaccone, A.; De Luca, A.; Carotenuto, M.; Frezzetti, D.; Martinelli, E.; et al. Genomic Profiling of KRAS/NRAS/BRAF/PIK3CA Wild-Type Metastatic Colorectal Cancer Patients Reveals Novel Mutations in Genes Potentially Associated with Resistance to Anti-EGFR Agents. Cancers 2019, 11, 859. https://doi.org/10.3390/cancers11060859
Rachiglio AM, Lambiase M, Fenizia F, Roma C, Cardone C, Iannaccone A, De Luca A, Carotenuto M, Frezzetti D, Martinelli E, et al. Genomic Profiling of KRAS/NRAS/BRAF/PIK3CA Wild-Type Metastatic Colorectal Cancer Patients Reveals Novel Mutations in Genes Potentially Associated with Resistance to Anti-EGFR Agents. Cancers. 2019; 11(6):859. https://doi.org/10.3390/cancers11060859
Chicago/Turabian StyleRachiglio, Anna Maria, Matilde Lambiase, Francesca Fenizia, Cristin Roma, Claudia Cardone, Alessia Iannaccone, Antonella De Luca, Marianeve Carotenuto, Daniela Frezzetti, Erika Martinelli, and et al. 2019. "Genomic Profiling of KRAS/NRAS/BRAF/PIK3CA Wild-Type Metastatic Colorectal Cancer Patients Reveals Novel Mutations in Genes Potentially Associated with Resistance to Anti-EGFR Agents" Cancers 11, no. 6: 859. https://doi.org/10.3390/cancers11060859